
Abstract
Background
In the sporadic model of Alzheimer's disease (AD), induced by intracerebroventricular streptozotocin (STZ) administration, cognitive impairment is accompanied by specific astrocytic changes in the hippocampus prior to amyloid deposition.
Objective
Hypothesizing that the synthesis of GABA, via MAO-B, contributes to ammonia elevation, thereby compromising antioxidant defense and ATP synthesis, and possibly contributing to cognitive damage, we determined the hippocampal levels of glutamine synthetase (GS), monoamine oxidase B (MAO-B) and other enzymes related to GABA metabolism.
Methods
Immunoblotting and RT-PCR assays were carried out in hippocampal samples of Wistar rats, at 4 and 16 weeks post-STZ, in the sporadic STZ-induced AD model, corresponding to the pre-amyloid and amyloid phases, respectively.
Results
We observed a reduction in GS activity and increased MAO-B content, both in 4 weeks and in 16 weeks, reinforcing the idea that astroglial dysfunction precedes the amyloid phase. These alterations were accompanied by an increase in the content of ornithine decarboxylase 1 (ODC1), which catalyzes the synthesis of putrescine (substrate for GABA synthesis, via MAO-B), and a reduction in the gene expression of arginine-glycine amidinotransferase (AGAT), an enzyme involved in the synthesis of creatine, and in the generation of GABA agonists. These changes were only seen in the amyloid phase of the AD model.
Conclusions
Our findings contribute to explain the greater damage that occurs in energy metabolism at this stage, in addition to the greater GABAergic loss. The changes reinforce the importance of the STZ model and further our understanding of the changes in both AD phases.
Introduction
Alzheimer's disease (AD) accounts for two-thirds of dementia cases and is characterized by extracellular amyloid deposition and neuronal fibrillary tangles. 1 However, the etiological basis of the disease remains unknown. The asymptomatic development of the disease precedes the dementia phase by decades and the hypothesis that astroglial dysfunction, often characterized by an increase in glial fibrillary acid protein (GFAP), precedes neuronal damage has gained more traction. 2 Although genetic models in rodents with increased amyloid precursor protein are widely used, the vast majority of cases of the disease are sporadic. 3 The intracerebroventricular (ICV) injection of streptozotocin (STZ) has been used to mimic and model sporadic AD in rodents and primates 4 and also to accelerate the development of the disease in genetic models. 5 This model displays many similarities to AD and is being used to evaluate preventive therapeutic pharmacological and non-pharmacological strategies for the disease. 6
We used, herein, the sporadic model of AD with STZ. In this model, cognitive loss is accompanied by specific astroglial changes in the hippocampus prior to amyloid deposition. These alterations include increases in GFAP 7 and in aquaporin-4 contents, 8 and a decrease in glutamine synthetase (GS) activity. 9
Glutamatergic excitotoxicity may underlie several alterations in neurotransmission in AD, including the cholinergic deficit, which is one of the first alterations reported in the disease.10,11 Consistent with the hypothesis that excitotoxicity modifies all synaptic communication in AD, reductions in glutamate uptake in the synaptic cleft (mainly mediated by astrocytic transporters) and in the activity of astrocytic GS, responsible for glutamine synthesis, are observed in the STZ-induced AD model. More recently, the imbalance in the glutamate/γ amino butyric acid (GABA) ratio has been addressed to justify this greater excitability. 12 Although not a consensus, reductions in hippocampal GABA levels have been found in both patients, and in animal models of AD.13,14 However, attempts to employ cerebrospinal fluid (CSF) levels of these neurotransmitters as markers of disease have failed. 15
Another indication of neurotransmission alteration in AD is the increase in monoamine oxidase B (MAO-B), a principally astroglial enzyme that is responsible for metabolizing catecholamines.16,17 Increased MAO-B activity underlies the dopaminergic deficit observed in AD. Moreover, enzyme can also be a source of GABA in astrocytes, via the oxidation of the putrescine polyamine, a metabolite of ornithine. 18 GAD67, the main isoform of the glutamate decarboxylase enzyme, which is responsible for the synthesis of GABA in neurons, could also be a source of GABA in reactive astrocytes. 19 This putative GABA increase, however, contradicts the idea of a reduction in GABA in AD and has led to the hypothesis that the increase in GABA occurs earlier on in the disease and that glutamatergic excitotoxicity occurs later. 12
In the STZ dementia model, as in AD, both a reduction in GS 9 and an increase in MAO-B 20 have been described, which potentially lead to increased brain ammonia. 21 Considering that the synthesis of GABA, via MAO-B, contributes to ammonia elevation, thereby compromising antioxidant defense and ATP synthesis, and possibly contributing to cognitive damage, we evaluated the hippocampal levels of GS and MAO-B, at 4 and 16 weeks post-STZ, in the sporadic STZ-induced AD model, corresponding to the pre-amyloid and amyloid phases. 22 Other parameters were also evaluated to understand the role and metabolism of GABA in the disease.
Materials and methods
Animals
Forty-eight male Wistar rats (90 days old; 300–400 g), were obtained from the vivarium of the Biochemistry Department of the Federal University of Rio Grande do Sul. The animals were kept in a 12-h/12-h light and dark cycle, at a constant temperature of 22°C ± 2°C, with free access to food and water, using the standard vivarium diet. The animals were housed in polyethylene boxes, on a bed of selected and autoclaved pine shavings. The handling and treatment of the animals complied with the recommendations of the Ethics Committee for the Use of Animals (CEUA) of the Federal University of Rio Grande do Sul, in addition to the federal legislation advocated by the Arouca Law (law n° 11.794, of October 8, 2008), with the approval of the CEUA under protocol CEUA 37872. The animals were randomly divided into 2 time points, with 24 animals each (4 and 16 weeks), and two groups of 12 animals (SHAM, STZ), according to the surgical procedure. See the experimental design in Figure 1.
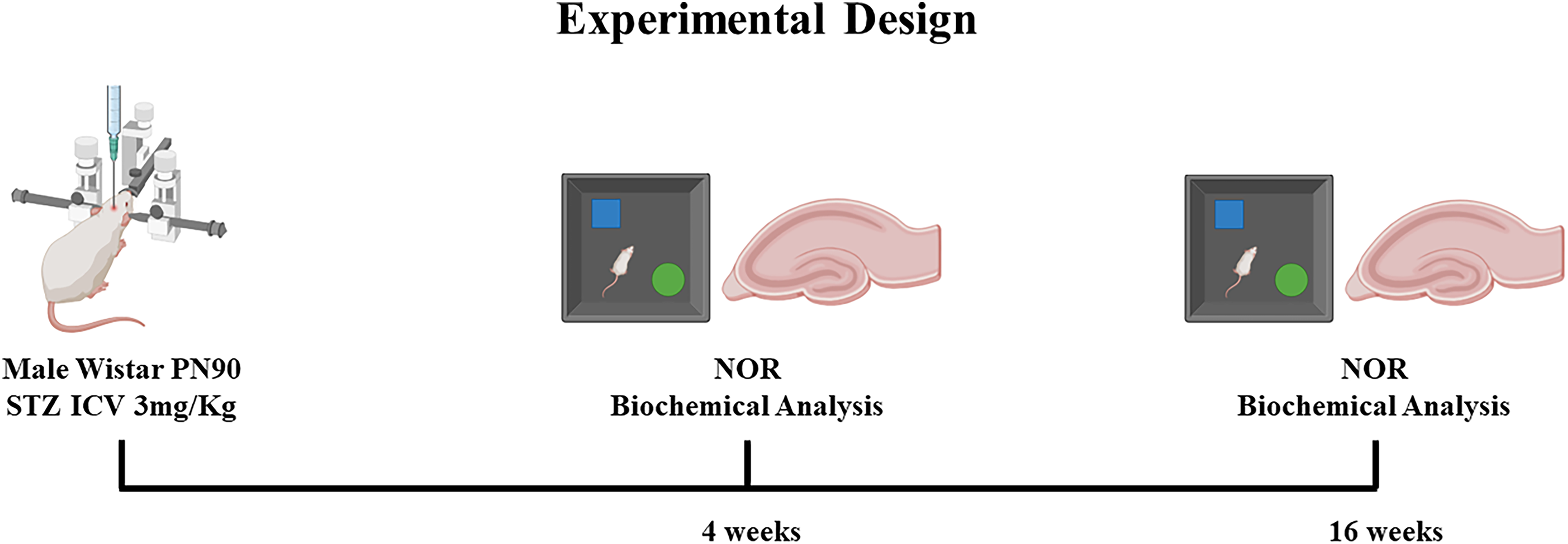
Illustration showing the experimental design. Forty-eight adult Wistar rats were submitted to the STZ-induced sporadic AD model and, after 4 or 16 weeks (12 animals per group, SHAM or STZ, in the two experimental times), their behavior was evaluated using the open field and novel object recognition (NOR) tasks. After behavioral tests, rats were euthanized, and hippocampal samples were collected for neurochemical analysis.
