
Abstract
The SWI/SNF chromatin remodeling complex uses the energy of ATP hydrolysis to alter contacts between DNA and nucleosomes, allowing regions of the genome to become accessible for biological processes such as transcription. The SWI/SNF chromatin remodeler is also one of the most frequently altered protein complexes in cancer, with upwards of 20% of all cancers carrying mutations in a SWI/SNF subunit. Intense studies over the last decade have probed the molecular events associated with SWI/SNF dysfunction in cancer and common themes are beginning to emerge in how tumor-associated SWI/SNF mutations promote malignancy. In this review, we summarize current understanding of SWI/SNF complexes, their alterations in cancer, and what is known about the impact of these mutations on tumor-relevant transcriptional events. We discuss how enhancer dysregulation is a common theme in SWI/SNF mutant cancers and describe how resultant alterations in enhancer and super-enhancer activity conspire to block development and differentiation while promoting stemness and self-renewal. We also identify a second emerging theme in which SWI/SNF perturbations intersect with potent oncoprotein transcription factors AP-1 and MYC to drive malignant transcriptional programs.
Keywords
Overview
In the last 10 years it has become clear that the SWI/SNF chromatin remodeling complex is one of the most important human tumor suppressors, with a mutational rate in cancer rivaling that of the guardian of the genome TP53.1,2 The impact of SWI/SNF mutations on malignancy is profound, both in terms of the number of cancers in which these mutations are observed and in the breadth of pro-tumorigenic consequences they can elicit. Fueled by a plethora of genetic and biochemical studies in diverse model systems, and turbocharged by the advent of modern Omics approaches, the story of SWI/SNF is an epic tale that not only illustrates the essentiality of basic—curiosity-driven—research to cancer discovery, but highlights how the complexities of storing, retrieving, and accessing the genetic material creates vulnerabilities in which cancers can flourish. As the first decade of cancer-forward SWI/SNF research closes, it is worth reflecting on the extraordinary progress made in deciphering the normal functions of SWI/SNF and the ways in which it is disrupted in cancer, and reexamining what we know about how perturbations in SWI/SNF engage downstream tumorigenic processes. In this review, we will recap fundamental aspects of the SWI/SNF-cancer nexus and describe how contemporary studies are coalescing on the idea that enhancer dysregulation is key to understanding SWI/SNF-driven cancers. 3 Looking ahead, we discuss a promising new line of thinking in which mutations in SWI/SNF can also unleash the pro-tumorigenic potential of oncoprotein transcription factors like AP-1 and MYC. 4 Together, these emerging themes in SWI/SNF are beginning to expose the ways in which alterations to this macromolecular machine impact transcriptional regulatory networks to initiate, progress, and maintain the malignant state.
SWI/SNF as a Gate-Keeper of the Genetic Information
DNA within eukaryotic cells is tightly compacted into chromatin, a hierarchical structure that is assembled from a basic repeat unit known as the nucleosome. Within each nucleosome, approximately 150 base-pairs of DNA is wrapped around an octamer of histones H2A, H2B, H3, and H4, as well as a linker histone H1. These nucleosomes, in turn, are then looped, coiled, and further condensed into higher order structures, an exercise that crams roughly 2 m of DNA into a nucleus less than 10 µm in diameter. The fact that chromosomes are 10 000 times shorter than the DNA molecule they contain not only illustrates the extraordinary capacity of chromatin to compact the genetic information, but like any long-term storage solution raises the critical issue of material availability. For DNA, which must be both deeply archived and dynamically accessible, one solution to this problem are chromatin remodelers—enzymes that use the energy of ATP hydrolysis to transiently alter histone-DNA contacts within the nucleosome, thereby making segments of DNA available for replication, repair, and transcription. There are 4 distinct families of ATP-dependent chromatin remodelers but the first to be discovered, and the subject of this review, is SWI/SNF.
The discovery of SWI/SNF has its origins far removed from cancer and occurred at a time when the full impact of chromatin structure on nuclear events was yet to be appreciated. In 1984, 2 yeast genetic studies introduced the terms “SWI” and “SNF” into the lexicon and set the focus squarely on the regulation of transcription. In one study, 5 unique “SWI” (switch-defective) genes were uncovered that altered proper mating-type switching in yeast, 5 likely by regulation of transcription of the enzyme that initiates mating-type interconversion in this species. In the other study, 5 novel “SNF” (sucrose-non-fermenting) genes were discovered that control sucrose fermentation, again via control of transcription of a key enzyme in the process. 6 The realization that SWI2 was identical to SNF2, together with subsequent genetic and biochemical studies, ultimately led to the term “SWI/SNF” as a handle for the multi-subunit complex composed of SWI and SNF gene products.
In the decade following discovery of yeast SWI/SNF, several high-impact papers reported that the yeast complex functions as a general positive regulator of transcription7,8 and linked this function to its ability to cause changes in chromatin structure.9,10 Seminal studies that identified and characterized the evolutionarily conserved human version of SWI/SNF corroborated these findings11-15 and cumulatively this work laid the important foundational concept that SWI/SNF is a multi-subunit complex that hydrolyses ATP to increase the accessibility of DNA within nucleosomes. Consistent with the idea that a primary function of SWI/SNF is to remodel histone-DNA contacts, realization of the impact of SWI/SNF subsequently expanded to intersect with factors that mediate histone modifications—including the Polycomb repressor complex16-20—and with other DNA-centric events including DNA replication and repair.21-23 For DNA repair, it is possible that SWI/SNF has functions independent of chromatin remodeling, 24 but the majority of evidence indicates that SWI/SNF is directly recruited to damaged DNA sites,25-28 where it remodels nucleosomes to facilitate the repair process.26,29,30
Collectively, these studies paint a simplistic, yet important, view of SWI/SNF as a gate-keeper of the genome, freeing DNA sequences from the confines of nucleosomes so they may be bound by other proteins, transcribed, or repaired. Although this concept continues to permeate contemporary thinking, more recent discoveries (described below) have detailed critical elaborations and intricacies of mammalian SWI/SNF that help us understand its far-reaching consequences on gene expression and its many and varied tumor suppressive properties.
