
Abstract
Extranodal NK/T-cell lymphoma (ENKTL) is a well-defined cytotoxic lymphoma strongly associated with Epstein–Barr virus (EBV) infection, commonly affecting the nasopharynx and upper aerodigestive tract. Primary central nervous system (CNS) involvement is rare, and only 17 cases were previously reported in the literature. Here, we report the case of a 44-year-old male admitted with a 3-month history of personality changes and progressive right leg weakness. Brain magnetic resonance imaging studies (MRIs) revealed multiple rim-enhancing brain lesions bilaterally. An extensive clinical and laboratory workup was unrevealing, and 2 brain biopsies were initially considered inconclusive. Pertinently, no systemic lymphoproliferative disorder was identified. The patient initially experienced remarkable clinical improvement with dexamethasone, pulse methylprednisolone, and rituximab therapy. However, he eventually had rapid clinical deterioration, was found to have increased brain lesions, and died nearly 6 months after the initial presentation. During this time, the second brain biopsy was found to show involvement by T-cell lymphoma of NK-cell lineage, which was EBV negative. No post-mortem examination was done to identify any systemic lymphoma. This case serves to expand the spectrum of lymphomas involving the CNS.
Keywords
Introduction
Primary central nervous system lymphoma (PCNSL) is relatively rare, constituting 4% of primary brain tumors and about 2% of non-Hodgkin lymphoma (NHL). Ninety percent of the PCNSL reported are diffuse large B-cell lymphomas (DLBCL) which are about 1.8% of NHL, and the other 0.2% are other types of B-cell lymphoma such as intravascular large B-cell lymphoma and marginal zone B-cell lymphoma. 1 T-cell lymphomas are comparatively rare, constituting about 2% of all PCNSL and 0.4% of all NHL. 2 Extranodal NK/T-cell lymphoma is typically associated with Epstein-Barr virus (EBV), and cases reported in the literature primarily occur in the paranasal region (80%), with a smaller proportion involving the skin (8%) and gastrointestinal (GI) tract (8%). CNS involvement is rare (2% of all extranodal NK/T-cell lymphoma), and the clinicopathological spectrum of NK/T-cell lymphoma involving the CNS is not well documented. 3 A significant risk factor for PCNSL is immunosuppression, especially acquired immunodeficiency syndrome (AIDS), despite introducing highly active antiretroviral therapy (HAART).4,5 To the best of our knowledge, there have been 17 reported cases of extranodal NK/T-cell lymphoma presenting as PCNSL. Here, we describe an additional rare case of extranodal NK/T-cell lymphoma not associated with immunocompromise or EBV infection, highlighting the significance of establishing this rare diagnosis.
Case Report
Clinical presentation
A 44-year-old American Caucasian male with hypothyroidism and bronchial asthma residing in Saudi Arabia presented with a 3-month history of low mood, anxiety, increasing social withdrawal, and difficulty sleeping, initially ascribed to a change in his employment status. The patient decided to seek medical advice at Riyadh’s hospital, King Fahad Medical City (KFMC). He had a brain biopsy from the left frontal lesion under craniotomy and then was discharged home on an oral tapering dose of dexamethasone, but soon as he started decreasing, he became weaker on the right side, worse in the leg. This occurred 4 months after the initial behavioral changes. Readmitted again 3 weeks after the discharge. He denied any history of fever, weight loss, night sweats, nausea, vomiting, raw milk ingestion, smoking, or drug and alcohol use and gave a history of contact with sick cats. The neurological examination revealed normal higher mental functions, language, speech, and cranial nerve function. He had a minimally spastic muscle tone on the right-sided limbs, with weak elbow and wrist flexion on the right side of 4+/5, but 0/5 power on the right lower limb, and normal power on the left, normal reflexes. He could stand and sit with support only.
