
Abstract
Erdheim Chester Disease (ECD) is a rare histiocytic neoplasm with heterogeneous features. It can frequently be misdiagnosed if proper clinical and radiological details are not available. Biopsy with confirmatory immunohistochemistry (IHC) for BRAFV600E or molecular analysis for the gene forms the mainstay of diagnosis. Most ECD patients require treatment and the choice of therapy must be individualized based on clinical characteristics. Herein is reported one such case of ECD in a 45-year-old male who remains disease-free after six months of follow-up.
Case Presentation
A 45-year-old man diabetic and hypertensive, presented with complaints of cough and shortness of breath of four months duration. He had a dry cough which was progressively worsening and associated with high-grade fever with chills and rigour. There was no history of any hemoptysis. The shortness of breath was progressive and worsened with physical activity. It was not present at rest. There was also a history of mild pedal oedema and associated weight loss of about five to seven kgs in the past one month with loss of weight, generalized weakness and myalgia. He also gave a history of intermittent headaches which got cured by taking medicine. He was recently diagnosed with Hypothyroidism. He had a history of cholecystectomy for gallstones around four years ago. There was no family history of any cancer. The patient was taken to a local hospital and put on oxygen support when his saturation levels were found to be 70% at home. Thereafter, he underwent an HRCT (high-resolution computed tomography) chest which revealed bilateral ground glass densities with inter-lobular septal thickening with perilymphatic nodules and air space densities with patchy areas of consolidation. There was also presence of pleura-pericardial effusion and marginal cardiomegaly with small mediastinal lymph nodes. A possibility of occupation interstitial lung disease or sarcoidosis was given. The patient was taken to another hospital and admitted. On further evaluation, CT thorax was suggestive of a lymphoproliferative disorder.
Patient underwent a biopsy when he was referred to our hospital and simultaneously underwent a PET-CT (positron emission tomography-computed tomography). PET-CT showed diffuse metabolically active septal thickening with subpleural ground glass infiltrates in both the lungs along with mild pericardial thickening and effusion. There was also presence of soft tissue thickenings around both adrenal glands and in bilateral perirenal spaces. There were diffuse metabolically active sclerotic lesions involving both axial and appendicular skeletal systems with periarticular soft tissue thickening around bilateral shoulder and hip joints.
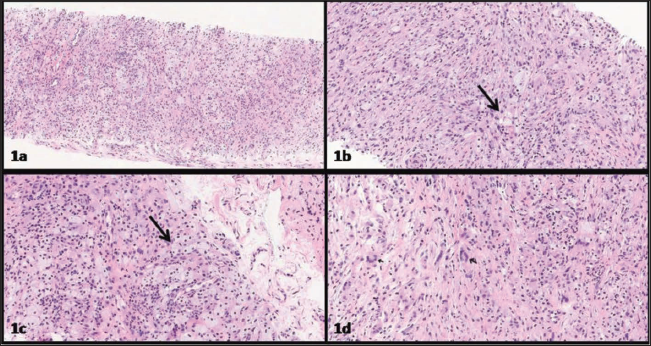
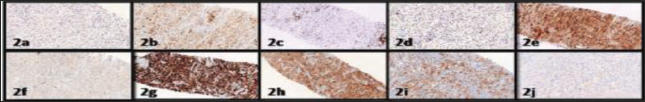
The biopsy was sent as ʻintramuscular lesion at the proximal end of right humerusʼ. Microscopic examination revealed a lesion composed of foamy histiocytes admixed with mononuclear plump spindled histiocytes dispersed in a fibro collagenous stroma (Figure 1a & b). Interspersed neutrophils and multinucleate giant cells (including Touton giant cells) were seen (Figure 1c & d). The lesional cells had small conspicuous nucleoli and no definite nuclear grooving was identified. A thin-walled vasculature was seen throughout the lesion. Mitosis was scarce with an absence of any significant atypia. No necrosis was seen. On immunohistochemistry (IHC), the lesional cells expressed LCA, CD68, CD163 and CD14 while being negative for CK, S100, CD3, CD20, Alk and CD1a. MIB 1 labelling index was approximately 2%–3%. Immunostain for BRAFV600E was positive (Figure 2a–j).
