
Abstract
Scleritis is a rare, potentially sight-threatening, painful eye disease. Based on its anatomical involvement, it can be categorized as anterior and posterior scleritis. There are several possible causes, among which infectious and noninfectious origins should be considered. From the therapeutic aspect, it is important to clarify the infectious origin, to provide target treatment, or to identify the possible underlying autoimmune disease. Corticosteroid therapy is considered to be the basis for the stepwise treatment of scleritis. In this article, we describe the management of three patients (investigations, stepwise approach of therapy, and treatment difficulties) who developed three different types of scleritis: anterior non-necrotizing scleritis, anterior necrotizing scleritis, and scleromalacia perforans. The differential diagnosis of scleritis and its management after diagnosis pose difficulties in clinical practice. In general, the therapeutic approach is based on the principle of early and individualized treatment, which depends on the nature and severity of the patient’s inflammatory eye disease and the presence or absence of associated systemic diseases.
Introduction
Scleritis is a predominantly unilateral inflammatory condition affecting the sclera, though bilateral involvement may occur. It can be divided into anterior and posterior scleritis based on anatomical localization. Based on the cause, it can be classified as infectious or noninfectious, that is, in the latter case, immune-mediated scleritis. In case of anterior scleritis, it is important to distinguish between necrotizing and non-necrotizing subtypes. The non-necrotizing subtype could be nodular or diffuse; the necrotizing subtype, a distinction is made between inflammatory and non-inflammatory conditions (scleromalacia perforans).1,2 Given its potential to significantly impair vision and quality of life, early recognition and appropriate management of scleritis are crucial in preventing serious ocular complications and improving patient outcomes.
Scleritis may be due to local or systemic infection. Local infection can be caused by a number of pathogens, such as herpes simplex, varicella zoster, acanthamoeba, bacteria, and even fungi, or other systemic infections such as syphilis or tuberculosis (TB) can cause scleritis.3,4
Scleritis of noninfectious origin may be idiopathic, but approximately half of the patients have underlying systemic diseases such as rheumatoid arthritis (RA), small vessel vasculitis (Wegener’s, Churg-Strauss), medium- and large-vessel vasculitis (polyarteritis nodosa), HLA-B27 spondylarthropathy (ankylosing spondylitis), polychondritis, or systemic lupus erythematosus (SLE). The surgically induced form, most often associated with pterygium or strabismus surgery, is a common cause. It is important to note that malignant ocular processes, such as uveal melanoma, conjunctival tumors, and lymphoma, can mimic scleritis. Scleritis induced by drugs (bisphosphonates, etanercept, and topiramate) may also occur.5,6
Scleritis can be associated with other ocular conditions: keratitis, uveitis, elevated intraocular pressure, vitritis, cystoid macular edema, papilledema, retinal or choroidal detachment associated with posterior scleritis.7,8 Necrotizing scleritis carries a higher risk for developing additional ocular complications. 9
Distinguishing between infectious and immune-mediated scleritis is crucial, given the significant divergence in their treatment protocols. The diagnostic approach for scleritis begins with a comprehensive collection of the patient’s ocular and general history, followed by an extensive ophthalmic examination, which includes a visual acuity assessment, intraocular pressure measurement, slit-lamp examination of the anterior and posterior segments, and an ultrasound evaluation of the posterior segment.
