
Abstract
Background
Germline mutations in high-penetrance genes contribute to the development of colorectal cancer with a >1% familial risk. The proband with a family history of cancer was at an increased risk for colorectal cancer development. However, data on the germline mutational landscape in non-Western populations, particularly in India, remain limited.
Objective
This study aimed to investigate the prevalence of germline mutations in individuals fulfilling clinical criteria for suspected Lynch Syndrome (LS) or familial adenomatous polyposis (FAP) using a targeted multigene next-generation sequencing (NGS) approach.
Method
Mutation analysis was performed for 110 suspected Lynch syndrome and 12 familial adenomatous polyposis (FAP) and attenuated FAP (AFAP) patients. Sanger sequencing was employed to validate pathogenic, likely pathogenic, and novel variants.
Results
Pathogenic mutations were identified in 20% of suspected Lynch syndrome cases and 66% of FAP cases, indicating a stronger genetic contribution in FAP. Notably, this is the first study to report a FANCD2 mutation in FAP. Most LS-associated mutations occurred in mismatch repair genes (MLH1, MSH2, MSH6), while APC and MUTYH mutations predominated in FAP. Four novel mutations were identified: MLH1 c.676delC, MLH1 c.378delC, MDC1 c.2839C>T, and RAD52 c.1215delG. Functional prediction of 100 VUS revealed several potentially disruptive variants, notably BLM P866L, which showed significant loss of protein–protein interactions. A pathogenic FANCD2 mutation was identified in one FAP family, suggesting a broader spectrum of contributing genes.
Conclusion
This study demonstrates the utility of multigene NGS panels in detecting clinically actionable germline mutations in suspected hereditary CRC cases in an Indian population. The identification of novel and ethnically distinct variants highlights the need for population-specific genetic databases and reinforces the role of genetic screening and counseling in cancer prevention and early diagnosis.
Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide and the second leading cause of cancer-related deaths in the world. 1 It accounts for 10% of all cancers, affecting approximately 1 million people worldwide every year. 2 In India, colon cancer is the fourth most common cancer in males and the fifth most common in females. 3 The etiology of CRC involves both environmental and genetic factors, with nearly 35% of cases attributed to hereditary predisposition. Approximately 70%-80% of CRC cases are considered sporadic, with no apparent hereditary component. In contrast, familial CRC—defined by the presence of two or more first-degree relatives affected by CRC—accounts for about 20%-30% of cases. Individuals with a family history of CRC have a three- to six-fold increased risk of developing the disease compared to those without such a history. 4 Among hereditary CRC syndromes, Lynch syndrome (previously referred to as hereditary non-polyposis colorectal cancer, HNPCC) and familial adenomatous polyposis (FAP) are the most prevalent (Figure 1). Other subtypes include MutY homolog (MUTYH) associated polyposis (MAP), Peutz-Jeghers syndrome (PJS), juvenile polyposis, and Cowden/PTEN hamartoma syndromes, among others. 5
Schematic Representation of Colorectal Cancer Risk.
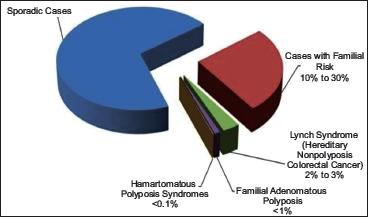
The Lynch Syndrome is an autosomal dominant disorder caused by pathogenic germline mutations in DNA mismatch repair (MMR) genes: MLH1, MSH2, MSH6, PMS2, and EPCAM. It is characterized by an increased predisposition to early-onset colorectal and endometrial (uterine) cancers, as well as other extracolonic tumors. 6 Approximately 70% of the mutations are known to be identified in MLH1 and MSH2 genes.7, 8 A mutation in any of these genes gives a person an increased lifetime risk of developing colorectal cancer and other related cancers.