Radiological and laboratory investigations
The initial basic laboratory investigations were all within normal limits including complete blood count with differential. Brain MRIs revealed multiple intracranial focal mass lesions involving the cortical-subcortical region of bilateral frontal lobes, showing rim enhancement and restricted diffusion in the periphery, and associated with perilesional vasogenic edema (Figure 1, images A, B, C, D, E, and F). Spine MRI showed no intramedullary lesions or leptomeningeal enhancement. Medical management was initiated with dexamethasone to reduce cerebral edema and levetiracetam for suspected seizures. For his behavioral concerns, we gave escitalopram and quetiapine. The infectious diseases team was consulted to look into possible infectious causes. Laboratory investigations were unrevealing for autoimmune and paraneoplastic auto-antibodies, lymphoma, zoonoses, Brucella, Bartonella, Q-fever, Treponema, Histoplasma, and HIV, and no systemic malignancy was identified despite extensive radiological evaluation. The LP (lumbar puncture) showed an opening pressure of 14 cmH2O with normal results, including cytology for the malignant cells and flow cytometry for the lymphoma panel. The slides of the first brain biopsy were sent for a second opinion and were reported to reveal a necroinflammatory process of uncertain etiology. While no acid-fast bacilli, fungi, or toxoplasma were identified on special stains, the lymphocytic infiltrate was notable for being exclusively composed of CD3-positive T-lymphocytes. A second brain biopsy was performed, 2 months after the first one, and the slides were again submitted for external consultation. The workup for occult malignancy was negative, including tumor markers, CT of the chest, abdomen, and pelvis, but multiple enlarged mesenteric lymph nodes on the CT abdomen with central necrosis were noted. Thyroid and scrotum ultrasound showed no malignancy. The whole-body FDG-PET (fluorodeoxyglucose-positron emission tomography) scan showed postoperative changes in the left frontal area with a right frontal hypometabolic lesion and no abnormal FDG uptake in the rest of the body to suggest a neoplastic or infectious process. In the interim, follow-up MRI of the brain showed minimal interval regression of bilateral supratentorial lesions with microhemorrhages (Figure 2, images A, B, and C). At the same time, MRI of the brain with perfusion, spectroscopy, and diffusion tensor imaging documented reduced to absent rCBV (relative cerebral blood volume), reduced rCBF (relative cerebral blood flow), and high rMTT (relative mean transit time), particularly on the left side, reduced NAA (N-acetyl aspartate) peak height, high Ch (choline) peak, high glutamate peak, and variable lipid lactate peaks with changes in fraction anisotropy and tractography (Figure 2, images D, G and H). Cerebral angiogram showed beading of small cortical branches of the anterior cerebral artery suggestive of an inflammatory process (Figure 2, images E and F).

Brain MRI. Image A, and B, DWI (diffusion weighted image) at the corona radiata and centrum semiovale, respectively, demonstrate 2 intra axial hyperintense lesions involving bilateral frontal lobes (arrow head). Image C, and D ADC (apparent diffusion coefficient) map at the same level demonstrate true restriction diffusion at the periphery of the aforementioned lesions (black curved arrow). Surrounding perilesional edema is seen on FLAIR (fluid attenuated inversion recovery) sequences at the same level (straight white arrow) image E, and F.

Brain MRI. Image A, B, and C. T1 post gadolinium enhancing brain MRI demonstrates bilateral supratentorial multifocal rim enhancing lesions at the subcortical region of bilateral frontal lobes (arrow head). Image D, MR (magnetic resonance) tractography at the level of centrum semiovale demonstrate subtle changes (curved white arrow) at the region correspond to the lesion at image A. Image E, and F. Digital subtraction angiography run through the right ICA (internal carotid artery) obtained in AP and lateral view respectively show minimal irregularities involving the cortical branches of the right ACA (anterior cerebral artery) with beaded appearance (white circle). Image G. multivoxel MR spectroscopy at the corona radiata shows area of reduced NAA (N-acetyl aspartate), high Choline and abnormal lipid/lactate peak at the right frontal lobe corona radiata. Image H. Dynamic susceptibility contrast MRI at the level of the corona radiata shows reduced rCBF (regional cerebral blood flow) and rCBV (regional cerebral blood volume) values corresponding to the enhancing lesion at image B.