Laboratory investigations, including complete blood count and inflammatory markers (CRP, ESR), form the cornerstone of diagnosis. Assessments of kidney and liver function further contribute to the diagnostic framework. Serological tests are imperative for identifying infectious agents, with specific tests targeting Treponema pallidum, such as the Rapid Plasma Reagin, Venereal Disease Research Laboratory, and Fluorescent Treponemal Antibody Absorption (FTA-Abs) Enzyme-Linked Immunosorbent Assay (ELISA) for Immunoglobulin G (IgG) and Immunoglobulin M (IgM), alongside tests for Borrelia burgdorferi and various viruses. The immunoserological landscape is delineated through markers such as HLA-B27, antinuclear antibodies (ANA), antineutrophil cytoplasmic antibodies, rheumatoid factor (RF), and cyclic citrullinated peptide antibodies. The Quantiferon test, indicative of tuberculosis, and the measurement of angiotensin-converting enzyme and serum lysozyme levels, suggestive of sarcoidosis, further refine the diagnostic approach. 5
In cases where infectious scleritis is suspected, microbiological cultures are essential to complement serological investigations. The presence of purulent exudates, conjunctival and scleral ulceration, or abscess formation, especially when accompanied by pus on the ocular surface or within the anterior chamber, underscores the necessity for these additional tests. 8
Diagnostic imaging, including chest and sacroiliac X-rays, B-scan ultrasound, optical coherence tomography of the anterior and posterior segments, and CT/MRI scans when B-scans are inconclusive, enhances the diagnostic accuracy. Collaboration with specialists in rheumatology, dermatology, and other relevant fields is recommended to address systemic symptoms and integrate findings from various tests.5,10
This comprehensive diagnostic approach not only elucidates the etiology of scleritis but also guides the selection of the most appropriate therapeutic regimen, thereby optimizing patient outcomes.
A wide spectrum of drugs is available for the treatment of scleritis to reduce the existing inflammation. Corticosteroids are the basis of therapy for scleritis and are very effective and rapid in reducing inflammation. However, long-term use may promote various side effects. In such cases, starting immunomodulatory therapy may be necessary to maintain inflammatory control without the side effects of corticosteroids.11,12
First-line treatment encompasses topical NSAIDs and corticosteroids, with targeted delivery through subconjunctival or sub-Tenon injections when indicated. For more targeted delivery, subconjunctival or sub-Tenon corticosteroid injections, utilizing agents such as dexamethasone and triamcinolone, are employed. Cyclosporine drops are particularly favored in cases presenting with corneal involvement or peripheral ulcerative keratitis (PUK). Systemic therapy introduces non-steroidal anti-inflammatory drugs (NSAIDs), with ibuprofen, indomethacin, and celecoxib as notable mentions, alongside corticosteroids like methylprednisolone administered orally or via pulse therapy for more severe cases. The transition to immunomodulatory agents is prompted by persistent inflammation or corticosteroid-induced adverse effects, underscoring the need for a balanced and responsive treatment approach.
When first-line therapies falter, second-line agents such as antimetabolites, T-cell inhibitors, and alkylating agents should be considered. Methotrexate (MTX), azathioprine, and mycophenolate mofetil (MMF) stand out among antimetabolites, with methotrexate gaining prominence in managing necrotizing scleritis, as evidenced by other retrospective case series.13,14 T-cell inhibitors such as cyclosporine and tacrolimus, along with alkylating agents such as cyclophosphamide, offer alternative pathways for immune modulation, catering to diverse patient needs and disease dynamics.
Third-line biological therapies, particularly TNF-alpha inhibitors (adalimumab, etanercept, and infliximab), are essential in managing refractory necrotizing scleritis. The anti-CD20 monoclonal antibody rituximab emerges as a viable option for vasculitis-associated scleritis, offering hope in the most challenging scenarios.11,12
This multilevel therapeutic strategy highlights the complexity of managing scleritis, emphasizing the necessity for personalized treatment plans that can adapt to the changing clinical presentation and patient response.
Several other biologics are under investigation that have been shown to be effective in other systemic inflammatory diseases. However, they have not been shown to be effective in the treatment of scleritis, but are emerging as options in refractory cases, such as Tocilizumab and Anakinra.11,15
In principle, scleritis as a disease is not an indication for surgery, but there are conditions such as severe scleral or corneal thinning, which are at high risk of perforation, or definite scleral or corneal perforation. For scleral involvement, there are different graft tissues, including preserved donor sclera, fascia lata, periosteum, aortic tissue, and synthetic Gore-Tex.16,17
Case 1: Non-necrotizing diffuse anterior scleritis
A 57-year-old female patient presented to our clinic in July 2019 with left eye light sensitivity, foreign body sensation, and eye pain. Her ophthalmological history included recurrent foreign body sensation and light sensitivity since 2014. Medical history was unremarkable. She was attending physiotherapy for musculoskeletal complaints, but no evidence of underlying inflammatory rheumatological disease was confirmed. Best corrected visual acuity (BCVA) was 1.0 (0.00 logMAR) OU.