Histopathological Images. (a–d) H&E Stained Slides Showing and Admixture of Foamy Histiocytes (Long Arrows) and Spindled Cells with a Sprinkling of Neutrophils (c) and Giant Cells (d).
Immunohistochemistry. (a) Pancytokeratin Negative. (b) Leucocyte Common Antigen (LCA) Positive. (c) CD 3 Negative. (d) CD 20 Negative. (e) CD 68: Diffuse Positivity. (f) S100 Negative. (g) CD 163: Diffuse Strong Positivity. (h) CD14 Positive. (i) BRAFV600E Positive. (j) ALK Negative.
Q1. What are your Differentials Based on Morphology and What is the Final Diagnosis?
Answer: (a) Xanthogranulomatous osteomyelitis, Lipid storage disorders, Histiocytic/dendritic cell neoplasms such as Langerhans cell histiocytosis (LCH), Rosai Dorfman, Adult xanthogranuloma and Erdheim Chester disease.
(b) Erdheim chester disease.
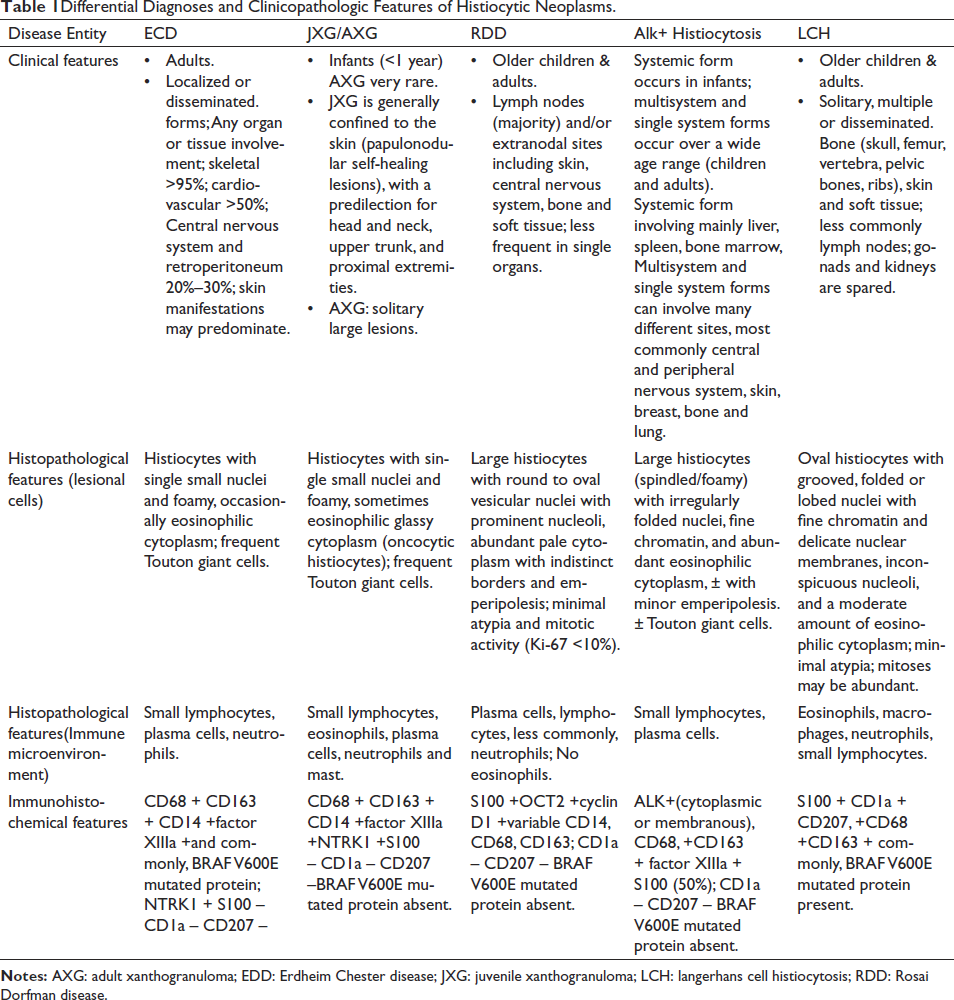
Erdheim Chester disease (ECD) is a rare histiocytic neoplasm with classic histopathologic findings including the presence of foamy histiocytes with small nuclei and surrounding fibrosis, multinucleated giant cells and Touton giant cells. It can have a spectrum of histopathological features within the xanthogranuloma family, often amidst a milieu of reactive lymphocytes, plasma cells, and rarely neutrophils. ECD may also display atypical features such as florid lymphohistiocytic infiltrates or fibrotic lamellae with only scattered foamy histiocytes and rare/absent Touton giant cells. 1 On morphology, due to the absence of any significant atypia/mitosis benign differentials also need to be ruled out by taking into account the relevant history and clinical findings. IHC for histiocytic markers such as CD 163, CD 68, CD 14 and Factor XIIIa can aid in the diagnosis. In this case, immunostains for LCA, CD 163, CD 68, CD 14 and BRAFV600E were positive. Table 1 summarises the clinicopathologic findings of the histiocytic neoplasms. 2
Differential Diagnoses and Clinicopathologic Features of Histiocytic Neoplasms.
Q2. What Clinicoradiological Finding will you Look for to Aid in the Diagnosis?
Answer: Bilateral symmetric involvement of the long bones.
Symmetrical osteosclerosis of meta diaphyses of leg bones is highly suggestive of ECD. 3 It is best visualized by FDG-PET but can also be seen on CT or MRI while often missed on plain X-rays.
Q3. Which Other Organ Systems/Manifestations are Commonly seen in this Disease and Which Organs are Generally Spared?