Characteristics of Mammalian SWI/SNF Complexes
In recent years, understanding of mammalian SWI/SNF has moved beyond the notion of a single complex that unpacks DNA to describe an elaborate set of SWI/SNF complexes that are capable of dealing with the complicated demands of genome access and gene regulation in mammals. It is now clear that there are 3 major mammalian SWI/SNF complexes, each containing over 10 subunits, and that these are combinatorially assembled from the products of 29 genes 31 (Figure 1). These 3 complexes are known as canonical BAF (cBAF), polybromo-associated BAF (pBAF), and non-canonical BAF (ncBAF, also known as GBAF32,33). Each complex contains a single ATPase subunit—either BRG1 or BRM1 (see Figure 1 legend for other subunit aliases)—a characteristic that led to the alternative nomenclature of “BAF” (BRG1/BRM1 associated factor) to describe these chromatin remodelers. Some subunits, including BAF155/BAF170 and BAF60A/B/C, which are essential for proper complex assembly, 34 are common to all 3 flavors of mammalian SWI/SNF. Some are shared between just 2 of the complexes, such as SNF5, which is found in both cBAF and pBAF. And other subunits are complex-specific, including ARID1A/B and DPF2 for cBAF, ARID2, PBRM1, and BRD7 for pBAF, and BRD9 and GLTSCR1/1L for ncBAF.31-34 This complexity is further amplified by the inclusion of tissue-specific SWI/SNF subunits, such as those selectively incorporated within subsets of neurons 35 or embryonic stem cells. 36 The elaborate nature of mammalian SWI/SNF composition raises the intriguing question of why, if these complexes ultimately perform the same biochemical function, has such diversity and plasticity evolved?
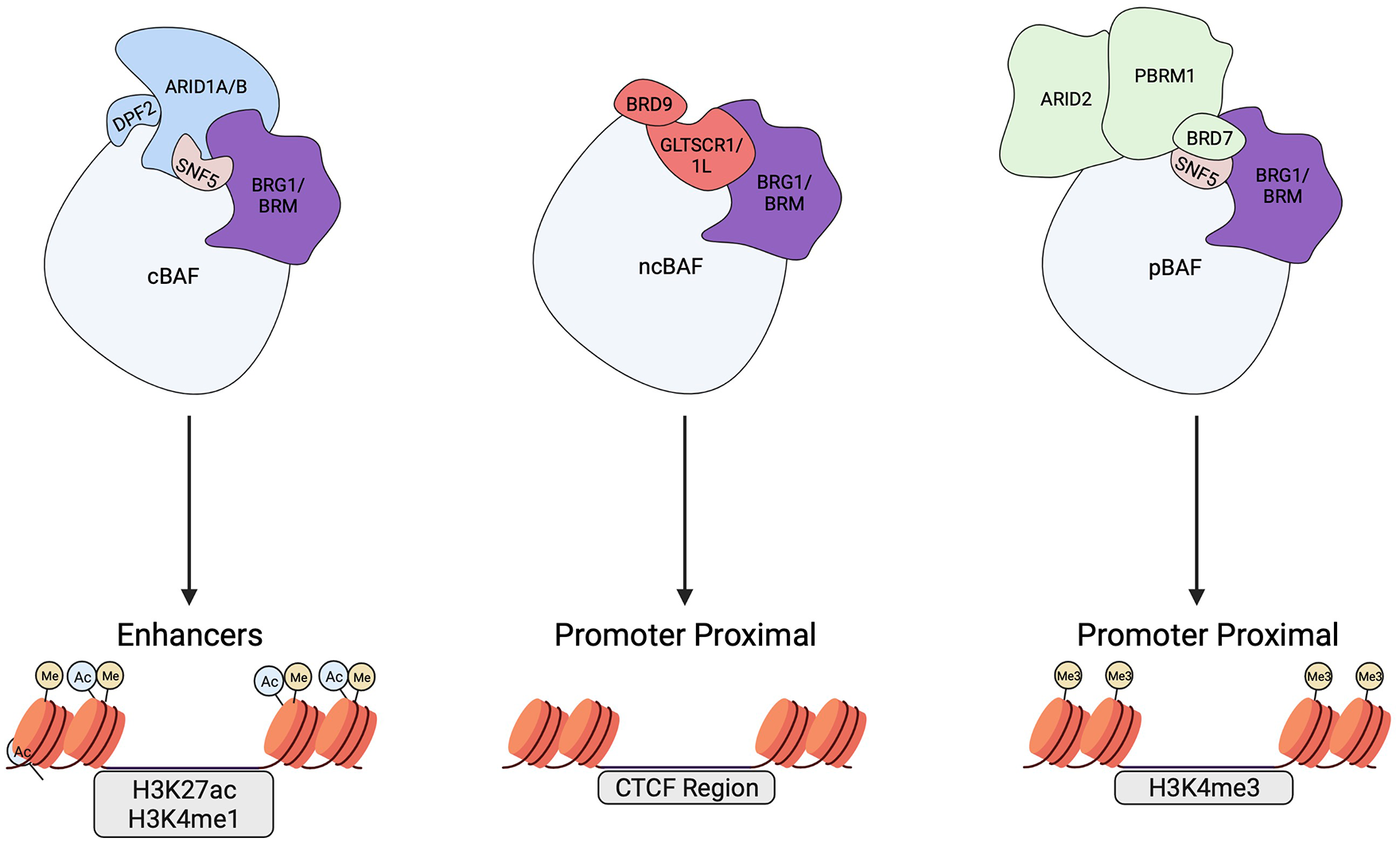
Three main mammalian SWI/SNF complexes. Illustration of the 3 main mammalian SWI/SNF complexes: cBAF, pBAF, and ncBAF. The core ATPase, BRG1 or BRM, and defining subunits are depicted. The SNF5 subunit which is only present in cBAF and pBAF is also shown. Arrows denote their respective typical genomic localization and genomic binding features. SNF5 is also called INI1, SMARCB1, and BAF47. BRG1 and BRM are also called SMARCA4 and SMARCA2, respectively. Additional SWI/SNF subunits such as BAF155 and BAF60 are not shown but are reviewed extensively elsewhere. Created with BioRender.com.