Slit-lamp examination revealed dilated episcleral and scleral vessels, most prominent in the superior and nasal region (Figure 1(a) and (b)). Non-necrotizing anterior scleritis was confirmed. Due to her joint complaints, repeated rheumatological consultation was recommended, where inflammatory rheumatological disease was still not confirmed. However, RA was suspected based on the non-typical intermittent small joint dominant arthralgias and RF positivity.
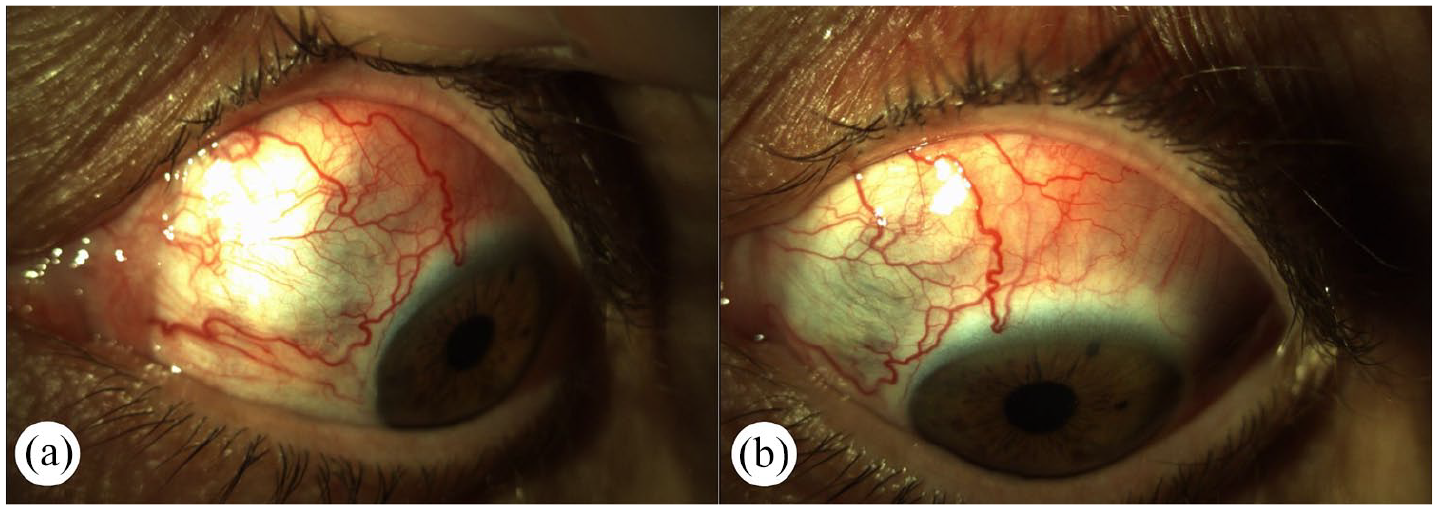
Slit-lamp photography of the left eye at the initial presentation demonstrates dilatation of the superficial and deep episcleral vessels markedly in the upper quadrant. (Pictures (a) and (b)) (Case 1.).
Initial therapy with topical and systemic NSAIDs proved ineffective, necessitating escalation to topical and systemic corticosteroids (methylprednisolone 64 mg) with comprehensive supplementation (gastric protection, potassium, vitamin D, calcium). Even with a 2-month duration of corticosteroid therapy, we could not achieve a completely non-inflammatory state, so we decided to start cyclosporine (125 mg daily), in addition to corticosteroid therapy, then the dose of cyclosporine was increased to the maximum dose (300 mg), with a decrease in the dose of corticosteroid. With careful reduction of cyclosporine to 250 mg, inflammatory symptoms recurred in December. Due to further therapeutic inadequacy, the corticosteroid dose was increased again as in agreement with rheumatologist colleagues, and we were planning to start cyclophosphamide. The further investigations confirmed a breast tumor, which was surgically resolved; this delayed the therapy. After treating her tumor, she was admitted to the rheumatology department due to recurrent and worsening joint complaints. RA was confirmed, and MTX and low-dose corticosteroids were started in April 2020.