Interim treatment
Given the possibility of primary CNS vasculitis, the patient was started on pulse steroids with intravenous methylprednisolone 1 g daily for 5 days. He experienced a significant improvement in mood and power on the right side and started walking with the help of a cane and wearing foot orthosis for the foot drop. However, his over anxiety, hyper-alertness, and low mood he would refer to as “terrible,” the right distal leg weakness with foot drop and polyuria persisted. Rituximab infusion of 1000 mg was started after completing pulse intravenous steroids, but only 1 dose was given as the patient developed a chest infection. He gradually deteriorated neurologically, experienced focal seizures, and became encephalopathic. A follow-up brain MRI showed an increase in enhancing lesions and increasing vasogenic edema.
Histopathology findings
Histologic sections showed extensively necrotic brain tissue involved by an atypical, angiocentric lymphoproliferative process consisting of small to medium-sized lymphocytes with slightly pleomorphic nuclei and clumped chromatin. The atypical lymphoid cells demonstrated CD2, CD3, CD7, granzyme B and TIA 1 expression by immunohistochemistry, and were negative for CD4, CD5, CD8, CD56, TCR beta F1, and TCR delta. Epstein-Barr virus (EBV) encoded RNA (EBER) in situ hybridization was also negative (Figure 3). The morphologic and immunophenotypic findings supported the diagnosis of peripheral T-cell lymphoma.
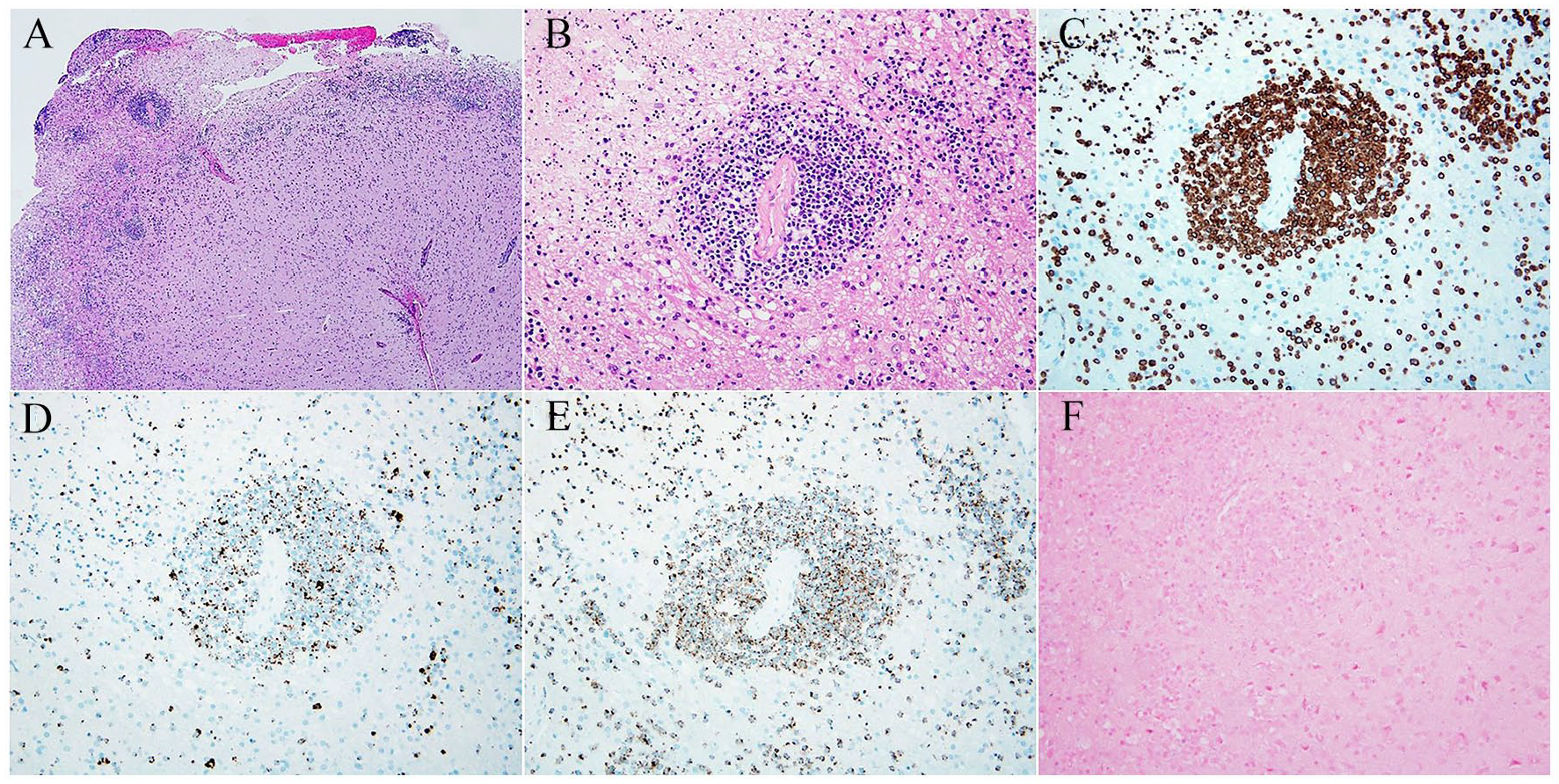
Histologic sections demonstrate a necrotic, infiltrative neoplasm with an angiocentric pattern involving the brain parenchyma, consisting of atypical, pleomorphic lymphoid cells (A, B). The lymphoma cells show CD3 (C), granzyme B (D) and TIA 1 (E) immunoexpression. In situ hybridization for EBV-encoded RNA (EBER) is negative (F).
Follow-up and clinical outcome
The patient developed pulmonary embolism, sepsis, and disseminated intravascular coagulation with multiorgan failure. On follow-up brain CT, he was found to have a large left frontoparietal intraparenchymal hematoma, associated with significant vasogenic edema, midline shift, and diffuse loss of gray-white differentiation throughout the brain parenchyma and effacement of sulci. Unfortunately, he passed away from cardiopulmonary arrest 6 months after his initial presentation.