The most obvious answer to this question is that each type of SWI/SNF complex is required for non-redundant and specific genomic functions, and that these elaborations are important in determining when, where, and how its chromatin remodeling activities will be used. Sites of genomic action, in particular, are a key point of divergence between the different SWI/SNF complex types. In general, chromatin-bound SWI/SNF is detected at tens of thousands of locations across the genome,19,31,37 commonly including critical regulatory elements such as transcription start site (TSS)-proximal promoters, CTCF insulator sites, RNA polymerase II and III transcription units, and promoter-distal (eg, enhancer) regions. 37 Within this broad framework, however, physical and functional distinctions between the different SWI/SNF complexes are apparent. Canonical BAF complexes, tracked by their defining subunits SNF519,31,38-40 and ARID1A or ARID1B,33,41-44 play an important role in maintaining active enhancers marked by histone H3 lysine 27 acetylation (H3K27ac) and histone H3 lysine 4 mono-methylation (H3K4me1)45-47 (Figure 1, left side). Polybromo-associated BAF, defined by ARID2, does show some promoter-distal action, 44 but is mostly detected at active promoters marked by histone H3 lysine 4 tri-methylation (H3K4me3) or bivalent promoters marked by H3K4me3 and histone H3 lysine 27 tri-methylation (H3K27me3)19,43,44 (Figure 1, right side). The same proclivity for promoter-proximal binding applies to ncBAF, defined by its unique BRD9 subunit31,33,34,48 (Figure 1, middle), although ncBAF has also been observed at enhancers,34,48 CTCF sites, 31 and promoter-distal intergenic/intragenic regions. 48
Evolution has clearly driven a “divide and conquer” strategy in terms of the functional significance of SWI/SNF complexity in mammals. This strategy enables the chromatin remodeling functions of SWI/SNF to be selectively deployed to one type of gene regulatory element versus another, vital for mediating complex gene expression programs that have tissue/cell-type selectivity or developmental transitions. All of this complexity, however, comes at a cost. Just as more elaborate machines have more points of failure, mammalian SWI/SNF has more failure points, and should a situation arise where a SWI/SNF subunit is no longer properly incorporated into its complex chances are high that selective genomic functions can be lost or new ones emerge—which is precisely what is seen in cancers defined by SWI/SNF mutation.
SWI/SNF Mutations in Cancer
Around the turn of this century, papers started appearing describing mutations in SWI/SNF subunits in a variety of human malignancies.49-59 The full extent of the phenomenon came into sharp focus in 2013, when 2 landmark publications systematically analyzed cancer genome/exome sequencing data and concluded that genes encoding SWI/SNF subunits are mutated in ~20% of all cancers,1,2 a frequency approaching that of the prominent tumor suppressor TP53. Non-genetic mechanisms, including epigenetic changes, drive the effective frequency of SWI/SNF dysregulation in cancer even higher. 60 Like TP53, SWI/SNF mutations occur in a staggering array of malignancies—including endometrioid, ovarian, bladder, gastric, liver, colorectal, pancreatic, breast, and oral cancers1,2—and are generally considered deleterious, 2 suggesting that loss of SWI/SNF function(s) is inherently pro-tumorigenic. Consistent with this notion, experiments in engineered mouse models confirmed that inactivation of just a single SWI/SNF subunit—including ARID1A, BRG1, ARID2, PBRM1, and SNF5—promotes cancer,52,61-67 earning multiple components of SWI/SNF their bona fide tumor suppressor credentials.
Perhaps the clearest example of how loss of a SWI/SNF subunit drives malignancy comes from the study of childhood rhabdoid tumors, so-named because the tumor cells resemble rhabdomyoblasts. Presenting in the brain, where they are called atypical teratoid rhabdoid tumors (AT/RT), or in soft tissue, where they are called malignant rhabdoid tumors (MRT), these cancers are rare, aggressive, and have dismal outcomes.68,69 Early on, rhabdoid tumors were recognized to be associated with loss of SMARCB1, which encodes the SNF5 component of the cBAF and pBAF complexes.49-51 Equally prescient, early modeling in mice confirmed that loss of SMARCB1 is sufficient to drive tumorigenesis in vivo.52,58,61-63 It is now clear that SMARCB1 is inactivated or lost in nearly 100% of rhabdoid tumors cases, and that SMARCB1 inactivation is often the only recurring mutation found in rhabdoid tumor genomes 70 —a striking demonstration of the potential of a change in SWI/SNF to propel malignancy.