Treatment with MTX (17.5 mg), corticosteroid was gradually tapered off and then discontinued. After this, the patient had no ophthalmological complaints, no inflammation in her eye (Figure 2(a) and (b)), and significant improvement of joint complaints were detected.

Slit-lamp photography of the left eye after gradually building up the therapy. Pictures (a) and (b) demonstrate scleral translucency following recurrent disease. (Case 1.).
This case illustrates several critical decision points: (1) the importance of persistent rheumatological evaluation despite initial negative findings, (2) the significance of comprehensive oncological screening before initiating immunosuppressive therapy, and (3) the successful resolution of scleritis following appropriate treatment of the underlying systemic condition (RA) with MTX.
Case 2: Necrotizing anterior scleritis
A 60-year-old female patient presented in May 2022 with right eye pain, light sensitivity, and tearing for 1 month. Her ophthalmologic history included endocrine orbitopathy, bilateral cataract surgery (2019, 2020), strabismus surgery for double vision and vertical strabismus (2020). Her medical history included SLE, hyperthyroidism, pulmonary embolism, and insomnia. She was receiving low-dose corticosteroid and chloroquine therapy when she presented to our Department. BCVA was 0.7 (0.155 logMAR) in her right eye, 1.0 (0.00 logMAR) in her left eye. On slit-lamp examination, diffuse conjunctival injection with dilated scleral vessels was seen. In the nasal region, conjunctival ulceration, scleral thinning, and several smaller ulcerations were detected superotemporally (Figure 3(a)). PUK developed in the nasal region of the cornea (Figure 3(b)). Scarred, echogenic orbital muscles were visible on ultrasound B-scan examination.

Slit-lamp photography of the right eye at initial presentation (Picture (a) and (b)) demonstrates conjunctival and ciliary congestion with ulceration of the conjunctiva and sclera- predominantly in the inferior and nasal quadrant—with scleral thinning, and several smaller ulcerations. Picture (b) shows the inferior quadrant, where cornea involvement, peripheral ulcerative keratitis (PUK), and conjunctival epithelial defects can be seen (Case 2).
After performing Quantiferon test for TB and infectious serologies (Borrelia burgdorferi, Treponema pallidum), which were all negative, we decided to admit her to our Department and started high-dose intravenous corticosteroid therapy (3 × 125 mg methylprednisolone, followed by 3 × 80 mg). Topical cyclosporine eye drops and antibiotic eye drops were added to the therapy due to PUK. Rheumatologist consultation did not find any underlying diseases and recommended the initiation of MTX/cyclosporine in case of scleritis flares following corticosteroid therapy discontinuation. Subsequently, systemic corticosteroid was gradually tapered (48, 32, 16, 8 mg) over 6 weeks. Her condition improved gradually (Figure 4(a) and (b)) for 1 month, then a recurrent inflammatory episode occurred while she was taking 8 mg methylprednisolone. MTX therapy was started in August at the starting dose of 16 mg/week. Her inflammatory symptoms increased (Figure 5(a)) again despite the use of MTX, so we decided to admit her to the ward to administer corticosteroid pulse therapy. After discharging her, inflammation decreased (Figure 5(b)). Due to her repeated inflammatory episodes, the MTX dose was increased to 20 mg/week, and the corticosteroid was gradually tapered to 8 mg/day.