Surgical procedure
For the induction of the animal model of sporadic AD, the animals were anesthetized with an intraperitoneal (IP) injection of ketamine (75 mg/Kg) and xylazine (10 mg/Kg). After confirmation of anesthesia, the animals were positioned in a stereotaxic apparatus to perform the surgical procedure. The stereotactic device was positioned on the bregma, and a sagittal incision was made in the midline of the scalp. The following coordinates were used to access the lateral ventricles: 0.9 mm posterior to the bregma, 1.5 mm lateral to the sagittal suture of 1.5 mm, 3.6 mm beneath the surface of the brain. Five 5 μL of solution containing 3 mg/Kg of STZ (Sigma Aldrich, S0130), or vehicle (Hanks’ balanced salt solution), were injected into each hemisphere, according to the experimental group.7,23 After the surgical procedure, the rats were placed on a heating pad to maintain the body temperature at 37.5 ± 0.5°C until recovery from anesthesia.
Novel object recognition
The novel object recognition (NOR) test was used to evaluate memory acquisition and consolidation processes. This test is performed in an open field apparatus (50 cm side) and consists of three phases; habituation, training and test. For the habituation stage, the animals were placed in the lateral of the apparatus and allowed to freely explore the open-field arena, in the absence of objects, for 10 min. Twenty-four hours after the habituation phase, the animals were exposed to a training phase during which the rats were returned to the apparatus, which contained two identical objects (A + A). At one hour and 24 h after the training phase, the rats were submitted to short-term memory (STM) and long-term memory (LTM) test, respectively. In the STM test, an object was replaced by a novel one (A + B). In the LTM test, object ‘B’ was replaced by a novel object ‘C’ (A + C). The exploration times were calculated based on the following equation (exploration time of the new object ×100/ exploration time for both objects), generating the Recognition Index (RI). 24
Sample collection
At four or sixteen weeks after surgery, the animals were anesthetized by IP injection of ketamine (75 mg/Kg) and xylazine (10 mg/Kg) to minimize any pain or discomfort during biological sample collection and euthanasia, carried out by decapitation. This procedure was performed in a quiet environment by a trained operator in the absence of other animals. Afterwards, the brains were removed and placed in sodium phosphate buffered solution (PBS), pH 7.4. The hippocampus was dissected and slices of 300 µm were obtained using a McIlwain Tissue Chopper. All samples were stored in a −80°C freezer until further analysis.
Glutamine synthetase activity assay
GS activity was determined based on Taday et al. (2024). 25 Briefly, homogenate (0.1 mL) was added to 0.1 mL of reaction mixture containing (in mM): 10 MgCl2; 50 L-glutamate; 100 imidazole–HCl buffer (pH 7.4); 10 2-mercaptoethanol; 50 hydroxylamine–HCl; 10 ATP and incubated for 15 min at 37°C. The reaction was stopped by the addition of 0.4 mL of a solution containing (in mM): 370 ferric chloride; 670 HCl; 200 trichloroacetic acid. After centrifugation, the supernatant was measured at 530 nm and compared to the absorbance generated by standard quantities of γ-glutamylhydroxamate treated with ferric chloride reagent. The results were presented as µmol/h/mg protein.
Reduced glutathione content
GSH content was determined as previously described by Browne and Armstrong with adaptations. 26 Hippocampal slices were homogenized in a sodium phosphate buffer (0.1 M, pH 8.0) containing 5 mM EDTA, and protein was precipitated with 1.7% metaphosphoric acid. Supernatant was collected and assayed with o-phthaldialdehyde (1 mg/mL in methanol) at room temperature for 15 min. Fluorescence was measured using excitation and emission wavelengths of 350 and 420 nm, respectively. A calibration curve was performed with standard glutathione solutions (0–500 mM). The GSH concentrations were expressed as nmol/mg protein.
Ammonia content
Ammonia content was measured in hippocampal slices using a urease-based assay commercial kit (Bioclin K047, Brazil). Briefly, tissue samples were homogenized and deproteinized with 1.7% metaphosphoric acid. The supernatant was then alkalized with phosphate buffer to pH 7.5, and in the presence of salicylate and sodium hypochlorite, ammonia produced a greenish compound (Berthelot reaction), whose color intensity is proportional to the concentration of ammonia in the sample analyzed. The results were presented as percentage of that of the SHAM group.
Western blotting
To perform western blotting, hippocampus samples were prepared as previously described. 8 After the determination of total proteins, equal amounts of each tissue sample (30 μg), in triplicate, were separated by SDS-PAGE. Separated proteins were transferred to a 0.45 µm nitrocellulose membrane. The transfer procedure was confirmed with Ponceau S staining. Membranes were blocked with 2% bovine serum albumin in T-TBS for 2 h at room temperature (RT). After blocking, membranes were incubated with the corresponding primary antibodies overnight at 4°C; 1:1000 anti-GAD67 (Elabscience E-AB-17390), 1:1000 anti-GAT-3 (Elabscience E-AB-17981), 1:1000 anti-GFAP (Sigma-Aldrich G9269), 1:1000 anti-MAO-B (Santa Cruz Biotechnology SC-515354), and 1:1000 anti-ODC1 (Elabscience E-AB-60518). After incubation with primary antibodies, the membranes were incubated with the respective peroxidase-conjugated secondary antibodies for 2 h at RT; anti-mouse IgG 1:10000 (Elabscience E-AB 1001), anti-rabbit IgG 1:10000 (Elabscience E-AB-1058). The chemiluminescence signal was obtained using ECL Super Signal West Pico Plus (Thermo Fisher) in a Chemidoc MP (Bio-Rad Laboratories). The optical densities of the images were calculated with ImageLab software (Bio-Rad Laboratories), and results were normalized using β-Actin density and presented as the percentage of that of the SHAM group.
RNA extraction and quantitative RT-PCR
Total RNA was isolated from hippocampal slices using TRIzol reagent. 27 cDNA was synthesized from extracted RNA (1 µg) using the High-Capacity cDNA Reverse Transcription Kit. Quantitative PCR determination of the messenger RNAs (mRNAs) encoding β-actin (Mm02619580_g1), L-arginine-glycine amidinotransferase (AGAT; Rn00578954_m1), and 4-aminobutyrate aminotransferase (GABA-T; Rn00578656_m1) were performed using the TaqMan real-time RT-PCR system with inventory primers and probes purchased from Applied Biosystems (Thermo Fisher Scientific), as indicated for each gene. Target mRNA levels were normalized to β-actin levels. Results were analyzed employing the 2−ΔΔCt method28,29 and presented as fold of change.