A variant called Muir Torre Syndrome is associated with an increased risk of certain skin tumors, such as sebaceous gland neoplasms (sebaceous adenomas and carcinomas) and/or neoplasms of the hair follicle (keratoacanthomas). Mutations in any of the MMR genes can be found in these patients, but the MSH2 mutation is most commonly seen. 9 A second variant, called Turcot Syndrome, is associated with certain brain tumors. These families may have LS (associated with glioblastomas) or familial adenomatous polyposis (associated with medulloblastomas). 10
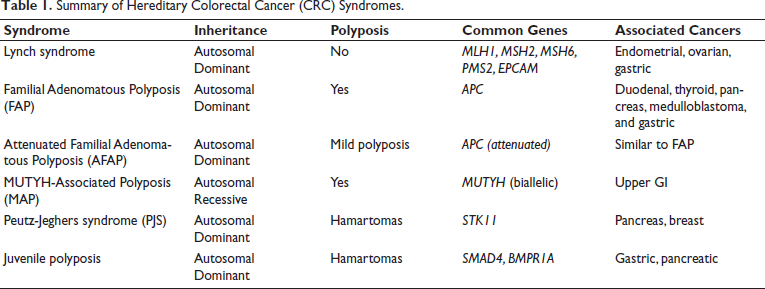
Familial adenomatous polyposis (FAP) is a polyposis syndrome caused by germline mutations in the APC gene, characterized by the development of hundreds to thousands of colorectal adenomas. Somatic mutations in APC are a key molecular event in sporadic colorectal cancer, present in about 80% of patients. Usually, about 25% of people with FAP harbor a de novo mutation in APC without any clinical or genetic evidence of FAP in the family. Attenuated FAP (AFAP) is a milder form of classic FAP with less polyps (<100) and a later age of polyp/cancer onset. 11 The summary of hereditary CRC syndromes is given in Table 1.
Summary of Hereditary Colorectal Cancer (CRC) Syndromes.
Advances in molecular genetics now allow a comprehensive evaluation of cancer susceptibility genes. Traditional methods like Sanger sequencing are limited by time and scalability. In contrast, Next Generation Sequencing (NGS) permits simultaneous analysis of multiple genes with high sensitivity and cost-effectiveness. This study aims to assess germline mutations in individuals with clinical suspicion of Lynch syndrome or FAP using a 56-gene multigene panel NGS approach in a South Indian cohort.
Materials and Methods
Patient Samples
A 10 ml peripheral blood sample was collected from 122 patients who attended the Hereditary Cancer Clinic at the Cancer Institute (WIA), Chennai, India, from the period of 2002 to 2016. A detailed questionnaire consisting of details regarding family history, existing medical conditions, and informed consent was obtained from each patient recruited in the study. Genetic testing was conducted for patients and families who met either the Amsterdam Criteria I or II or the Bethesda guidelines. In our study, almost all of the patients and/or the families met the Bethesda criteria.
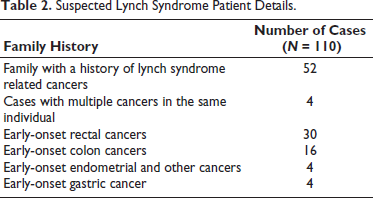
The patient details for all suspected lynch syndrome (N = 110) cases and FAP (N = 12) cases are given in Tables 2 and 3.
Suspected Lynch Syndrome Patient Details.
FAP/AFAP Patient Details.

Methods
Lymphocytes were isolated from the collected peripheral blood sample and stored at –20 °C until DNA extraction. Genomic DNA was isolated using QIAamp DNA Easy Kit (Qiagen, GmbH, Germany) according to the manufacturer’s protocol. Briefly, cells were lysed using AL buffer and proteinase K. DNA was precipitated with ethanol and purified using spin columns and wash buffers. Finally, DNA was eluted in 100-200 µL of AE buffer.
NGS Library Preparation
Approximately 1.0 µg of the genomic DNA was fragmented using the Bioruptor sonication system to a target size of 150 base pairs. The library was prepared using Agilent Sure select XT2 kit (Agilent Technologies Inc., USA) according to the manufacturer’s instruction. The libraries were pooled and hybridized with a custom gene panel. The Sureselect custom enrichment kit was used to capture 2.4 Mb targeted sequences, which includes 2 kb upstream and downstream of the 56 genes.