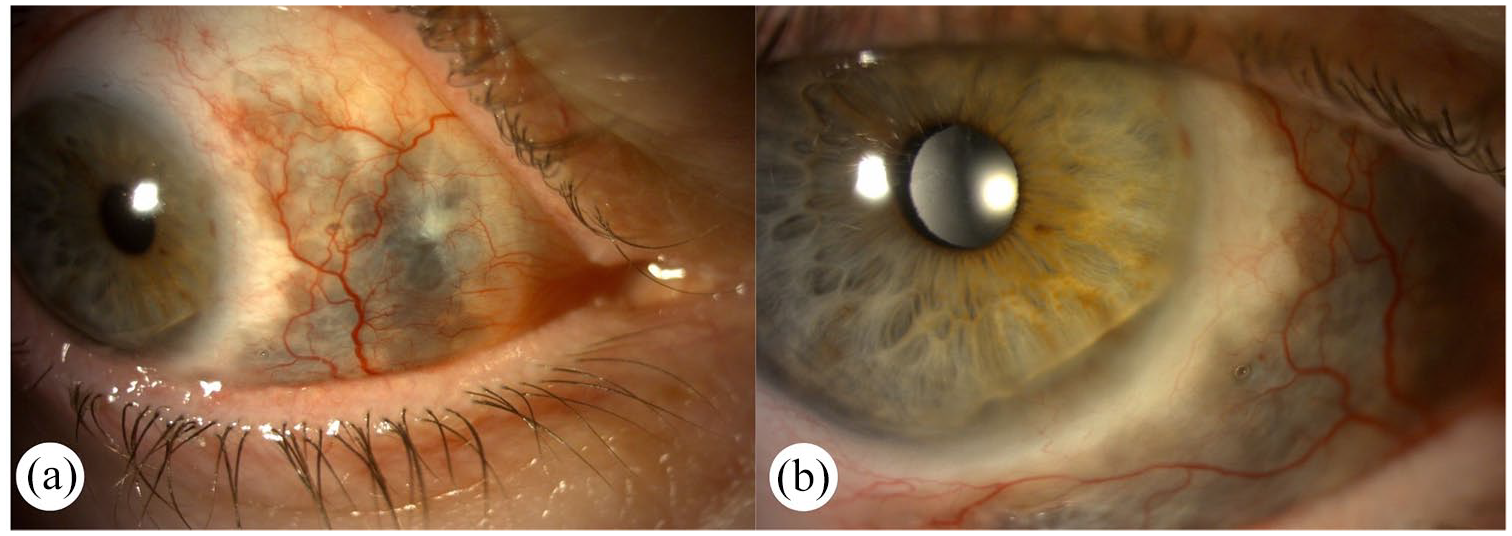
Slit-lamp photography of the left eye after administering topical cyclosporine and systemic corticosteroid treatment. Picture (a) shows that superficial and deep episcleral vessels dilatation markedly improved, and conjunctival and scleral ulcerations have ceased. Picture (b) shows a bigger magnification, the nasal conjunctival defect is epithelialized, in the inferonasal quadrant of the cornea, PUK can be seen in stable condition (Case 2).

Picture (a) demonstrated a slit-lamp photography of the right eye that shows a recurrent inflammatory episode after tapering down corticosteroid treatment. Multiple conjunctival pustules and severe scleral inflammation are seen according to the area previously affected. Picture (b) shows that the conjunctival injection is markedly improved, and pustules have disappeared. Dark pigmentation represents direct visualization of the uvea due to loss of scleral tissue from the inflammatory process (Case 2).
Combined low-dose corticosteroid and antimetabolite therapy maintained stability with BCVA 0.7 (0.155 logMAR). This case demonstrates key therapeutic considerations in necrotizing scleritis management: (1) the necessity of infectious disease exclusion before immunosuppression, (2) the challenge of balancing corticosteroid tapering with disease control, and (3) the importance of MTX dose optimization in maintaining remission despite preexisting SLE.