Statistical analysis
Descriptive statistical analysis and the assessment of data normality were performed by Shapiro-Wilk test. Results are presented as means ± standard deviation (SD). For NOR analysis, the one sample t test was used to evaluate differences between the Recognition Index from a chance performance (50%). For the other results, differences between SHAM and STZ groups were statistically analyzed using Student's t-test, where the value of p < 0.05 was used as a criterion for statistical significance. All analyses were performed using the GraphPad Prism 9 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Cognitive impairment at two time points in STZ-induced dementia
Cognitive processes were evaluated using the novel object recognition (NOR) task at 4 and 16 weeks after ICV STZ administration (Figure 2). During the discrimination phase, the STZ group (at 4 weeks) exhibited decreased recognition index values for short-term memory (STM, measured at 1 h, Figure 2A, p = 0.0003 for SHAM and p = 0.1324 for STZ) and for long-term memory (LTM, measured at 24 h, Figure 2B, p < 0.0001 for SHAM and p = 0.8803 for STZ). Furthermore, in another experimental set, at 16 weeks after ICV STZ administration, cognitive impairment in the NOR task was also observed at both 1 h (Figure 2C, p = 0.0001 for SHAM and p = 0.2531 for STZ) and 24 h (Figure 2D, p = 0.0004 for SHAM and p = 0.1612 for STZ).
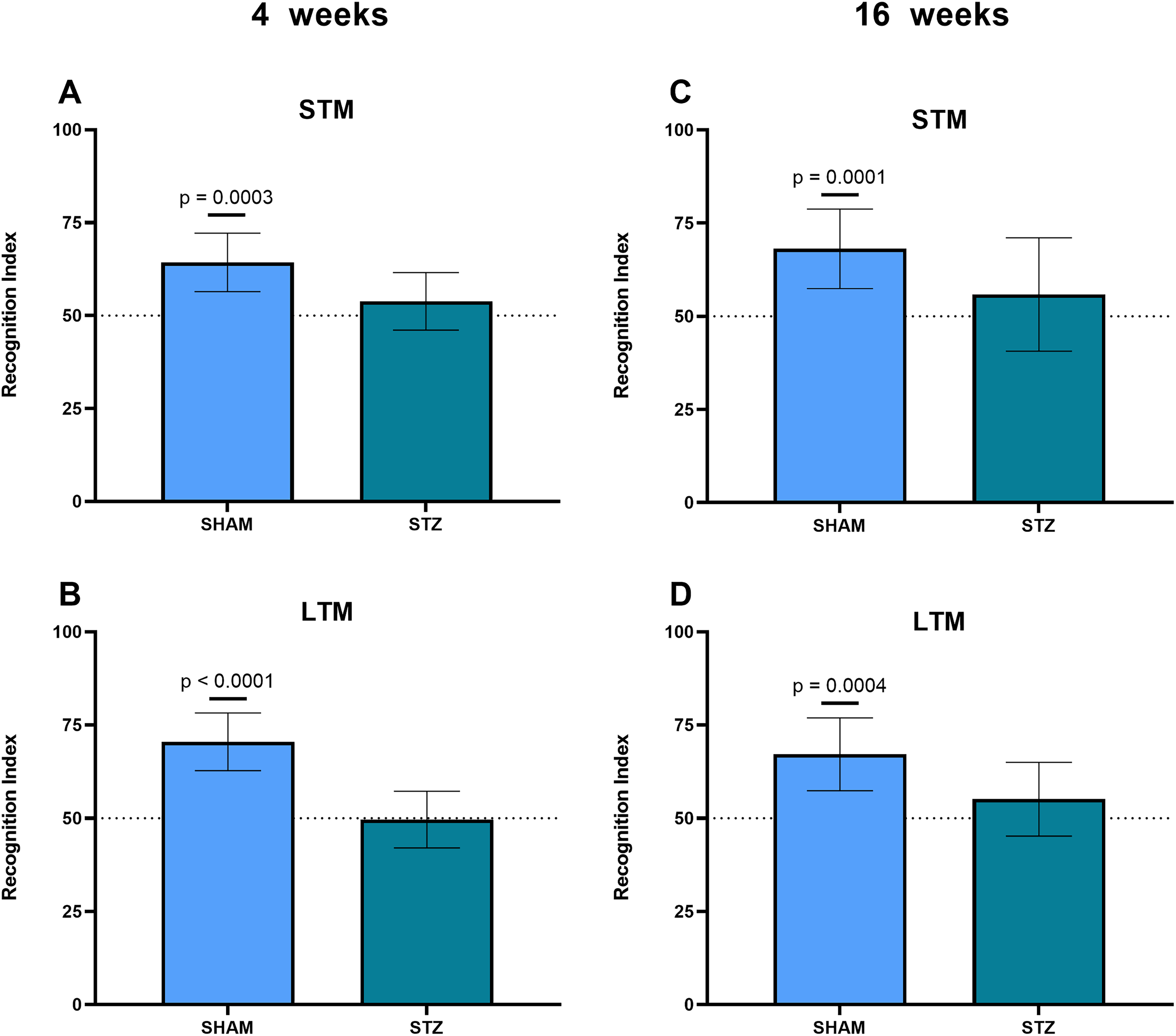
Novel object-recognition task for rats submitted to the sporadic AD model induced by STZ. (A, B) Cognitive performance was assessed at 4 weeks by NOR tests, carried out at 1 h and 24 h after the training session, corresponding to short-term and long-term memory, respectively. (C, D) Recognition performance was assessed at 16 weeks by the NOR tests, carried at 1 h and 24 h after the training session, corresponding to short-term and long-term memory, respectively. All data are reported as means ± SD (11 animals per group in panels (A-D) and Student's t-test was used. The p value indicates statistical significance, when compared to chance level - 50% of novel object investigation in the discrimination phase.
Astroglial dysfunction is observed at both time points in the AD model
Hippocampal astroglial immunoreactivity, based on GFAP content increment, was observed at 4 weeks (Figure 3A, p = 0.0018), and at 16 weeks (Figure 3B, p = 0.0243), after ICV STZ administration. Moreover, hippocampal GS activity was decreased at 4 weeks (Figure 3C, p = 0.0194), and at 16 weeks (Figure 3D, p = 0.0433). Based on the reduction in the GS/GFAP ratio, we can infer astroglial dysfunction, at both time points.