Discussion
Primary central nervous system lymphoma (PCNSL) is rare, although the incidence appears to be increasing even among immunocompetent patients, possibly due to the advancement in diagnosis. 6 Based on the Central Brain Tumor Registry of the United States (CBTRUS), the average annual age-adjusted incidence rate of all primary malignant and non-malignant brain and other CNS tumors from 2013 to 2017 was 23.79 per 100 000 person-years with an incidence of the PCNSL 1.9/100 000 person-years. 7 Lukes and Collins 8 were the first to recognize non-Hodgkin lymphoma (NHL) as an immune system tumor in the 1970s, and the subsequent Kiel classification proposed by Lennert et al. 9 has since dominated clinical practice. In 1994, the International Lymphoma Study Group (ILSG) determined the most common type of NHL to be diffuse large B-cell lymphoma (DLBCL), which constituted about 90% of PCNSL, while T-cell and natural killer (NK) cell disorders were only 12%. 10 Swerdlow et al 11 reported extranodal NK/T-cell lymphoma (ENTCL) as constituting only 2% of all NHL, most of which affect the nose and paranasal sinuses, and a smaller proportion involves extra-nasal sites, including the skin and gastrointestinal tract. The most common type of PCNSL is DLBCL, while T-cell PCNSL is the least common and remains poorly understood and difficult to diagnose and treat. Based on reported cases of NK/T-cell PCNSL (summarized in Table 1), the age at presentation ranged from 13 to 77 years, with a median age of 46.2 years, predominantly affecting Asian and African American patients. The presenting symptoms included headaches, cognitive impairment, and weakness. It showed multiple different locations and diagnoses confirmed by the detection of histopathology of NK-cell lymphoma. All but 2 reported cases were immunocompetent, and all tested cases were shown to be associated with Epstein Barr Virus infection. The diagnoses were established either on biopsy or by the detection of malignant cells in the cerebrospinal fluid.12-25
Seventeen reported cases of CNS NK/T-cell lymphoma with their characteristics.