Case 3: Scleromalacia perforans
An 85-year-old female patient presented to our clinic in July 2021 with moderate right eye pain, discharge, and visual impairment for 3 months. Her ophthalmic history included bilateral cataract surgery and left eye trabeculectomy. In her medical history, hypertension was included. Her BCVA was counting fingers at 1 m (1.85 logMAR) in her right eye at the first presentation. On slit-lamp examination of the right eye, nasal scleral and conjunctival thinning was observed between the 1 and 4 o’clock positions, along with the absence of episcleral and conjunctival blood vessels (Figure 6(a) and (b)). In the nasal quadrant, PUK with corneal perforation was noted, with the iris plugging the perforation. The anterior chamber depth was variable, but no inflammation was present. Ultrasonography showed circular choroidal ablation.
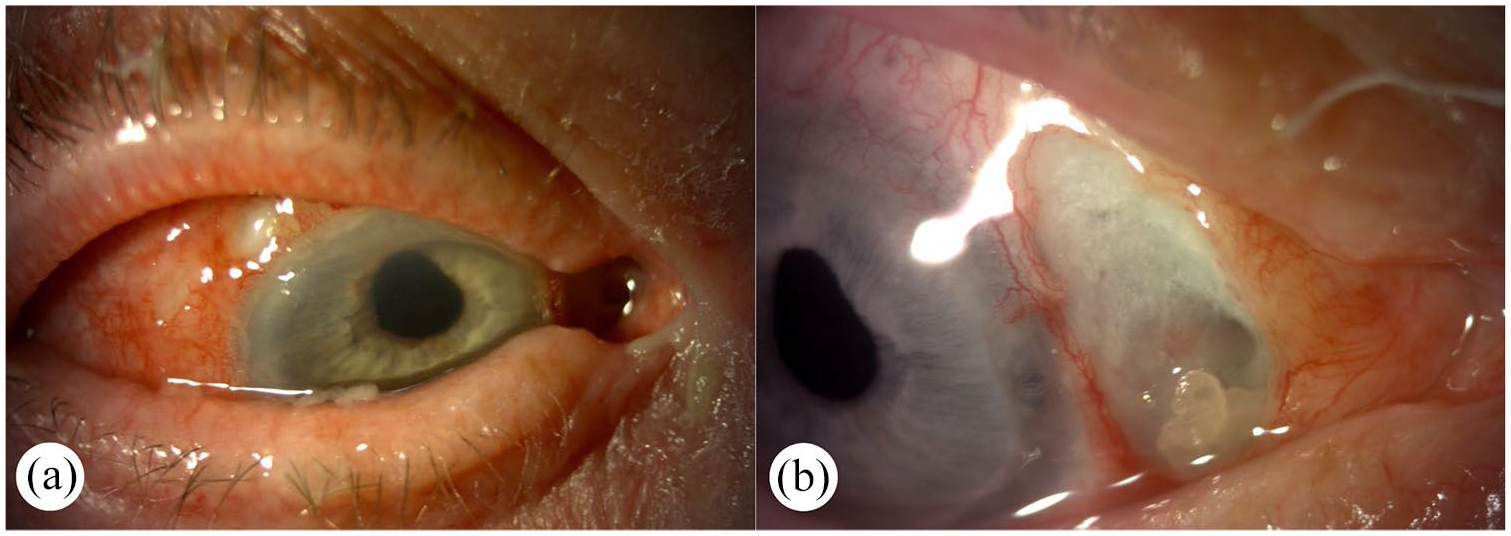
Slit-lamp photography of the right eye at initial presentation. Picture (a) shows conjunctival congestion and focal conjunctival epithelial defect in the superotemporal quadrant. Picture (b) shows a focal epithelial and scleral thinning (3 × 7 mm) nasally and severe PUK in the nasal part of the cornea (Case 3).