The unusually simple genetic profile of rhabdoid tumors, and the astonishing ability of a single genetic lesion to drive cancer in this instance, has made rhabdoid tumors popular and productive territory in which to dissect how SWI/SNF subunit loss results in tumor formation. But SMARCB1-deficient cancers are rare, and as such the high frequency of SWI/SNF mutations in cancer is driven largely by changes in genes encoding other SWI/SNF components. Mutations in ARID1A, for example, occur in neuroblastomas as well as colorectal, bladder, gastric, lung, and liver cancers,1,71,72 and are particularly prevalent in ovarian clear cell and endometrioid carcinomas, where they are found in between 30 and 60% of all cases.1,54,55 Truncation mutations in the gene encoding PBRM1 (a defining subunit of pBAF) are observed in over half of all clear cell renal cell carcinomas (ccRCC), making PBRM1 the second most highly-mutated gene in ccRCC.2,57,73 And ARID2, the gene that encodes the ARID2 subunit of pBAF, is frequently inactivated in melanoma 74 and hepatocellular carcinoma, 56 adding to the broad range of malignancies in which inactivation of a SWI/SNF subunit is observed.
Although the above examples involve mutations in non-ATPase components of SWI/SNF, it is worth pointing out that even the ATP hydrolyzing functions of SWI/SNF, core to its chromatin remodeling activities, are not immune to disruption in cancer. Recall that all 3 SWI/SNF variants carry 1 of 2 mutually exclusive ATPases, BRG1 (encoded by the SMARCA4 gene) or BRM (encoded by SMARCA2). In a rare and aggressive ovarian cancer called small cell carcinoma of the ovary, hypercalcemic type, (SCCOHT), inactivating mutations in SMARCA4 lead to a complete loss of the BRG1 protein.75-77 Interestingly, these cancers do not express BRM, 78 meaning that these malignancies thrive in the absence of ATP-dependent SWI/SNF chromatin remodeling activity. A similar scenario plays out in BRG1-null thoracic cancers, 79 subsets of non-small cell lung cancers, 80 and 10% to 20% of other cancers including bladder, pancreas, colon, and breast tumors. 81 We will return to the intriguing issue of how residual SWI/SNF complexes that remain following dual ATPase loss 82 can impact cancer-driving processes later in this review.
In sum, data collected in tumors, cell lines, and mouse models have solidified the concept that the SWI/SNF chromatin remodeler is a preeminent human tumor suppressor complex and have paved the way for researchers to begin to unravel the mechanisms by which loss of SWI/SNF subunits drives tumorigenesis.
Mechanisms of Tumorigenesis
The mechanisms of tumor suppression by SWI/SNF, and the ways in which mutations in SWI/SNF subunits promote cancer, are active areas of research and there is still much to learn. That said, evidence from multiple studies is beginning to point to common themes by which SWI/SNF subunit mutations drive oncogenesis. One dominant mechanistic theme involves dysregulation of enhancer-mediated gene expression; a combination of disrupting the function of SWI/SNF at enhancers linked to cell differentiation and development, while retaining or promoting new actions of SWI/SNF at enhancers driving pro-tumorigenic gene expression. Although invocation of enhancer dysfunction can explain how mutations in SWI/SNF induce widespread changes in gene expression programs, this mechanism is unlikely to be the sole way in which these mutations act. Indeed, more recent studies have started to expose a second theme with pointed connections to established oncogenic processes and involving a direct intersection with oncoprotein transcription factors. We discuss the evidence for, and implications of, each theme below.
Enhancer dysregulation
The normal actions of SWI/SNF are, in large part, tied to the proper selection and maintenance of enhancers. SWI/SNF has been shown to increase enhancer-associated gene expression programs through chromatin remodeling and nucleosome shifting,83,84 to mediate enhancer-driven gene expression patterns essential for differentiation and lineage commitment,85-87 and to collaborate with signal-responsive transcription factors to activate cell type-specific enhancer function. 88 It is not surprising, therefore, that much of the impact of SWI/SNF mutations is directed toward enhancer dysregulation, particularly with respect to enhancers that govern cell identity.
In rhabdoid tumor cell lines which lack SNF5, reintroduction of SNF5 results in prominent changes in enhancer function, evidenced by an increase in TSS-distal binding of SWI/SNF subunits to chromatin and a concomitant boost in histone marks associated with active enhancers: H3K4me1 and H3K27ac. 39 Notably, a majority of active enhancer sites gained upon SNF5 reintroduction are associated with transcriptional control of differentiation and development genes.19,88 Together with the idea that a normal function of SWI/SNF is to regulate enhancer-mediated gene expression during development, these studies make a strong case for the notion that loss of SNF5 drives tumorigenesis, at least in part, by causing the collapse of enhancers that maintain a less stem-like, more differentiated, cellular state.