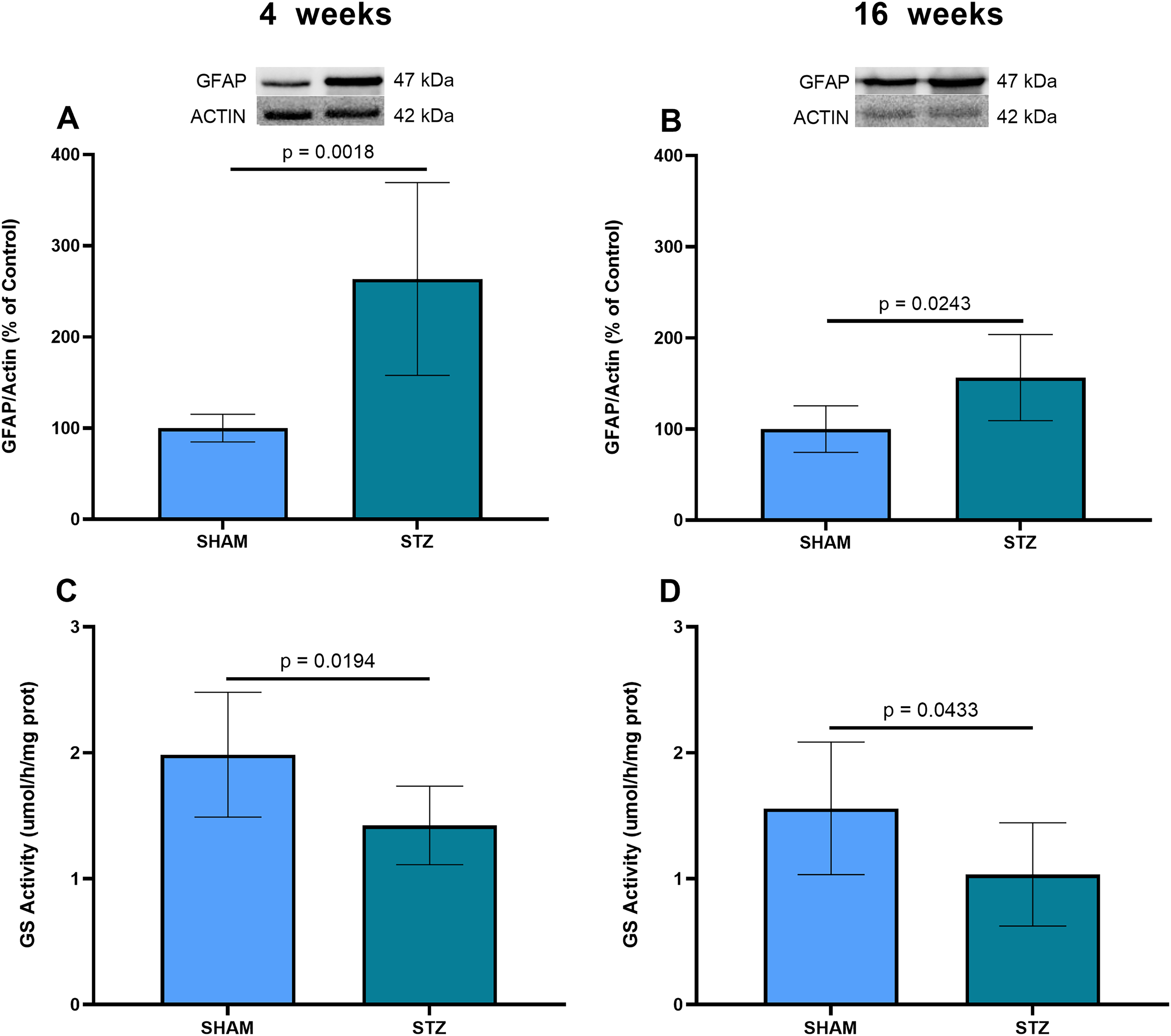
Hippocampal GFAP content GS activity in the STZ-induced model of sporadic AD, at 4 and 16 weeks. (A, B) Hippocampal samples were used to measure the GFAP content by western blotting, at 4 weeks and 16 weeks, respectively. Representative immunoblots of these proteins are shown, as well as actin content. (C, D) Tissue samples were used to measure GS activity, expressed in μmol of product formed/h/mg of protein, at 4 weeks and 16 weeks, respectively. Data are expressed as means ± SD (6 animals per group in panels (A-B) and 8 animals in panels (C-D). The p value indicates statistical significance, analyzed by Student's t-test.
Increases in MAO-B and ODC1 are observed in the AD model
Increased hippocampal MAO-B was observed both at 4 weeks after ICV administration of STZ (Figure 4 A, p = 0.0483), and at 16 weeks (Figure 4B, p = 0.0365). This enzyme degrades catecholamines but can also form GABA from putrescine. Therefore, we investigated the ODC1 content at these time points; this enzyme generates putrescine from ornithine for MAO-B, to catalyze the synthesis of GABA. Notably, while ODC1 was not significantly increased at 4 weeks (Figure 4C, p = 0.1684), an increase was observed at 16 weeks in the STZ-induced animals (Figure 4D, p = 0.0118).
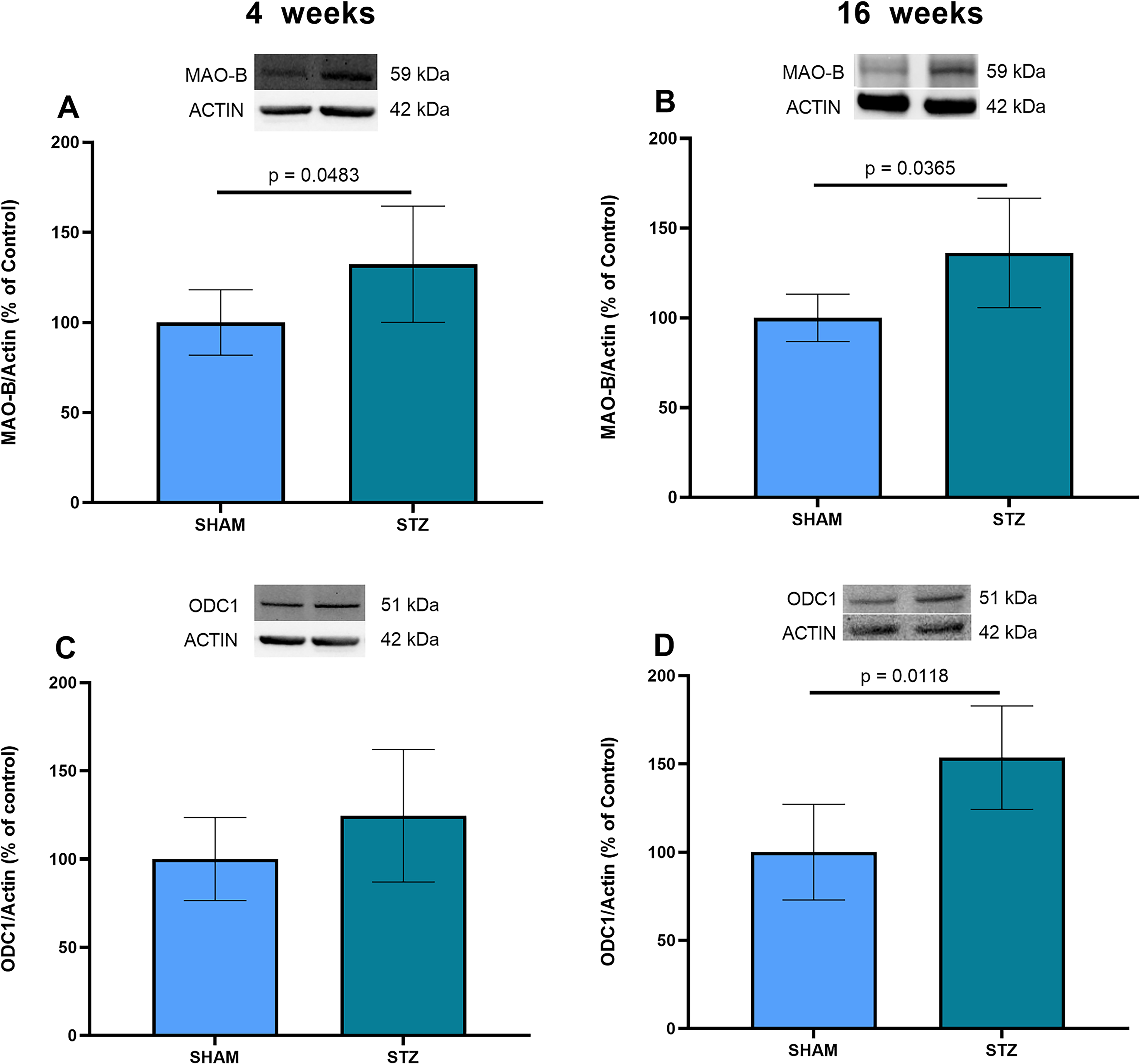
MAO-B and ODC1 contents at 4 and 16 weeks, in the STZ-induced model of sporadic AD. (A and B) Hippocampal samples were used to measure the MAO-B content by western blotting at 4 and 16 weeks, respectively. (C and D) Tissue samples were used to measure the ODC1 content by western blotting at 4 and 16 weeks, respectively. Representative immunoblots of these proteins are shown, as well as actin content in all panels. Data are presented as means ± SD (6 animals per group in panels A-D). The p value indicates statistical significance, analyzed by Student's t-test.