Cluster Generation and Sequencing
The concentration of the prepared DNA templates was adjusted to 2 nM and then denatured by adding 0.1 N NaOH. Template DNA at a final concentration of 8 PM was subjected to cluster generation using cBOT instrument (Illumina, Inc., USA), on a paired-end flow cell v.3.0. Sequencing was performed using SBS version 3.0 reagents, on a HiScan SQ instrument (Illumina, Inc., USA).
Data Analysis
The raw .bcl files generated by the sequencer were converted to FASTq files using the Casava software (Illumina, Inc., USA). CLC Bio suite (CLC Bio, Aarhus, Denmark) version 5.5 was used for further processing of the data. Alignment was performed using the complete human reference genome (GRch37).
Validation of the Identified Mutations by PCR-Sanger Sequencing
Sanger sequencing was performed only for validation of truncating (nonsense, frameshift), splice-site, Pathogenic missense, and novel variants detected by NGS. Benign missense variants, VUS, or known polymorphisms were not routinely validated unless deemed clinically significant. PCR was performed for the genes in which deleterious mutations were identified. Specific gene primers were used to amplify the DNA for 35 cycles using the following cycling conditions: initial denaturation at 95 °C for 5 min, denaturation at 94 °C for 10 s, annealing at 60 °C for 30 s, extension at 72 °C for 45s, and final extension at 72 °C for 5 minutes. Amplified products were resolved on 2% agarose gel and visualized.
PCR products intended for sequencing were purified using AmPure XP beads to remove residual primers and dNTPs, following the manufacturer’s instructions. Sequencing was performed bi-directionally using Big Dye terminator kit v3.1 Cycle sequencing kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol.
Prediction of Functional Effects of VUS Mutation
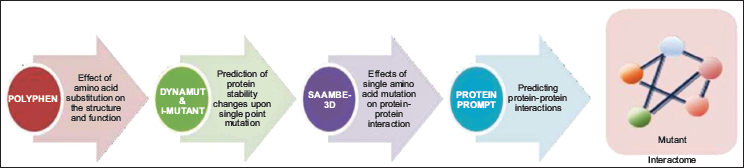
Variants were classified into 5 categories: Pathogenic, likely pathogenic, variant of unknown significance (VUS), likely benign, and benign according to ACMG Guidelines. The structural and functional prediction for about 100 VUS that conferred single amino acid change were predicted using PolyPhen-2 (Polymorphism Phenotyping v 2.0) and DynaMut. PolyPhen-2 predicts the potential impact of an amino acid substitution on the structure and function of a human protein, while DynaMut evaluates the protein stability validated by i-mutant. Additionally, SAAMBE-3D predicts the effect of single amino acid modification on protein–protein interaction. Protein prompt predicts the list of proteins that interact with the mutated protein12–16 (Figure 2).
Results
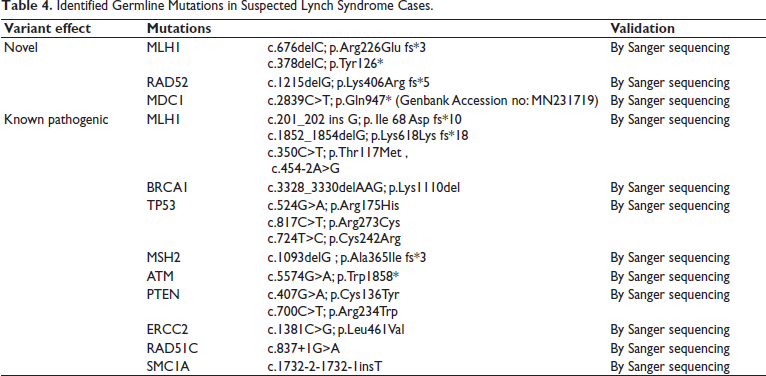