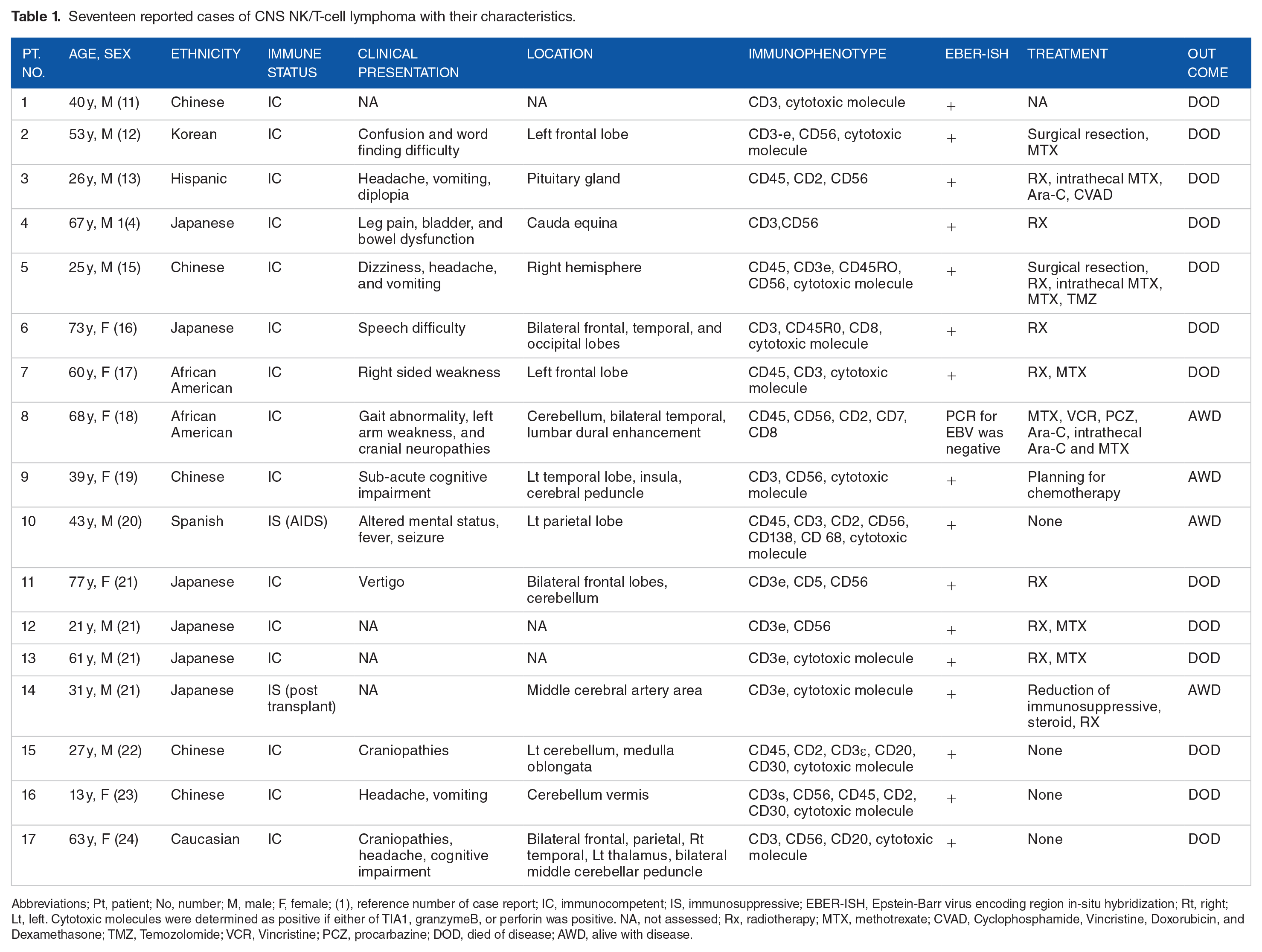
Abbreviations; Pt, patient; No, number; M, male; F, female; (1), reference number of case report; IC, immunocompetent; IS, immunosuppressive; EBER-ISH, Epstein-Barr virus encoding region in-situ hybridization; Rt, right; Lt, left. Cytotoxic molecules were determined as positive if either of TIA1, granzymeB, or perforin was positive. NA, not assessed; Rx, radiotherapy; MTX, methotrexate; CVAD, Cyclophosphamide, Vincristine, Doxorubicin, and Dexamethasone; TMZ, Temozolomide; VCR, Vincristine; PCZ, procarbazine; DOD, died of disease; AWD, alive with disease.