This concept is further amplified by studies of a second cBAF complex member, ARID1A. Experiments to determine how ARID1A inactivation contributes to colorectal cancer have shown that removal of ARID1A causes a loss of SWI/SNF binding at enhancers marked by H3K27ac and H3K4me1. 65 Sites that lose chromatin-bound SWI/SNF also show reduced H3K27ac signal and reduced expression of linked developmental genes, 65 echoing the idea that, as for SNF5, the most conspicuous effects of loss of ARID1A is collapse of cell identity enhancers. A separate study performed in the context of ovarian cancer reached a similar set of conclusions, 89 but interestingly also reported induction of promoter-proximal H3K27ac levels upon ARID1A loss, reminding us that SWI/SNF perturbations can have context-specific effects and that any broad generalizations—such as those we draw here—inevitably break down as studies expand.
Additional ties to enhancer dysregulation have come from studies of ARID1B—a subunit that integrates into SWI/SNF in a mutually-exclusive way with ARID1A. ARID1B has received considerable attention as a potential therapeutic vulnerability in ARID1A-mutant cancers, 90 as loss of ARID1B is synthetically lethal with loss of ARID1A. 90 Consistent with this concept, ARID1B knockdown in cell lines replete with ARID1A has little if any effect on open chromatin status or enhancer function. 41 In contrast, its loss in ARID1A-null cells leads to prominent changes in chromatin accessibility—most of which occur at enhancers. Interestingly, genes with the largest reduction in expression upon dual ARID1A/ARID1B loss, compared to ARID1A or ARID1B loss alone, are linked to growth factor signaling or are oncogenes (eg, JUN, FOSB, MYC), suggesting that the enhancers controlling their expression only become ARID1B-dependent in an oncogenic setting. It appears, therefore, that SWI/SNF can select between enhancers that govern cell identity and those that govern cell growth and proliferation, and that this selection is determined by the mutagenic background and by the specific subunits that are incorporated into the SWI/SNF complex.
Discussion of enhancers in this context would not be complete without a description of super-enhancers, which are clusters of enhancers discernible by their size, transcriptional potency, and density of regulatory proteins.91,92 Super-enhancers are near the top of the hierarchy in terms of transcriptional mechanisms controlling cell identity, and their de novo appearance or repurposing on the route to tumor formation is thought to lock in the identity of a cell as malignant.91,93 Oncogenic super-enhancers are implicated in a wide-variety of cancers,44,91,93,94 including those marked by SWI/SNF loss. In rhabdoid tumors, loss of SNF5 is inferred to cause depletion of residual SWI/SNF complexes from a majority of traditional enhancers, but at the same time permits binding of the remaining complexes to a set of super-enhancers that are essential for rhabdoid tumor survival and plasticity. 39 Similarly, loss of ARID1A results in the widespread collapse of traditional enhancers, but also promotes H3K27ac, increased chromatin accessibility and increased activity at super-enhancers important for malignant invasion. 95 Arguably, the ability of SWI/SNF mutations to discriminate between traditional enhancers and super-enhancers contributes to the regulatory mayhem they induce, promoting cellular identity as oncogenic while at the same time attenuating the ability of cancer cells to undergo development or differentiation.
Oncogene activation
The loss of enhancers controlling expression of genes linked to differentiation and development, combined with activation of super-enhancers linked to cell survival and invasion, makes a tidy argument for how mutations in a single SWI/SNF subunit can promote cancer. And there are compelling reasons to accept this mechanism as a malignant driver in these tumors. But more recently, evidence has emerged of a deeper tumorigenic mechanism at play in at least some SWI/SNF mutant cancers. This mechanism is not mutually exclusive with the concept of enhancer dysregulation, but it does add an important new layer to our understanding of how these cancers form. As we describe for the rest of this review, there is provocative evidence that at least some cancer-driving mechanisms at work in SWI/SNF-altered cancers result from physical and functional interactions of SWI/SNF with oncoprotein transcription factors AP-1 and MYC.
Activator protein-1 (AP-1) refers to a collection of basic leucine zipper transcription factors that function as dimers, made up of assemblies of various members of the JUN, FOS, ATF, and MAF family of proteins. 96 Some AP-1 proteins have tumor-suppressive function, 97 while others (including c-FOS, c-JUN, and FOSB) are encoded by bona fide oncogenes.97,98 Oncogenic AP-1 proteins drive transformation, are overexpressed in tumors such as breast, skin, and liver cancer,97,98 and can be thought of as signal-responsive transcription factors that connect RAS/MAPK signaling to the regulation of genes important for proliferation, migration, apoptosis, and differentiation. 98 AP-1 proteins directly interact with subunits of SWI/SNF88,99,100 and recruit SWI/SNF to sites bound by lineage-specific transcription factors to facilitate gene expression programs that drive differentiation during development.88,101 The positioning of AP-1 between RAS/MAPK and SWI/SNF has obvious advantages in terms of enforcing signal-responsive developmental programs, but also sandwiches AP-1 between a major oncoprotein and a major tumor suppressor, raising the question of what happens in a cell that has ectopic RAS signaling or altered SWI/SNF function?
