
Abstract
Objective:
To report the safety, tolerability, and pharmacokinetics/pharmacodynamics (PK/PD) of eptinezumab using intravenous (IV) infusion compared to other routes of administration from two phase 1 trials.
Methods:
Study 1 (NCT01579383) and Study 2 (ACTRN12615000531516) were double-blind, placebo-controlled, randomized trials. Study 1 singly administered ascending doses of eptinezumab 1–1000 mg IV infusion or 100 mg subcutaneous (SC) injection to healthy adults on day 1 (n = 60); in a second part, eptinezumab 300 mg IV + sumatriptan 6 mg SC was administered to healthy adults and patients with migraine (n = 18). Study 2 administered eptinezumab 100 or 300 mg intramuscular (IM), 100 mg SC, or 100 mg IV to healthy adults on days 1 and 84 (n = 60).
Results:
No withdrawals due to treatment-emergent adverse events (TEAEs) were reported due to IV administration, with IV generally reporting TEAEs similar to placebo. The pharmacokinetics of eptinezumab were as expected for a monoclonal antibody, with the 100 mg and 300 mg IV doses exhibiting higher C max and shorter t max compared to identical SC and IM doses.
Discussion:
These phase 1 safety and tolerability data supported eptinezumab intravenous infusions at 100 and 300 mg; both were approved for migraine prevention, were well tolerated, had low immunogenicity and rapid attainment of high plasma concentrations.
Keywords
Introduction
Migraine is one of the major contributors to disability worldwide, particularly among young adults and women. 1 Specifically, this highly prevalent disorder is related to significant personal and societal burden; migraine affects not only the individual with the disease, but also impacts interpersonal relationships at home, work, or school through interictal burden and disease costs. 2,3 In addition, patients with migraine report a higher burden or impact compared to patients without headache and worse outcomes that are consistent with headache frequency. 2
Many studies suggest that the release of calcitonin gene-related peptide (CGRP) increases during acute migraine attacks, making CGRP a likely contributor to migraine pathophysiology. 4,5 Overall, clinical studies utilizing small molecule CGRP antagonists or monoclonal antibodies (mAbs) that neutralize CGRP or bind to its receptor have suggested that targeting the CGRP pathway may provide clinical benefit for patients with migraine. 5,6 Due to the overall efficacy of CGRP-blocking therapeutics, the “capsaicin model” was developed and validated to evaluate CGRP-therapeutic target engagement and assess these potential therapeutics in early clinical development as a measure of pharmacodynamics. 6 This pharmacodynamic model involves topical application of capsaicin to the human forearm skin, where capsaicin may induce the local release of CGRP, resulting in increases in dermal blood flow. 6 This model has been previously demonstrated to be a robust model allowing for repeated, reproducible measurements that permit the early clinical evaluation of CGRP-blocking therapies. 6,7
Binding to CGRP with high affinity, 8 eptinezumab is a humanized IgG1 mAb approved for the preventive treatment of migraine. 9 –11 Two pivotal phase 3 trials, PROMISE-1 in patients with episodic migraine (EM) and PROMISE-2 in patients with chronic migraine (CM), 12,13 demonstrated that intravenous (IV) infusion of 100 mg or 300 mg of eptinezumab administered every 12 weeks achieved its primary efficacy endpoint, which was a statistically significant decrease in mean monthly migraine days over weeks 1–12. 12,13 Further, the preventive effect of eptinezumab in both trials was shown to be rapid and sustained, with the onset of preventive efficacy established on the day following the initial dose. 14 Finally, the phase 3 RELIEF trial, which evaluated the efficacy of eptinezumab when initiated during a migraine attack, indicated that patients treated with eptinezumab achieved statistically significantly faster headache pain freedom and resolution of the most bothersome symptom relative to placebo. 15
These pivotal phase 3 trials were informed by the phase 1 clinical trials that examined the safety of eptinezumab by comparing IV, intramuscular (IM), and subcutaneous (SC) routes of administration, as well as dose levels between 1 mg and 1000 mg and concomitant use of sumatriptan, in healthy adults and patients with migraine. The objective of this manuscript is to report the results of two phase 1 trials that provided support for the IV route of eptinezumab administration and dose levels used for phase 3 trials. In each study, the safety, pharmacokinetics (PK), pharmacodynamics (PD) using the capsaicin model, and immunogenicity were assessed.