Glial dysfunction is confirmed by increased ammonia and reduced GSH in hippocampal tissue
Higher MAO-B activity should result in ammonia and peroxide generation, which may contribute to increased ammonia content and reduced levels of reduced glutathione in the hippocampal tissue. Decreased GSH was observed both at 4 weeks (Figure 5A, p = 0.0177), and at 16 weeks (Figure 5B, p = 0.0297). Hippocampal ammonia levels did not increase significantly at 4 weeks (Figure 5C, p = 0.7298), but a clear increase was observed at 16 weeks (Figure 5D, p = 0.0051).
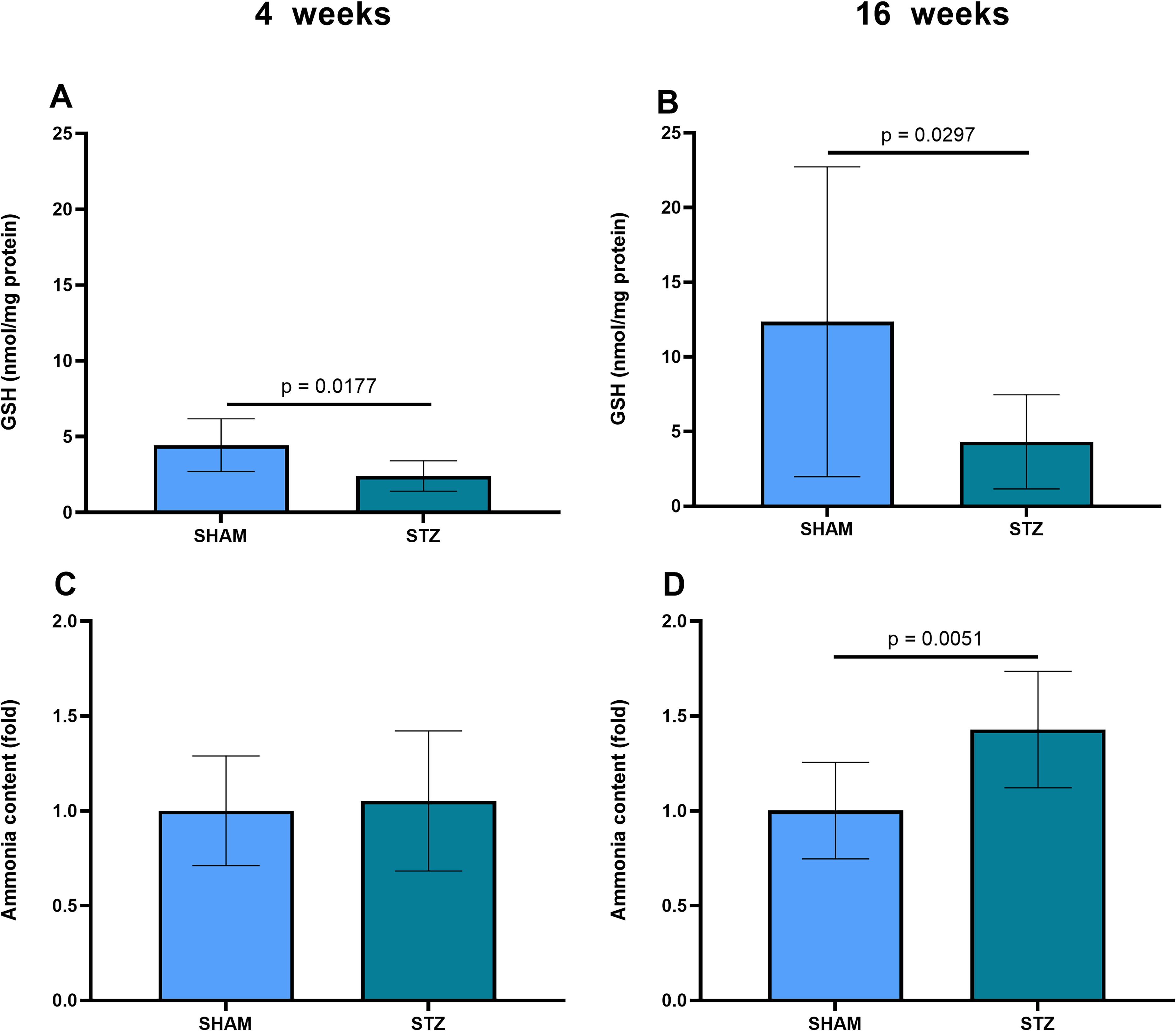
GSH and ammonia levels in the hippocampus of rats submitted to the STZ- induced model of sporadic AD. (A and B) Levels of GSH measured by a fluorimetric assay with o-phthaldialdehyde, at 4 and 16 weeks after STZ administration, respectively. (C and D) Levels of ammonia, measured by a colorimetric assay with salycilate/hypochlorite, at 4 and 16 weeks after STZ administration, respectively. Data are expressed as means ± SD (8 animals per group in panels (A-B) and 10 animals in panels (C-D)). The p value indicates statistical significance in relation to the SHAM group, analyzed by Student's t-test.
GAD67 content is decreased in STZ-induced dementia
GAD67 is commonly the main source of neuronal GABA, but an increase in the enzyme can be observed in reactive astrocytes in some situations of damage. However, no change in this enzyme was observed in the STZ-induced AD model at 4 weeks (Figure 6A, p = 0.5780); in contrast, a decrease occurred at 16 weeks (Figure 6B, p = 0.0025). This same profile was observed for the main glial GABA transporter. No change in GAT-3 was observed at 4 weeks (Figure 6C, p = 0.2586), but a reduction occurred at 16 weeks in the STZ-treated animals (Figure 6D, p = 0.0006).