An obvious connection between SWI/SNF mutations and RAS signaling is seen in pancreatic ductal adenocarcinoma (PDAC)—subsets of pancreatic cancers marked by KRAS mutations and frequent (~20%) alterations in SWI/SNF. Pancreas-specific inactivation of Arid1a in developing mice is sufficient to cause pancreatic inflammation and intraepithelial neoplasia, and synergizes with KRAS activation to accelerate development of intraductal papillary mucinous neoplasms. 102 Similar findings were reported upon suppression of ARID1A in KRAS-mutant adult mouse pancreatic acinar cells, 103 and are connected to reduced chromatin accessibility at acinar-specific enhancers enriched with AP-1 binding motifs. Neither of these studies looked directly at the impact of SWI/SNF and KRAS mutations on AP-1 binding to chromatin, but parallel work in colorectal cancer cells filled this gap. In ARID1A-null colorectal cancer cell lines, knockdown of ARID1B results in widespread enhancer collapse and loss of AP-1 binding at these enhancers, likely because of altered nucleosome spacing around AP-1 motifs. Importantly, enhancers that lose AP-1 binding are linked to genes that mediate signaling events within the RAS/MAPK and PI3K pathways. 41 Although it is unknown if the SWI/SNF–ARID1A/1B connection in this instance is due to a direct physical interaction between AP-1 and SWI/SNF, this example illustrates how loss of a SWI/SNF subunit can promote a functional association between AP-1 and residual SWI/SNF complexes that drives an overtly oncogenic transcriptional program.
AP-1 also features in cancer types carrying mutations in additional SWI/SNF components. In BRG1-null SCCOHT tumor cells, reintroduction of BRG1 promotes an epithelial gene signature that is AP-1-dependent, 104 implying that loss of BRG1 drives an epithelial–mesenchymal transition (EMT) in these cancers, in part, by robbing AP-1 of its ability to sustain epithelial-like gene expression programs. In SNF5-null rhabdoid tumor cells, introduction of SNF5 leads to activation of enhancers enriched in AP-1 binding motifs,19,39,105 and conversely loss of SNF5 in mouse embryo fibroblasts results in collapse of enhancers (decreased H3K27ac) marked by AP-1 motif enrichment. 38 In both cases, these enhancers are linked to development and differentiation, reinforcing this recurring theme in tumorigenesis by SWI/SNF and highlighting AP-1 as a prime target for dysregulation in these malignancies.
Our groups recently reported yet another connection between SWI/SNF and AP-1. 4 Seeking to understand how residual SWI/SNF complexes in rhabdoid tumor cells sustain oncogenic transcriptional events, we performed gene expression and chromatin accessibility profiling following acute depletion of BRG1. Despite the low levels of residual SWI/SNF complexes in these cells, 39 depletion of BRG1 reduces chromatin accessibility at more than 6000 sites across the genome, revealing that residual SWI/SNF complexes maintain chromatin accessibility even in the absence of SNF5. A majority of these sites are promoter-distal, including histone-marked enhancers carrying AP-1 binding motifs. The genes associated with reduced chromatin accessibility upon BRG1 depletion show a commensurate decrease in expression and are overtly pro-tumorigenic, enriched in genes controlling known cancer hallmarks such as migration, angiogenesis, and signaling. These findings suggest that when SWI/SNF complex function is altered by SNF5 loss, an AP-1–SWI/SNF interaction at cancer-driving genes is either created or selectively preserved (Figure 2). Amidst the backdrop of widespread loss of SWI/SNF function at enhancers controlling differentiation and development, the coalescence of residual SWI/SNF function at these AP-1 sites further strengthens the idea that AP-1 is a prominent player in tumor-promoting mechanisms of SWI/SNF mutant cancers.

Enhancer dysregulation by SWI/SNF subunit loss. (A) Normally, SWI/SNF binds the AP-1 transcription factor to regulate open chromatin at enhancer regions. These enhancers are associated with genes involved in cell lineage and differentiation (left). At the same, the binding of SWI/SNF at these enhancers prevents the complex from acting at enhancers that control the expression of genes that promote cell growth and tumorigenesis (right). (B) Upon loss of the SNF5 or ARID1A subunits in cancer, cell lineage/differentiation enhancer regions are no longer accessible and enhancer histone marks are reduced, while other enhancer regions, some of which are super-enhancers, are activated to promote tumorigenesis. Although not formally demonstrated, this process may involve direct AP-1–SWI/SNF interactions as denoted by the question mark on AP-1. Created with BioRender.com.
Beyond the AP-1-SWI/SNF connection, another transcription factor with ties to SWI/SNF is MYC, a family of 3 related oncoproteins (c-, N-, and L-MYC) that collectively are overexpressed in more than half of all malignancies. 106 The oncogenic functions of MYC flow from its ability to control the expression of genes linked to protein synthesis, cell growth, the tumor microenvironment, angiogenesis, invasion, and metabolism, actions that in turn are dependent on its interactions with MAX 107 and recognition of DNA sequence motifs—E-boxes—in the regulatory elements of its target genes (Figure 3). MYC is invariably bound at the promoters of the genes it regulates, which typically contain high affinity E-box sequences, but can occupy lower affinity E-box (and non-E-box) sequences at enhancers when its levels rise. 108 Overexpression of MYC in cancer can be driven by changes in the MYC loci, such as amplification or translocation.106,109 MYC can also be overexpressed in cancer by activation of other oncogenic, or loss of other tumor-suppressive, pathways, including RAS, 110 APC, 111 and NOTCH. 112 The myriad ways in which MYC levels can be induced by oncogenic events undoubtedly underlies its pervasive overexpression in cancer. But MYC can also be activated in a less conspicuous manner by events that leave its expression unchanged but nonetheless unleash its oncogenic function—as our recent work on MYC and SNF5 105 revealed.