We developed a comprehensive 56-gene panel (Supplementary 1) covering most of the genes involved in DNA repair mechanisms. These genes have been previously implicated in various hereditary cancer syndromes. This panel enabled us to identify both commonly and rarely mutated genes, including those associated with lower cancer risk. All the patients undergoing clinical care at the Cancer Institute (WIA) for GI cancer were referred to the hereditary cancer clinic, and blood samples were collected after fulfillment of the Bethesda criteria. A total of 122 unrelated families were recruited into this study, which included 110 cases that had suspected Lynch syndrome associated disease and 12 patients with FAP and attenuated FAP. Among the 110 suspected Lynch syndrome cases, 52 (47%) patients had a family history of cancer. Deleterious mutations in at least one of the 56 screened genes were identified in 16 of these 52 patients (32%). Six patients with early-onset colorectal cancer harbored a deleterious mutation. Some unrelated families shared identical mutations, while a few individuals carried multiple deleterious variants. Overall, 20% (22 of 110) of the patients carried deleterious mutations, and the rest 80% (88 of 110) did not carry any deleterious mutation. Among the 56 genes, MLH1, MSH2, MSH6, PMS2, BRCA1, BRCA2, APC, MUTYH (biallelic), and TP53 were classified as high-penetrance genes. Germline mutations identified in these genes are given in Table 4. Notably, four novel mutations were identified in the genes MLH1 c.676delC; p.Arg226Glu frameshift and c.378delC; p.Tyr126*: MDC1 c.2839C>T; p.Gln947* and RAD52 c.1215delG; p.Lys406Arg frameshift. Out of the four novel mutations, MDC1 p.Gln947* was submitted to the NCBI-Genbank tool, whereas the other three mutations were not submitted due to their limitation of nucleotide size in the submission tool. Splice-site variants were found in RAD51C, MLH1, SMC1A, and APC across three families with a suspected Lynch syndrome and one early-onset rectal cancer case without a family history. Our study did not observe any mutations in PMS1 and MSH3 genes, which are also known to be mutated in Lynch syndrome.
Identified Germline Mutations in Suspected Lynch Syndrome Cases.
In total, 295 known single nucleotide polymorphisms and 100 variants of uncertain significance were identified in the 110 patients (Table 5). More than one Variant of uncertain significance (VUS) was found in PMS2, ERCC5, MDC1, FANCD2, MSH3, MSH6, BRIP1, MSH2, MRE11A, SDHA, PALB2, POLH, and RAD50 genes (Figures 3 and 4).
Germ Line VUS (N = 100) Identified by NGS 56 Gene Panel.
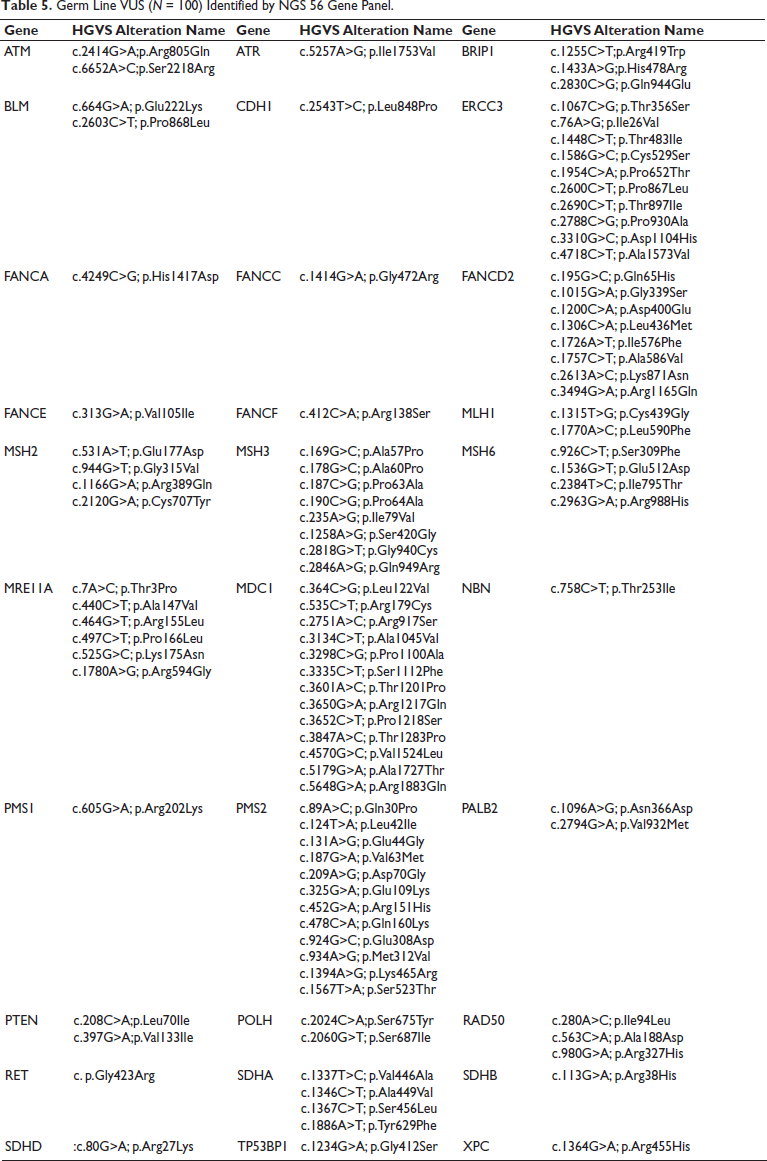
Graphical Representation of Number of SNPs Identified.