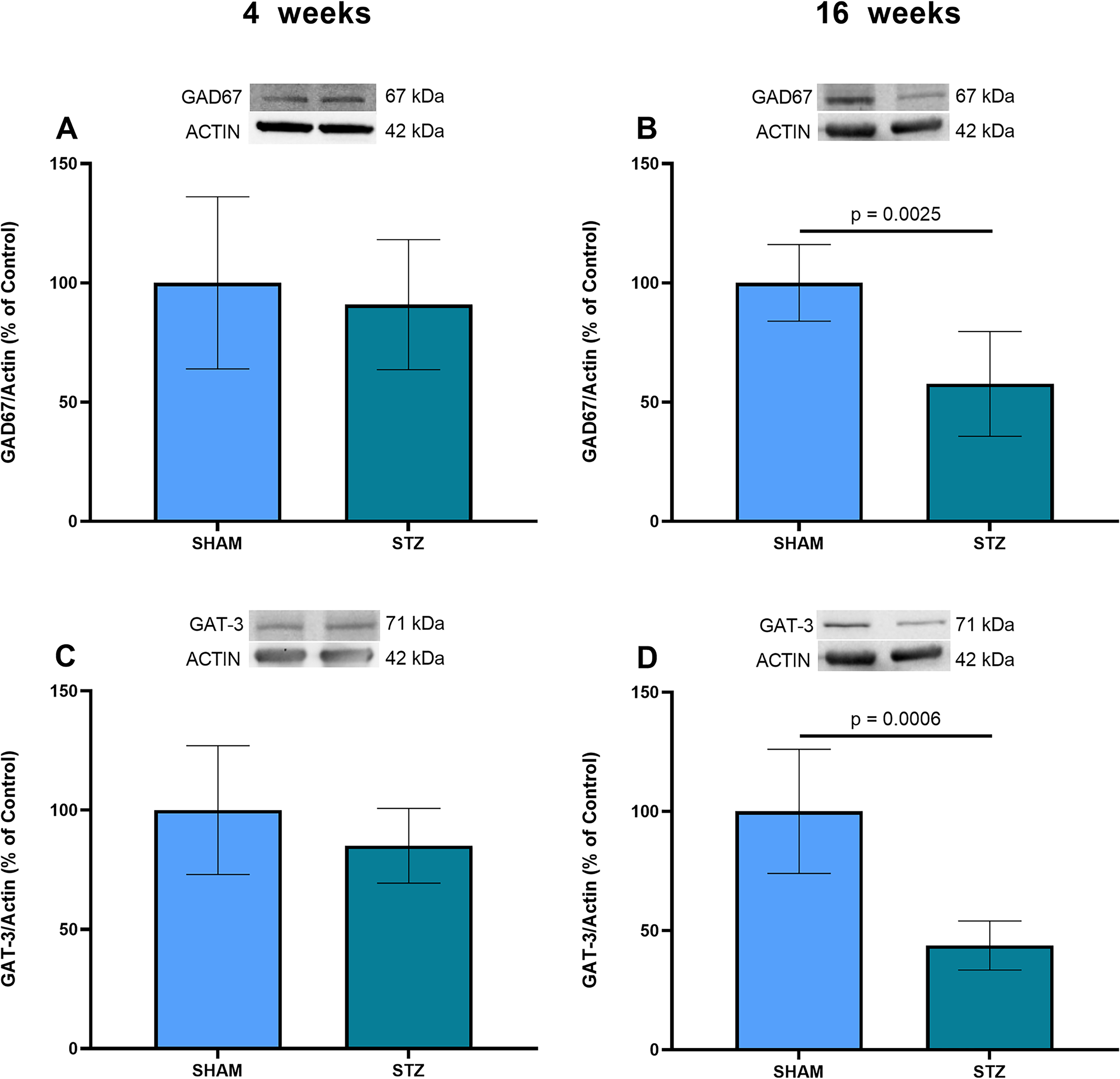
GAD67 and GAT-3 contents at 4 and 16 weeks in the STZ-induced model of sporadic AD. (A, B) Hippocampal samples were used to measure the GAD67 content by western blotting at 4 and 16 weeks, respectively. (C, D) Tissue samples were used to measure the GAT-3 content by western blotting at 4 and 16 weeks, respectively. Representative immunoblots of these proteins are shown, as well as actin content in all panels. Data are presented as means ± SD (6 animals per group in panels (A-D)). The p value indicates statistical significance, analyzed by Student´s t-test.
AGAT gene expression changes in the STZ-AD model
To understand other metabolic fates of GABA, we investigated the gene expressions of two enzymes; AGAT, the enzyme that converts GABA into gamma-guanidinobutyric acid (γ-GBA - another GABA agonist), and GABA-T, the transaminase that initiates the metabolism of GABA, destined for the Krebs cycle. Expression of the gene encoding AGAT was increased at 4 weeks (Figure 7A, p = 0.0407), but a reduction in gene expression occurred later at 16 weeks (Figure 7B, p = 0.0072). However, GABA-T gene expression does not change at 4 weeks (Figure 7C, p = 0.4425), or at 16 weeks (Figure 7D, p = 0.0956).
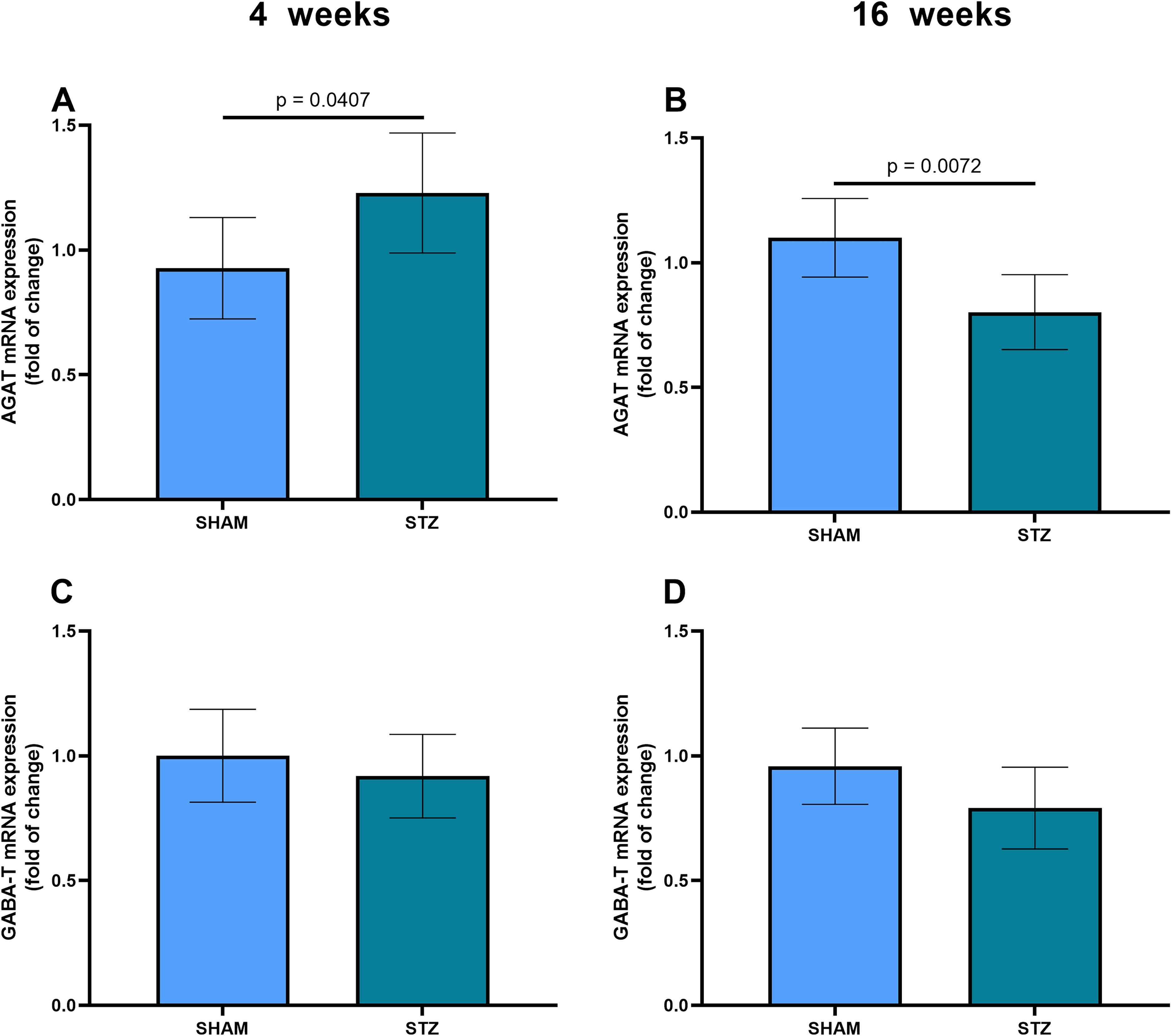
Gene expressions of the AGAT and GABA-T enzymes in the STZ-induced model of sporadic AD. (A, B) The mRNA expression of the AGAT enzyme, measured by RT-PCR in hippocampal samples from SHAM and STZ animals, at 4 and 16 weeks, respectively. (C, D) The mRNA expression of the GABA-T enzyme, measured by RT-PCR in hippocampal samples, at 4 and 16 weeks, respectively. The data represent means ± SD (6 animals per group in panels (A-D)). The p value indicates statistical significance, analyzed by Student's t-test.