She was admitted to our Department, where investigations were started: lab tests, immunoserology, infectious serology, chest X-ray, consultation with a rheumatologist, and an ear, nose, and throat specialist. No systemic autoimmune diseases were confirmed. During the hospitalization, systemic corticosteroid pulse therapy was started (3 × 250 mg methylprednisolone, then gradually reduced to 64 mg) at dismissal. Topical antibiotics and cyclosporine eye drops were initiated for PUK. Subsequently, the corneal perforation closed spontaneously, and choroidal ablation showed a gradual decrease. BCVA improved to 0.9 (0.046 logMAR), complaints decreased, and choroidal ablation resolved. After 2 months of stable condition on low-dose corticosteroid (methylprednisolone) therapy, the patient experienced moderate pain and a recurrent visual loss again. BCVA was 0.02 (1.699 logMAR) on her right eye. Slit-lamp examination showed significant thinning of the sclera, so we admitted her to the Department. She was started on intravenous corticosteroid therapy again (3 × 250 mg), followed by gradual tapering. In addition to corticosteroid therapy, we decided to start MTX (20 mg weekly) after consultation with rheumatologists in October. Prophylactic virostatic medication was started along with MTX in consultation with hepatologists due to anti-HBc IgG positivity. The patient’s vision improved to 0.1 (1.000 logMAR). However, during the gradual withdrawal of corticosteroids, her condition started to progressively deteriorate, so we decided to add cyclophosphamide to her therapy. Between November and January 2021, the patient received a total of 1800 mg of cyclophosphamide infusion for scleritis, but in addition to this, the patient developed anterior uveitis, and her vision decreased again to 0.04 (1.398 logMAR). Since no autoimmune underlying disease was confirmed in the background, the therapy was supplemented with autologous serum eye drops and subconjunctival autologous blood injections. Due to the ineffectiveness of first- and second-line therapies, TNF-alpha inhibitor (adalimumab) was started in April (the therapeutic delay was due to the authorization process), and anterior segment inflammation decreased, and BCVA gradually improved to 0.45 (0.347 logMAR).
Combined therapy (TNF-alpha inhibitor, low-dose corticosteroid, MTX) achieved stability, though scleral covering surgery remains under consideration (Figure 7(a) and (b)).
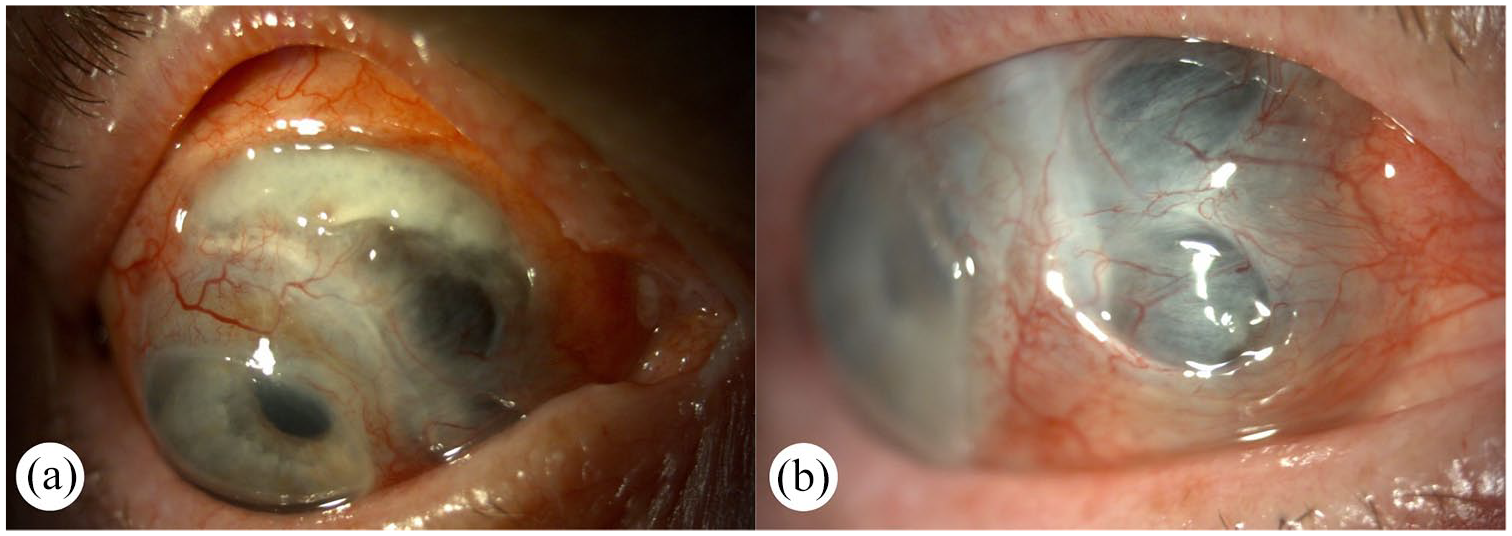
Slit-lamp photography taken of the right eye during the administration of immunomodulatory therapy. Pictures (a) and (b) show conjunctivalization of the peripheral cornea and severe thinning and exposure of underlying uvea (Case 3).