A recent review reported a collection of 18 cases of primary CNS T-cell lymphomas. 26 Fifteen cases were classified as peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS), 2 of which were of γδ T-cell derivation and 1 was TCR silent; there was 1 anaplastic large cell lymphoma (ALCL), ALK-positive and 2 ALCL, ALK-negative. 26
Our patient had an unusual clinical presentation, with psychological and cognitive symptoms, until a brain MRI showed multiple brain lesions, which were thought to be an NHL until the biopsy was non-conclusive. The patient had not experienced B-symptoms, which is consistent with most published cases of NK/T-cell lymphoma. In 1 study evaluating 14 cases (4 newly reported and 10 reported reviewed), none of the patients had B symptoms; however, just like our patient’s clinical course, all patients exhibited aggressive clinical behaviors. 22
In our case, the patient received a short course of dexamethasone, which might have complicated the pathological evaluation. The presence of CD3-positive T-lymphocytes is not sufficient for diagnosing T-cell lymphoma, as these may reflect an inflammatory or infectious process. Also unusual in our case is the absence of EBV infection and lack of immunosuppression, associated with the most reported T-cell PCNSL. 22 There are no specific radiographic features or locations related to the CNS NK/T-cell lymphoma. In the study mentioned above, a brain lesion was found throughout the brain in all patients, involving the temporal lobe, the frontal, 1 case of a pituitary gland 2 of the leptomeningeal lesion. 22 There is no consistency in the reported treatment regimens for NK/T-cell PCNSL, extensive variability in the treatment regimens when reported, and the detailed treatment protocols have not been mentioned in about half of the cases reported. The overall survival (OS) of the published cases was 4-29 months (median, 19 months), and our patient was deceased 6 months after initial diagnosis, in keeping with the reported dismal prognosis for these patients.
In summary, our report of a rare case of T-cell PCNSL serves to add to the small number of cases reported in the literature thus far. Further, possibly multi-institution studies evaluating more extensive series of these patients may help develop more comprehensive management guidelines and possibly help identify targeted therapies.
Learning Points
Primary CNS lymphoma of NK/T-cell types is extremely rare, but has an aggressive clinical course and high mortality.
A high index of suspicion is needed to evaluate infiltrating T-lymphocytes seen in the biopsy, mandating relevant ancillary testing, as well as extensive systemic evaluation to exclude inflammatory processes.
The use of pre-operative steroid therapy may alter the biopsy findings, further making the diagnosis challenging.
A multidisciplinary approach is needed to establish an accurate diagnosis and treatment regimen.
Footnotes
Acknowledgements
We are deeply indebted to King Fahad Medical City’s nursing staff for their excellent care provided to the patients. Furthermore, we appreciate all the specialties involved in this patient’s multidisciplinary care, especially neurology, neurosurgery, ID, endocrinology, rheumatology, Neuro Critical Care Unit, physical and occupational therapists, and ancillary services of Medical Imaging and Laboratory Departments.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Naim I Kajtazi has contributed with case report design, planning, scanned files review, writing patient history, investigations, treatment, follow up, interpretation of data, the intellectual content of the manuscript, coordinated work with other co-authors, and submission on behalf of all of them. Mohammed Bafaquh has contributed with literature review, discussion part, and whole manuscript revision with the intellectual content of the manuscript. Juman Al Ghamdi has contributed with case report design, review, and editing of images and intellectual content of the manuscript. Zahra AlEissa has contributed to the literature review, discussion part, and intellectual content of the manuscript. Arwa Al Shmeikh, has contributed to the intellectual content of the manuscript. Ali Alsaeed has contributed to the literature review. Tarek Sulaiman has contributed to the intellectual content of the manuscript. M Adelita Vizcaino has contributed to the manuscript content and with the review and illustration of the histopathologic findings. Majed Al Hameed has contributed to the intellectual content of the manuscript. Aditya Raghunathan has contributed with case report design, interpretation, and review of histopathology data, literature review with discussion part, and supervised the work with other co-authors.