Discussion
In the sporadic AD model induced by ICV administration of STZ, 4 weeks post-STZ corresponds to a time within the pre-amyloid phase and 16 weeks corresponds to a time within the amyloid phase. 22 From a cognitive point of view, as assessed by the NOR task, cognitive impairment was observed at these two times, both for STM, assessed at 1 h, and for LTM, assessed at 24 h. In addition, we evaluated the astroglial changes particularly associated with GABAergic neurotransmission, and ammonia metabolism, at these two time points of the model.
Recent evidence indicates that, in the presence of amyloid beta peptides, astrocytes increase their expression of urea cycle enzymes (e.g., ODC1 and arginase1, see Figure 8), possibly in order to buffer the ammonia toxicity observed in AD. 30 The reduction in GS activity contributes to this ammonia toxicity, due to reduced removal of ammonia, while increased MAO-B activity can generate ammonia. Understanding these pathophysiological changes in the STZ model further validates the model and increases our understanding of the alterations that occur in AD in both phases of the disease.
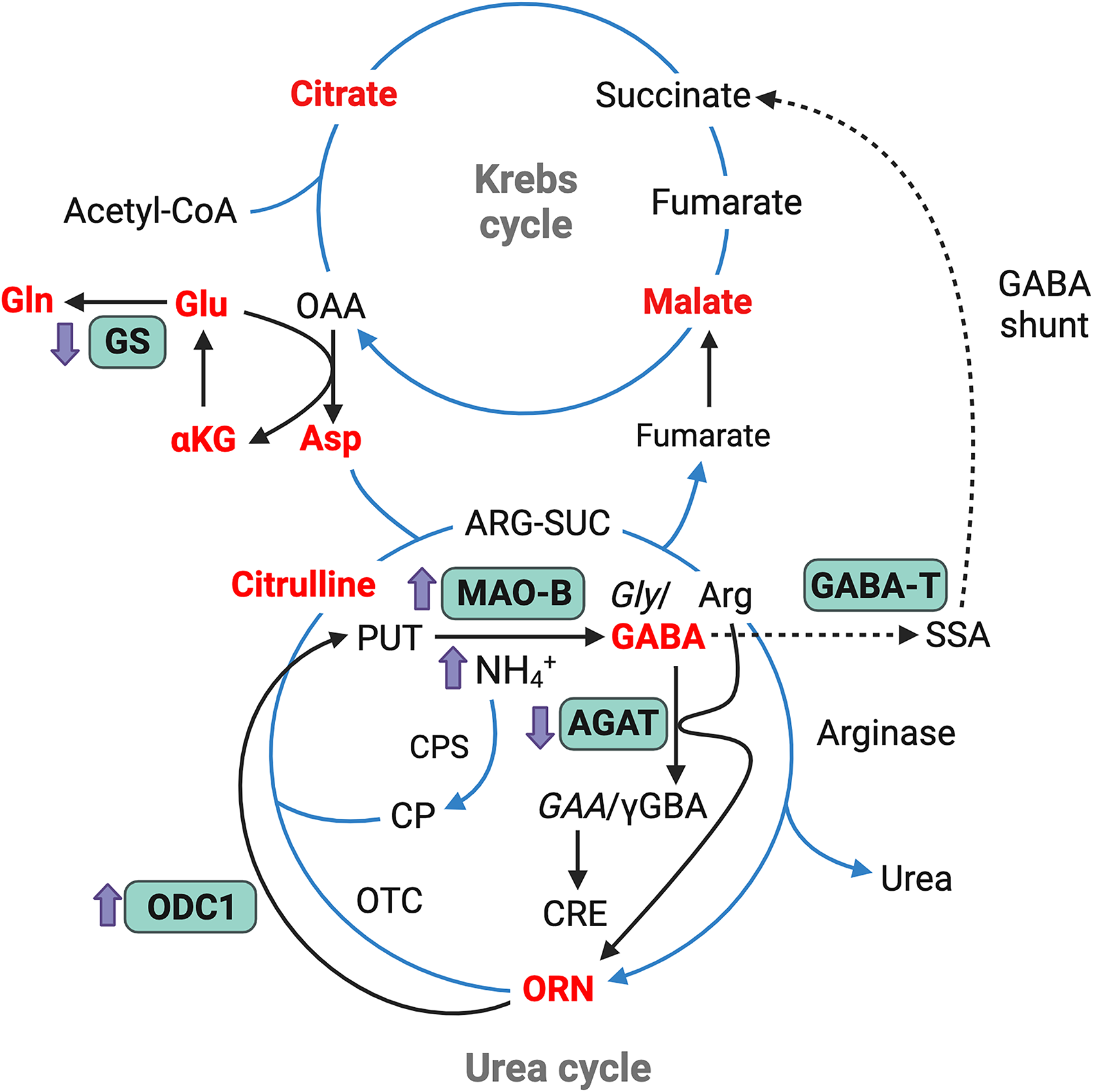
Illustration of metabolic pathways and findings in the amyloid phase of the STZ-induced sporadic AD model. The scheme shows the link between the Krebs cycle and the urea cycle, operating in astrocytes, emphasizing their roles and associations with the enzymes analyzed in this study (highlighted in green). Changes in the amyloid phase of the model (16 weeks post-STZ), up or down, are indicated by small purple arrows, next to these enzymes. Note that changes in the GS and MAO-B enzymes are already present from the pre-amyloid phase (4 weeks post-STZ). As the cycles take place in different compartments, the compounds in red are those that have a transporter in the mitochondrial membrane. Note that arginine is both a substrate for AGAT and arginase, just as ornithine is a substrate for both mitochondrial ornithine transcarbomylase and ODC1. AGAT, arginine-glycine amidinotransaminase; Arg, arginine; ARG-SUC, argininosuccinate; Asp, aspartate; CIT, citrulline; CP, carbamoyl phosphate; CPS, Carbamoyl phosphate synthetase; CRE, creatine; GAA, guanidinoacetate; GABA-T, GABA transaminase; Gln, glutamine; Glu, glutamate; Gly, glycine; GS, glutamine synthetase; MAO-B, monoamine-oxidase; OAA, oxaloacetate; ODC1, ornithine decarboxilase 1; ORN, ornithine; OTC, ornithine transcarbamylase; PUT, putrescine; SSA, succinic semialdehyde; αKG, alpha ketoglutarate; γGBA, gamma guaninidinobutyrate. Figure created with BioRender.