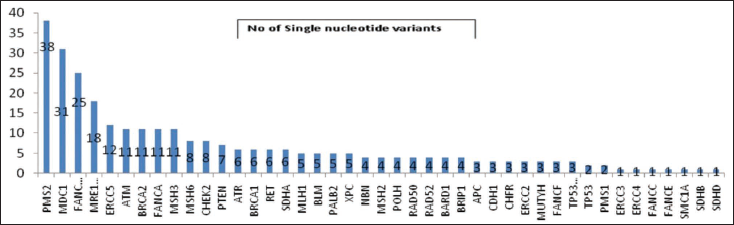
Graphical Representation of Number of VUS Identified.
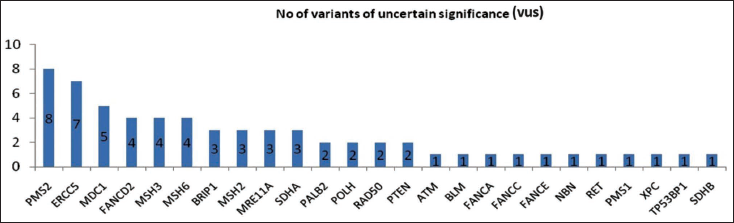
Among the 12 patients with FAP or attenuated FAP, 9 (75%) had a family history of cancer. Deleterious mutations were detected in 5 of these individuals (55%), while 3 additional cases with no family history also carried mutations, totaling 8 patients (66%) with pathogenic variants (Table 6). Two frameshift and one nonsense deleterious mutations were seen in APC gene: c.4605_4606insG; p.Glu1536Gly frameshift, c.4313delC; p.Thr1438Asn frameshift, and c.1987C>T; p.Q663*. Biallelic mutations in MUTYH gene-c.721C>T; p.Arg241Trp and c.1396G>T; p.Glu466* were observed in two attenuated FAP patients. One patient with FAP carried splice variants in APC c.645+1G>A and ATR c.7349+2T>C genes.
Identified Germline Mutation in FAP or Attenuated FAP.

A notable case involved a patient with FAP diagnosed at 29 who later developed rectal cancer at 38 and harbored a FANCD2 insertion (c.919_920insC; p.Leu307del), classified as a VUS in ClinVar. This mutation was also found in two first-degree relatives. Predictive testing revealed that the elder son (who carried the mutation), developed FAP at age 19, while the younger son (non-carrier), remained unaffected. This is the first report linking a FANCD2 mutation with FAP.