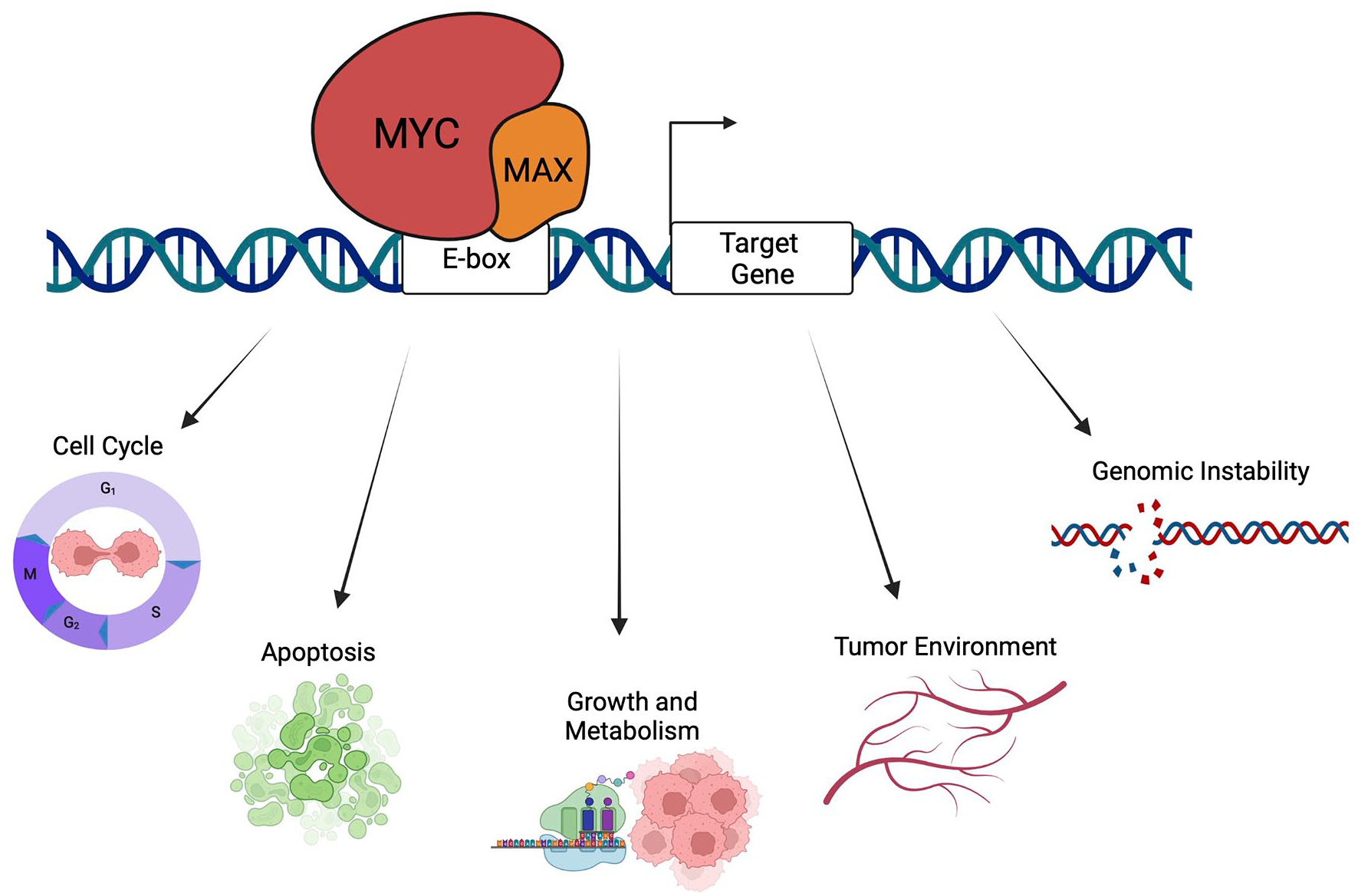
MYC regulate expression of genes linked to cancer hallmarks. MYC binds to DNA as a heterodimer with its obligate binding partner MAX at specific DNA sequences across the genome. MYC can impact expression of genes involved in fundamental cell processes such as translation, cell growth, cell cycle, metabolism, and apoptosis. In addition, MYC can regulate genes that control angiogenesis, tumor environment, metastasis and has known roles in controlling genomic stability. Created with BioRender.com.
A high-profile study in the 1990s showed that the carboxy-terminal DNA binding domain of MYC interacts directly with SNF5, 113 and it was originally proposed that this interaction promotes the ability of MYC to drive oncogenic transcriptional programs. Subsequent work confirmed a direct physical interaction between the 2 proteins.105,114,115 Although the idea that SNF5 is a co-activator for MYC is at odds with its tumor-suppressive functions,52,63 to be fair this discovery was made prior to realization of the impact of SNF5 loss in cancer, and over the ensuing years it became clear that the relationship between MYC and SNF5 is not collaborative but rather antagonistic. Multiple lines of evidence—from tumors as diverse as MRT and PDAC—reveal that loss of SNF5 is associated with activation of MYC target gene signatures.58,116,117 In addition, reintroduction of SNF5 into rhabdoid tumor cell lines suppresses MYC target gene expression and MYC-dependent transformation, 114 genetic inhibition of MYC suppresses AT/RT tumor cell growth in vitro and in vivo, 118 and structural studies went on to show that interaction with SNF5 is fundamentally incompatible with the ability of MYC to bind E-box DNA in vitro. 115
To dissect the underlying mechanisms at play, we used a combination of genetic, genomic, and biochemical techniques to examine how SNF5 regulates the transcriptional activities of MYC. 105 We confirmed that SNF5 inhibits the DNA binding ability of MYC/MAX dimers in vitro, and showed that acute depletion of SNF5 from cells increases the interaction of MYC with target gene chromatin without altering MYC levels. We also found that reintroduction of SNF5 into MRT cells causes a decrease in chromatin binding by MYC, resulting in an inhibition of RNA polymerase II pause release at MYC target genes. Indeed, the transcriptional consequences of SNF5 re-expression in MRT cells mirrors that of MYC inhibition—revealing that a significant portion of the transcriptional actions of SNF5 in this setting are directed toward MYC. Thus, independent of any changes in MYC protein expression, oncogenic loss of SNF5 can activate MYC at a functional level, providing a simple rationale for the frequent involvement of MYC target gene signatures in rhabdoid tumors.