Answer: (a) Bone involvement, perinephric infiltrates, periaortic infiltrates, pituitary and neurologic involvement, lung heart and skin infiltrates (xanthelasma) may be seen.
(b) Lymph nodes, spleen and liver are usually spared.
ECD is a multisystem disease and can affect virtually any organ, typically causing central diabetes insipidus, restrictive pericarditis, perinephric fibrosis, and sclerotic bone lesions while lymph nodes, spleen, and liver are generally spared. 4 Bilateral symmetric involvement of the long bones is characteristic and detected in 80%–95% of cases. Perinephric (58%) infiltrates leading to ʻhairy kidneyʼ appearance and periaortic (46%) infiltrations leading to ʻcoated aortaʼ appearance are also frequent. Pituitary and neurologic involvement occurs in 20%–50% of cases. Infiltration of lungs with interstitial lung disease 5 along with infiltrates in heart, serosa and skin (xanthelasma or papular lesions) can also occur. Upto 15% of ECD cases are associated with different myeloid neoplasms including chronic myelomonocytic leukaemia. Multiple histiocytoses can also occur with presence of other histiocytic neoplasms such as LCH or Rosai Dorfman disease (RDD) in the same patient alongside ECD.
Q4. What is the most Common Molecular Alteration and the Methods of Detecting it?
Answer: (a) BRAFV600E.
(b) IHC, Polymerase chain reaction (PCR), pyrosequencing, droplet digital PCR (dd PCR) and targeted capture next-generation sequencing.
ECD was recently recognized as a neoplastic disorder owing to the discovery of recurrent activating MAPK (RAS-RAF-MEK-ERK) pathway mutations amongst which BRAFV600E mutation is the commonest; present in 50%–60% of the cases. 6
IHC is a cost-effective method of testing the mutation and presence of diffuse strong cytoplasmic staining with VE1 monoclonal antibody may signal a BRAF p.V600E mutation, but should always be confirmed with molecular analysis. IHC is also considered a less sensitive method 7 and, all negative or equivocal IHC tests should be confirmed by a sensitive sequencing technique on the same or alternative tissue specimens. ddPCR is considered the most sensitive method of testing. 8
Q5. What is the Diagnostic Criteria and what Baseline Investigations will you do in a Newly Diagnosed Case?
Answer: (a) Essential criteria: typical histology with collections of bland foamy histiocytes in the appropriate clinical and radiological context.