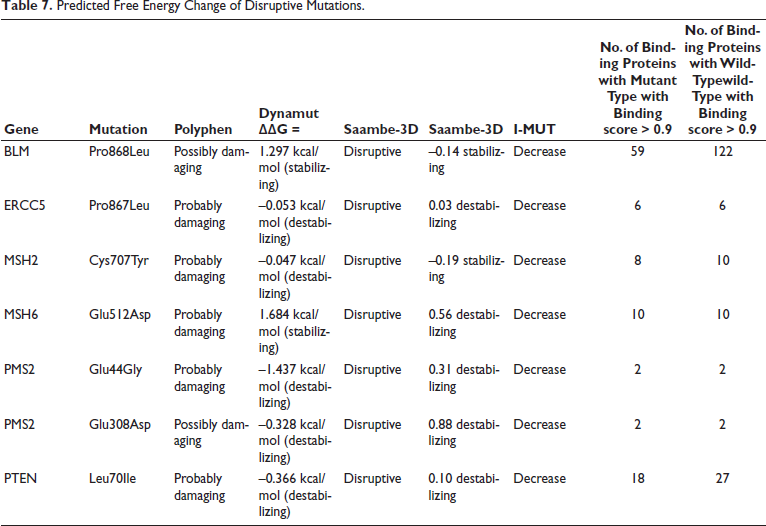
The functional impact of the identified VUS mutations was predicted using Polyphen2. Among the 100 VUS mutations, Polyphen2 results showed that 40 were probably damaging mutations, 16 were possibly damaging mutations, and the remaining 44 were benign missense mutations. Only 35 out of 56 damaging mutations with PDB structures qualified for DynaMut analysis. The free energy (∇∇G) of the predicted mutant protein structure was used to determine the stability of the mutant protein structure, which showed 19 stabilizing and 16 destabilizing structures. SAAMBE-3D analysis showed 7 disruptive mutations and 23 non-disruptive mutations out of 35 predicted protein structures (Table 7). Among the 7 disruptive mutants, 5 mutants in PTEN, ERCC5, MSH6, and PMS2 were observed to have destabilizing structures, and 2 variants in BLM and MSH2 had a stabilizing protein structure. The protein prompt prediction showed that BLM mutant had a significant loss of protein–protein interactions, that is, BLM wild-type protein interacts with 100 other proteins (binding score > 0.9), whereas the mutant BLM P866L interacts with only 59 proteins (binding score >0.9). The major structural variation of BLM P866L was identified using DynaMut (Figures 5a and 5b). The mutant MSH2 resulted in loss of interaction with only 2 proteins, and the PTEN variant, although it showed a loss of 9 protein–protein interactions, is still considered insignificant due to a destabilizing structure.
Predicted Free Energy Change of Disruptive Mutations.
The clinico-pathological characteristics of the individuals with suspected Lynch syndrome associated cancers are given in Table 8. A total of 110 patients were included in the evaluable cohort. The age at diagnosis ranged widely, with the majority of patients between 31-40 years (38%), followed by 21-30 years (24%), and 41-50 years (20%). Only 6% of patients were younger than 20 years, and 12% were older than 50 years at diagnosis. Stage II was the most frequent disease status at presentation, accounting for 35 patients (31%), while Stages III and IV were observed in 18% each. A positive family history of cancer was reported in 48% of patients (n = 52), while 54% (n = 58) had no known family history. At diagnosis and during follow-up, synchronous tumors were identified in 8 patients (7%), while metachronous tumors were seen in 24 (22%). The remaining 78 patients (71%) did not develop another primary tumor.
Description of the Cohort – Suspected Lynch Syndrome Associated Cancer.
Out of the total cohort, 31 patients were reported to have colon tumors. Among these, 61% (n = 19) had tumors located in the distal colon, while 39% (n = 12) had proximal colon tumors. Regarding disease status, 44 patients (40%) were disease-free at follow-up, whereas 22 (20%) had died due to the disease. A notable portion (35%) was defaulted for treatment, and 5 patients (4%) were alive with active disease. We identified 25% (10 out of 40) of individuals with deleterious mutations were found to be free of disease, while the remaining 75% (30 out of 40) had persistent or progressive disease. Among those with deleterious mutations, 22% (5 out of 22) had died due to the disease. Of the five patients who died, three had been diagnosed with more than one primary cancer, and the remaining two experienced disease relapse.