Based on this initial study, we took a step back and looked more broadly at the association of MYC with SWI/SNF components. 119 We discovered that, in addition to SNF5, MYC also interacts directly with the pan-SWI/SNF subunit BAF155. Intriguingly, we also learned that MYC and SNF5 compete for interaction with BAF155, revealing that SNF5 effectively blocks the ability of MYC to recognize its docking site on the BAF155 protein. The implication of these findings is that SNF5 can have 2 independent inhibitory actions against MYC: One that impacts the ability of MYC to bind DNA and another that prevents access of MYC to BAF155. By extension, this concept further predicts that MYC has unrestricted access to both its target genes and to residual SWI/SNF complexes in rhabdoid tumor cells, and may use these complexes to drive malignant gene expression programs in this setting. In agreement with this notion, we more recently discovered that inhibition of residual SWI/SNF function through acute degradation of BRG1 impairs MYC-target gene expression in rhabdoid tumor cells. 4 Taken together, these data reveal that there are at least 2 anti-MYC functions of SNF5, and show that residual SWI/SNF complexes in rhabdoid tumor cells actively support the pro-tumorigenic transcriptional functions of MYC (Figure 4).

Dual mode of MYC inhibition by SNF5. (A) SNF5 acts as a tumor suppressor through 2 direct modes of inhibition that converge directly on MYC: antagonizing MYC binding to DNA and repressing MYC interactions with other SWI/SNF subunits. (B) When SNF5 is not present, MYC has unrestricted access to target genes and interactions with SWI/SNF complexes that remain following SNF5 loss, called residual SWI/SNF. The dual loss of SNF5-mediated repression causes increased MYC-target gene expression. Created with BioRender.com.
The SWI/SNF–MYC connection is not solely centered on SNF5 and rhabdoid tumors. MYC target gene signatures are activated in SCCOHT tumor samples, 120 and the BAF155 subunit of a SCCOHT-specific residual SWI/SNF complex 82 co-localizes with MYC at conserved target genes, 119 hinting at yet another functional interaction of MYC with SWI/SNF that is yet to be explored. In neuroblastomas, there is a strong genetic association between MYCN amplification and ARID1A deletion 121 and overexpression of N-MYC in mouse neural crest cells (NCC) drives tumor formation in a way that preferentially selects for NCC bereft of Arid1a. 121 Loss of ARID1A in MYCN-amplified cells has also been shown to result in a reduced differentiation ability and an increased resistance to cisplatin 44 —a major point of failure for treating neuroblastomas in the clinic. The strong implication here is that that ARID1A-containing SWI/SNF complexes, like those containing SNF5, are normally potent suppressors of MYC activity. Precisely how ARID1A loss activates MYC, and if and how MYC is making use of residual SWI/SNF in these cancers, remains to be determined.
The intersection of SWI/SNF with prominent oncoprotein transcription factors such as AP-1 and MYC is an intriguing development in the tale of SWI/SNF and cancer, and has clear implications for how these cancers can be better understood and perhaps one day treated. 122 But this may just be the tip of the iceberg. Additional interactions between SWI/SNF components and oncogenic transcription factors have been reported,123-126 and it will be interesting in the future to see how these interactions are altered by tumor-associated SWI/SNF mutations and how these alterations contribute to malignant progression.
Conclusion
Not too long ago, the received wisdom was that the cellular apparatus connected to core nuclear events such as packing and unpacking chromatin was too big to fail in cancer, and that there would be limited opportunities for mutations to arise that could drive malignancy without also compromising essential nuclear processes. But like the discovery of oncohistones, 127 the realization of potent and varied tumor suppressive actions of SWI/SNF has shattered that view, and shown how the complexities of regulating gene expression in mammals create vulnerabilities that are readily exploited by cancer. The last decade has witnessed an explosion in our appreciation of the role of SWI/SNF in malignancy and an understanding of the underlying molecular mechanisms. Transcriptional dysregulation has been at the heart of these efforts, and revealed that enhancers and super-enhancers, as well as oncoprotein transcription factors, are prime targets for corruption in cancers driven by SWI/SNF loss. The next decade will surely bring more surprises, hopefully those that shine a light on new ways that SWI/SNF mutant cancers can be treated. Given the sheer breadth of cancers linked to SWI/SNF, the impact of future work on targeting residual SWI/SNF complexes or disabling the downstream oncogenic pathways on the landscape of human cancer therapy will likely be profound.
Footnotes
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research on SWI/SNF in our laboratories is or has been supported by a Tennessee Louis Stokes Alliance for Minority Participation award to C.A.J., a Rally Independent Investigator award from the Rally Foundation for Childhood Cancer Research to A.M.W, Alex’s Lemonade Stand Foundation (W.P.T.), St. Baldrick’s Foundation (W.P.T.), and grant CA247833 from the NIH/NCI to A.M.W. and W.P.T.
Author Contributions
CAJ, WPT, and AMW wrote the manuscript.