Astrocytes are very abundant and heterogeneous cells in the central nervous system, where they participate in responses to a variety of noxious stimuli. In the case of AD, astroglial reactivity and dysfunction seem to precede amyloid damage. 2 Indeed, the most commonly-used marker of astroglial reactivity, GFAP, is increased centrally in AD, 31 as well as peripherally in the blood, in the pre-symptomatic phase. 32 We herein observed an increase in GFAP in hippocampal tissue at 4 and 16 weeks post-STZ, as well as increases in the enzymes, GS and MAO-B, which are expressed by astrocytes. Reduced GS has been associated with both glutamatergic toxicity and ammonia toxicity,21,33 while increased MAO-B is associated with increased GABAegic activity and ammonia toxicity.12,18
As the elevation in ammonia observed in AD affects mitochondrial ATP synthesis 21 and may contribute to reduced astroglial antioxidant defense, 34 we measured both the GSH content and ammonia levels directly. The decrease in GSH at both times highlights astroglial dysfunction, since the synthesis and recycling of this compound depends on the activity of astrocytes. 35 Measurement of hippocampal ammonia content indicates a worsening of this dysfunction within 16 weeks, during the amyloid phase.
The imbalance in the glutamate/GABA ratio has been addressed to justify the greater excitability observed in AD, as supported by reduced levels of GABA in brain tissue, in AD patients and in animal models.13–15 However, this is inconsistent with the potentially greater production of GABA, via MAO-B. To address this discrepancy, we investigated the sources and destinations of GABA, in the STZ-induced AD model, in the pre-amyloid and amyloid phases. Part of the ornithine in the brain can generate putrescine via a reaction catalyzed by ODC1. In turn, putrescine is the substrate that generates GABA through the action of MAO-B (see Figure 8). ODC1 levels increase significantly at 16 weeks in the STZ-induced AD model and an increase in urea is also observed, corroborating the hypothesis of an increase in urea cycle enzymes, as recently proposed during the amyloid phase of AD. 30 However, it is worth noting that the assay used here to measure ammonia includes urease and, therefore, does not allow us to distinguish urea and free ammonia in the hippocampal tissue.
Given that GABA may be synthesized via MAO-B, we also investigated the levels of GAD67, the neuronal enzyme responsible for the synthesis of GABA. No change was observed in the amount of this enzyme at 4 weeks post-STZ, but a reduction was observed at 16 weeks post-STZ. Considering that GAD67 can also be expressed in reactive astrocytes, 19 we cannot rule out the possibility of a neuronal reduction as early as 4 weeks. Nevertheless, it appears that, in the amyloid phase, MAO-B could be the main source of GABA. The astroglial GABA transporter (GAT-3) is responsible for the uptake of GABA from the synaptic cleft but can invert this gradient by acting as a GABA releaser in the cleft. The content of this transporter was not changed at 4 weeks post-STZ, but as occurred with GAD67, a decrease was observed in the amyloid phase. The decreases observed in GAD67 and GAT-3, in the amyloid phase, suggest a progression of the damage to GABAergic communication, but do not explain the reduction of GABA in the brain tissue.
We then investigated the gene expressions of two enzymes associated with GABA metabolism: GABA transaminase (GABA-T) and AGAT, in an attempt to explain the reduction in GABA levels. GABA transaminase initiates the use of GABA as an intermediate in the Krebs cycle, through succinic semialdehyde and then succinate, in accordance with the hypothesis that glucose hypometabolism in AD could be compensated by greater use of glutamate and GABA (GABA shunt, see Figure 8) as energetic substrates/intermediates of the Krebs cycle. 36 GABA-T expression did not change at 4 or 16 weeks post-STZ. However, at present we cannot exclude the possibility of further catabolism of newly synthesized GABA to succinate.
Finally, we evaluated the gene expression of AGAT, an enzyme that participates in the pathway for synthesizing creatine from glycine and arginine and that may provide information regarding the energetic situation in the STZ-induced AD model. In fact, the metabolic energy deficit observed in AD is also associated with a reduction in hippocampal creatine levels. 37 The AGAT reaction generates ornithine and guanidinoacetate (GAA). GAA is destined for creatine synthesis, but is also a GABA agonist. 38 Moreover, AGAT is also capable of catalyzing the reaction from GABA (instead of glycine) and arginine, generating γGBA (another GABA agonist) and ornithine. Ornithine could feedback the synthesis of GABA (via ODC1/MAO-B), or favor the synthesis of astroglial urea. 30 Note that, at 4 weeks after ICV- STZ, there is an increase in AGAT expression, perhaps suggesting an attempt to resist glucose hypometabolism. 39 However, at 16 weeks, there is a reduction in AGAT, which may accentuate the metabolic energy deficit in the amyloid phase. Possible increases in GAA and γGBA synthesis in the pre-amyloid phase (due to increased AGAT) support the idea of an increase in GABAergic activity earlier on in AD, 12 followed by an increase in excitability later, in the amyloid phase. However, this possibility should be confirmed by measuring the compound levels in brain tissue, as well as evaluating their exit to the synaptic cleft.
An important limitation of this work needs to be mentioned. Our interpretations about glutamatergic and GABAergic miscommunications in the brain tissue in the STZ-induced AD model are based on the content of enzymes and transporters, whose cellular and subcellular localizations have already been characterized in the literature. However, we did not measure the products of these enzymes, such as Glu, GABA or γGBA, directly. This is partly because measurements of these compounds in cerebrospinal fluid (which represent weighted brain averages) are inconclusive in demonstrating excitotoxicity, for example. Additionally, appropriate extracellular measurements in specific sites, such as hippocampal subregions, would require in vivo microdialysis assays (e.g., 40 ), which are not available and standardized at this time in our laboratory. We are aware that such measurements will be important in the future to validate our observations.
Conclusions
The increase in glial reactivity, based on increased GFAP, together with reduced GS and increased MAO-B, in the STZ-induced AD model, in both the pre-amyloid and amyloid phases, supports the hypothesis that astroglial dysfunction and ammonia neurotoxicity contribute to neurochemical and cognitive damage in the disease. The reduction in GSH levels and the increase in ammonia levels also reinforce this possibility. The increase in ODC1 (with a consequent increase in ammonia) and the decrease in AGAT (possibly coupled with a reduction in creatine) in the amyloid phase of the model may reflect the greater metabolic and energetic commitment of this phase. The reductions in the levels of GAD67 and GAT-3, also in the amyloid phase, demonstrate the progression of GABAergic impairment. Taken together, these changes highlight the importance of the ICV STZ-induced AD model and provide further knowledge of the pathophysiological mechanisms of AD in both phases of the disease.
Footnotes
Acknowledgments
The authors have no acknowledgments to report.
ORCID iDs
Author contributions
Éverton Carlos Brezolin (Conceptualization; Formal analysis; Investigation; Writing – review & editing); Vitor Gayger-Dias (Conceptualization; Formal analysis; Investigation; Writing – review & editing); Vanessa-Fernanda Da Silva (Formal analysis; Writing – review & editing); Anderson Cigerce (Formal analysis; Writing – review & editing); Bruna Schultz (Investigation; Writing – review & editing); Thomas Michel Sobottka (Formal analysis; Writing – review & editing); Patrícia Nardin (Writing – review & editing); Adriano Martimbianco de Assis (Writing – review & editing); Marina Concli Leite (Writing – review & editing); André Quincozes-Santos (Conceptualization; Funding acquisition; Writing – review & editing); Larissa Daniele Bobermin (Conceptualization; Formal analysis; Methodology; Writing – review & editing); Carlos-Alberto Goncalves, MD, PhD (Conceptualization; Funding acquisition; Project administration; Supervision; Writing – review & editing).
Funding
This research was supported by public research funding agencies in Brazil: Conselho Nacional de Desenvolvimento Científico Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS), and Instituto Nacional de Ciência e Tecnologia (INCT) para Saúde Cerebral (406020/2022-1) e para Excitotoxicidade e Neuroproteção (17/0516-3).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.