Discussion
Using NGS targeted resequencing, we identified pathogenic germline mutations in high and moderate penetrance cancer-susceptible genes in approximately 20% of patients with suspected Lynch syndrome and 67% of those with FAP or AFAP. These detection rates align with global studies, where germline mutation rates in Lynch syndrome cohorts vary between 15%-30%, depending on population genetics, screening criteria, and the number of genes analyzed.17, 18
A total of 72% of identified germline mutations were found in mismatch repair genes, which are known to cause Lynch syndrome. This is consistent with the Mismatch repair gene variant database (InSiGHT), where approximately 90% mutations were identified in MLH1 and MSH2 genes, and the rest 10% were found in MSH6 gene. 19 Mismatch repair (MMR) mechanism plays a key role in maintaining genomic stability by repairing post-replication errors in DNA. MMR proteins: MLH1, MSH2, MSH6, MSH3, PMS1, and PMS2 are involved in diverse genetic pathways through interaction with different proteins. Mutation in MMR genes alters the genomic stability and results in microsatellite instability in many human cancers, such as lynch syndrome and sporadic colorectal cancers. 20 MMR proteins suppress homologous recombination and are found to be involved in DNA damage response by regulating the ATM and TP53 proteins.21, 22
Among the three MLH1 frameshift mutations, c.676delC; p. Arg226Glu frameshift was identified as novel, that is, it has not been reported in any of the public databases such as ClinVar, LOVD, Insight, UMD, and ExAc databases. The two known mutations c.201_202insG; p. Ile68Aspframeshift and c.1852_1854delG; p.Lys618Lys occur within functional domains critical for ATPase activity and interaction with PMS2/MLH3/ PMS1, potentially impairing MLH1 protein. 23 One novel frameshift mutation was identified in MSH2 gene at codon 365. This mutation truncates the protein chain of the MSH2 gene. Though the mutation does not fall in any of the functional domain regions of the gene, it can be considered as pathogenic due to the truncating nature of the mutation. In MSH6, a codon 540 deletion mutation was reported as a variant of uncertain significance in the databases. This mutation was seen in a proband with endometrial cancer diagnosed at the age of 59 years. Our result is in concordance with other studies, which have reported that the risk for endometrial or ovarian cancer in women with MSH6 mutation is more compared to the risk for colorectal cancer. 24 The proband with MSH6 mutation underwent staging laparotomy followed by six cycles of adjuvant chemotherapy and then pelvic brachytherapy. The proband had a good clinical response to treatment and remains disease free.
Of all the variants identified, 58% were found in the MMR gene PMS2, with 17% of these classified as VUS. Due to the high frequency of pseudogenes near to the PMS2 gene, it is very difficult to assess its significance. 25 Microsatellite instability analyses performed for suspected lynch syndrome cases showed high MSI for probands with pathogenic mutations in MMR genes. In our earlier study by Rajkumar et al., wherein MSI analysis was performed for 31 colorectal cancer patients, 12 patients were found to be MSI high, but only five of them had the pathogenic MMR mutation. 26 Thus, to enhance the diagnostic sensitivity, integrating MSI and mutational analysis is important. In this study, 1% of the mutation is in BRCA1/2 gene in colorectal cancer, which is similar to a study by Yurgelun et al., in Lynch syndrome cohort. 27 The patient with a germline BRCA1 mutation also had a pathogenic APC c.1987C>T; p.Q663* mutation and was diagnosed to have FAP, who later developed CRC. In some patients, the risk of developing CRC increases due to the presence of co-occurring mutations in moderate penetrance genes. 27
Consistent with previous studies, a biallelic MUTYH inherited mutation was seen in 16% (2 of 12) of FAP/attenuated FAP cases, and they did not harbor germline APC mutations.28–31 Therefore, the autosomal recessive inheritance pattern of MUTYH mutation increases the risk for CRC. 32 We also identified six deleterious mutations in the APC gene exclusively among FAP cases. Given our small sample size (n = 12), no statistically significant genotype-phenotype correlation could be established. However, the discovery of a FANCD2 mutation in a patient with FAP underscores the expanding genetic landscape of polyposis syndromes. FANCD2, part of the Fanconi anemia pathway, participates in DNA cross-link repair, and its dysfunction may synergize with APC-related tumorigenesis.33, 34
In a study by Akbari et al., 656 women with malignant ovarian cancer were screened for MSI and alterations in MMR genes of which only 0.6% carried pathogenic MMR mutations. 35 In concordance with their study, only one pathogenic mutation in MMR gene (MLH1) was identified in a patient with ovarian cancer. 35 This supports the notion that Lynch syndrome accounts for <1% of ovarian cancer cases globally. 36