Desirable criteria: mutation analysis of the MAPK pathway.
(b) FDG PET-CT imaging including the brain and distal extremities, MRI of the brain with gadolinium, and cardiac MRI along with CT of chest, abdomen and pelvis. Laboratory studies needed to assess endocrinopathies, peripheral blood count abnormalities, renal/ hepatic function, immunological assessment, and the degree of inflammation also have to be done.
The goal of the evaluation in newly diagnosed patients is to define the extent of disease involvement, assess subsequent risk of end-organ compromise, and define a plan of treatment and surveillance. FDG PET-CT is recommended for initial evaluation to assess organ involvement and as a tool for guiding biopsy targets. Dedicated CT of the chest, abdomen, and pelvis is recommended to demonstrate pulmonary, periaortic, and perinephric infiltrates.
Q6. What is the Treatment for this Disease and what Targeted Therapy can you Offer to the Patient?
Answer: (a) Targeted therapy, Immunosuppressive therapy, cytokine-directed therapy, chemotherapy, and corticosteroids.
(b) BRAF inhibitors, MEK inhibitors, mTOR inhibitors and tyrosine kinase inhibitors.
For patients with multisystem BRAF-V600-mutant ECD with life-threatening cardiac or neurologic involvement, the first-line recommendation is to consider BRAF inhibitors such as vemurafenib or dabrafenib. For BRAF-V600-mutated ECD without end-organ dysfunction, it is appropriate to consider either BRAF-inhibitor or immunosuppressive/cytotoxic therapy. For patients without BRAF-V600-ECD, MEK inhibitors such as Cobemetinib are recommended. Conventional immunosuppressive agents like Interferon-a-2a (IFN-a) and pegylated interferon-a-2a (PEG–IFN-a) have long been used for the treatment of ECD. Cytokine-directed therapy such as interleukin-1 (IL-1) receptor antagonists (anakinra, canakinumab), IL-6 receptor antagonists (tocilizumab), and tumour necrosis factor-a (TNF-a) inhibitors (infliximab, etanercept) may also be used.
Q7. Which Imaging Modality is used for Response Assessment?
Answer: FDG-PET-CT.
Although there are no prospectively validated response criteria for ECD, FDG-PET-CT is considered the optimal modality for ECD response assessment. 9 FDG PET imaging should be obtained three to six months after initiation of therapy to assess metabolic response. Complete metabolic response, that is, normalization of lesion FDG avidity to that of the surrounding organ background, is considered the optimal response to ECD therapy. A sustained partial metabolic response with clinical improvement represents a favourable outcome. Organ-specific imaging such as CT or MRI (e.g., heart, brain, orbit) should be performed every three to six months initially, and every 6–12 months once the disease stabilizes.
Additionally, monitoring for pituitary hormone abnormalities every one to two years is recommended as endocrinopathies may develop during the course of treatment as well. A decline in the levels of C-reactive protein with treatment suggests a favourable response. Clinical examination and other lab tests such as peripheral blood counts and bone marrow may also be done if needed.
Q8. Name some Poor Prognostic Factors?
Answer: CNS, lung and retroperitoneum involvement, advanced age at presentation.
Prognosis depends on site and extent of the involvement and advanced age, CNS, lung and retroperitoneum involvement have been considered as poor prognostic factors. 10 CNS involvement requires higher doses of targeted (BRAF- or MEK-inhibitor) therapies or dual therapy for BRAF-V600-mutated ECD to attain a robust response.
Conclusion
ECD is a rare histiocytic neoplasm with heterogeneous features, posing significant diagnostic and treatment challenges to clinicians. Lesional tissue should be biopsied in all cases to confirm ECD diagnosis and establish mutational status to potentially guide therapy. Most ECD patients require treatment and the choice of therapy must be individualized based on clinical characteristics.
Footnotes
Acknowledgements
The authors thank the technical staff of the laboratory medicine department for their skilful assistance.
Authors’ Contributions
SP and PC performed the histological examination. PC and AM were major contributors in drafting the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author received no financial support for the research, authorship and/or publication of this article.