This case highlights crucial aspects of managing scleromalacia perforans: (1) the complex decision-making in elderly patients with multiple comorbidities, (2) the systematic approach to therapy escalation from conventional immunosuppression to biologics, and (3) the importance of multidisciplinary management, particularly regarding HBV prophylaxis during immunosuppression.
Discussion
The presented cases demonstrate the complex therapeutic challenges we face in managing different types of scleritis. Although corticosteroid therapy remains the first choice, introducing steroid-sparing agents early in selected cases may result in better outcomes. Each case required a different therapeutic strategy depending on disease severity.
In our first case, we encountered the importance of thorough systemic evaluation before initiating immunosuppression. The patient’s breast cancer diagnosis necessitated a careful approach to MTX treatment. Our second case, however, demonstrated that in necrotizing inflammation with PUK, rapid therapeutic escalation is essential to prevent sight-threatening complications. Our third case with scleromalacia perforans presented unique challenges in an elderly patient. The progression from conventional immunosuppression to biological therapy requires careful monitoring, especially considering the patient’s age and comorbidities.
Based on our experience with these cases, we found that standard therapeutic protocols need individual adaptation. When deciding on immunomodulatory therapy timing, we considered multiple factors: disease severity, risk of complications, patient-specific factors, initial treatment response, and underlying systemic conditions.
Recent literature supports our findings regarding early immunosuppressive therapy in necrotizing scleritis cases. According to current research, early aggressive immunosuppression helps prevent disease progression and reduces complications. 13 Our second case particularly demonstrates the importance of prompt therapeutic intervention in severe presentations.
Despite the successful outcomes, we encountered several limitations in managing these cases. Current debates in literature focus on the optimal timing of biological therapy initiation. 14 The third case highlighted specific challenges in elderly patients with multiple comorbidities, reflecting similar observations in recent publications. 9
Our experience raises several questions about future scleritis management. Further research is needed regarding the role of newer biological agents in refractory cases. Advanced imaging techniques might help predict disease progression and therapeutic response. In addition, studies focusing on early markers of disease severity could improve treatment strategies.
Conclusion
Scleritis, a rare and potentially sight-threatening ocular condition, presents significant diagnostic and therapeutic challenges in clinical practice. We emphasize the importance of differentiating between infectious and noninfectious, particularly immune-mediated, etiologies, as this distinction is crucially informative of treatment strategies. The case reports illustrate the diverse manifestations of scleritis—from anterior non-necrotizing and necrotizing forms to scleromalacia perforans—and underscore the complexity of management, which often requires a multidisciplinary approach involving rheumatology, dermatology, and other specialties. Corticosteroid therapy forms the cornerstone of treatment, but its long-term use necessitates consideration of side effects and potential transition to immunomodulatory agents. Our treatment algorithm highlights the importance of a stepwise approach, starting with local and systemic therapies and advancing to immunomodulatory and biologic agents in refractory cases. The differential diagnosis is essential, particularly in distinguishing from other “red eye” conditions, which have different treatment protocols.
Ultimately, this comprehensive overview of scleritis, from diagnosis to management, underscores the need for individualized treatment plans and ongoing research to optimize outcomes for patients suffering from this complex eye disease.