Thus, it is recommended to identify the deleterious mutations in intermediate risk genes that either directly or indirectly increase the risk of developing cancer during their lifetime. It is important to do risk-factor surveillance by genetic testing and suggest the required screening methods, such as colonoscopy and sigmoidoscopy, as a preventive measure for high-risk individuals. It is also equally important to give the recommended genetic counseling to deleterious mutation carriers, which may prevent or help in the early diagnosis of hereditary cancers. 37
A notable finding in this cohort was that the majority of patients were between 31 and 40 years of age. This skew toward younger adults may indicate a higher burden of early-onset cancer in the population studied. Importantly, early-onset cancers have been increasingly recognized for their unique molecular profiles and familial associations, which is consistent with the 48% reporting a positive family history of cancer in this cohort. 38 The presence of metachronous tumors in 22% of patients and synchronous tumors in 7% reflects the importance of long-term surveillance in cancer survivors, especially those with familial risk. 39 Among the 31 patients with colon tumors, a distal tumor site was more common (61%) than proximal (39%), aligning with global data and suggesting higher rates of distal colorectal cancer, particularly in younger populations. This supports the growing evidence of shifting colorectal cancer patterns, which may have implications for screening strategies and molecular testing.40, 41
Predicting the functional effects of VUS mutations has revealed the possible protein–protein interactions with the mutant protein. Among the 7 disruptive mutations, the protein prompt results showed that 4 mutant proteins of ERCC5, MSH6, and PMS2 do not lose any interaction compared with the wild type, whereas 3 disruptive mutations in BLM, MSH2, and PTEN lose interaction with the protein partners compared to the corresponding wild type. BLM mutant significantly loses interaction with 41 proteins interacting with its wild type. BLM P866L was shown to interact with less protein compared to wild type. BLM is one of the RecQ DNA helicases that is involved in DNA replication and repair by unwinding the double helix DNA. BLM mutations may lead to genomic instability and increased cancer susceptibility. 42 The variant is associated with increased tumor mutation burden, a potential immunotherapy efficacy biomarker, and useful in patient stratification across multiple cancer types. 43 Experimental validation of the disruptive VUS can identify the functional role of the variant and its contribution to tumorigenesis. Considering and studying the function can identify the novel pathogenic role of disruptive VUS mutations and the development of promising biomarker for Lynch syndrome.
Though the study focuses on the use of a comprehensive 56-gene panel analysis and in-silico tools for variant classification, it has several limitations. The sample size for FAP and AFAP was relatively small; hence, genotype-phenotype correlation could not be established, especially for the novel FANCD2 mutation. Functional assays were not conducted in this study. Though in-silico tools were employed to predict the impact of VUS, these are not a substitute for experimental validation. Further studies should include a larger, ethnically diverse population, functional assays for VUS, and longitudinal clinical follow-up to correlate genetic findings with treatment response.
Conclusion
Our study demonstrated that targeting next-generation sequencing using a 56 gene-panel effectively identifies deleterious mutations in suspected Lynch syndrome and FAP cases, which are known to predispose individuals to hereditary cancer syndromes. Due to the lack of early diagnosis, identifying germline deleterious mutations in an individual with a family history of cancer helps in predicting the risk of cancer development. Thus, screening procedures can be offered to high-risk mutation-positive individuals so that we can help in preventing or diagnosing the disease much earlier. It is important to identify other gene mutations for an individual who has deficient MMR due to microsatellite instability, which in turn leads to tumor formation. Targeting the DNA repair pathway defects associated with FANCD2 mutations could open new avenues for therapeutic interventions. Further studies are required to assess the prevalence and functional impact of FANCD2 mutations in FAP and other familial CRC syndromes. Evaluating VUS through in-silico methods. offers insights into potential structural and functional effects, particularly in cases where experimental data are lacking. Overall, our study supports the use of multigene panel testing as an accurate and cost-effective tool for diagnosing hereditary CRC syndromes. To assess the prevalence and functional impact of these mutations, further large-scale studies are warranted.
Footnotes
Acknowledgment
We thank the Department of Science & Technology for the infrastructural facility at the Department of Molecular Oncology, Cancer Institute (WIA).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
NA.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Department of Science & Technology, Research and development grant [grant numbers SR/S9/Z-08, 2010], Government of India.
Patient Consent
NA.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.