Methods
Design
An overview of the study details for Studies 1 and 2 are described in Table 1, which includes dosing frequency, populations sampled, dosing days, and number of included participants. Both studies were conducted in accordance with standards of Good Clinical Practice as defined by International Conference on Harmonisation and all applicable national and local regulations. Study 1 was the first in human study and the main objectives were to determine the safety, tolerability, and PK of single ascending IV infusions ranging from 1 mg to 1000 mg; to compare 100 mg SC injection of eptinezumab to placebo in healthy men and women; and to determine the safety and tolerability of the co-administration of eptinezumab and sumatriptan versus sumatriptan alone in healthy women and/or migraine patients (men and women). For Study 2, the objective was to determine the safety, tolerability, and PK of IM injections of eptinezumab compared to SC injection and IV infusion of eptinezumab and placebo in healthy adults.
Eptinezumab phase 1 clinical studies.

IM: intramuscular; IV: intravenous; SC: subcutaneous.
a Dose escalation was dependent upon review of safety data through to Week 2 (day 12) from the prior dose level. The interval between dosing cohorts in healthy subjects was approximately 14 days. The first two subjects randomized into each dosing cohort formed a sentinel cohort; one subject received placebo and one received eptinezumab. The sentinel cohort was dosed 24 hours prior to the dosing of the other subjects of that cohort. Healthy subjects were confined to the unit from day –1 to day 3. Prior to commencement of Part B, all available safety data for Cohorts A–E to the week 4 post-dose time underwent an interim analysis by the Principal Investigator, the Medical Monitor, and Sponsor representatives.
b Sumatriptan was dosed in combination with eptinezumab or placebo. The first two subjects randomized formed a sentinel cohort; one subject received placebo plus sumatriptan and one received eptinezumab plus sumatriptan. The sentinel cohort was dosed 24 hours prior to dosing the remaining 10 subjects. Subjects were confined to the unit from day –1 to day 3.
c Each subject received three injections of eptinezumab or placebo on two different dosing days: days 1 and 84 (total of six injections). The order of administration for each dosing day was IV, SC, IM. IV infusions (100 mL) were administered over 1 hour (± 15 minutes); SC injections (1 mL) were made to the anterior abdominal wall; IM injections (3 mL) were made to the anterior thigh (vastus lateralis muscle). All three dose administrations were to be completed within approximately 90 minutes (± 15 minutes) on dosing days.
Eligibility criteria
The key inclusion criteria for the studies were that subjects were healthy men or women aged 18–65 years; willing to comply with protocol visits and procedures, willing to use effective contraception, had normal renal function, body mass index (BMI) between 18 kg/m2 and 30 kg/m2, and total body weight between 50 kg and 100 kg; and were free from clinically significant illness or disease. Study 1 also included patients with confirmed diagnosis of migraine for more than 1 year with one to eight moderate or severe attacks of migraine per month in the 2 months prior to trial. Study 2 had the additional requirement that subjects were responsive (>50% relative to vehicle) to capsaicin. Full inclusion and exclusion criteria for both studies are in Supplemental Methods.
Outcome measures
For both studies, the safety endpoints included the incidence, nature and severity of adverse events (AEs), as well as physical examinations, vital signs, and clinical laboratory test results. An AE was defined as any untoward medical occurrence in a subject who was administered a pharmaceutical product that did not necessarily have a causal relationship with the treatment. Treatment-emergent adverse events (TEAEs) were defined as those reported during or after eptinezumab or placebo infusion or injection. AEs without an onset date or time were defined as treatment emergent except if an incomplete date clearly indicated that an event started prior to treatment. The severity of TEAEs were graded as mild (grade 1), moderate (grade 2), severe (grade 3), life-threatening or disabling (grade 4), or fatal (grade 5). A serious adverse event was defined as any untoward medical occurrence that at any dose resulted in death, was life-threatening, required or prolonged hospitalization, was a congenital anomaly or birth defect, or was an important medical event that did not otherwise meet the criteria for seriousness. TEAEs were considered related to treatment (TRAEs) if the investigator, using medical judgment, determined that the AE was likely related to eptinezumab considering all relevant factors, including (not limited to) relevant history, concomitant illness, and concomitant medications. AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA), were grouped by MedDRA system organ class, and were summarized overall and by treatment group.
Subjects in Study 1 receiving SC injections and all subjects in Study 2 were monitored for injection site reactions; if an injection site reaction was present, the term “injection site reaction” and the National Cancer Institute Common Terminology Criteria for AEs grade was recorded on the AE case report form.
The timing of blood samples taken to quantify PK, PD, and immunogenicity properties for both studies are included in Table 2. In both studies, standard PK parameters were calculated using the concentrations of free eptinezumab in plasma from all subjects and total eptinezumab from selected subjects, when possible, using validated assay methods. For Study 1, the concentrations of sumatriptan were also assessed. In both studies, PD was measured through changes in the right volar forearm skin blood flow induced by topical capsaicin application, and the ratio of capsaicin/vehicle dermal perfusion values were compared to the pre-treatment values for each subject. Finally, in both trials, immunogenicity was measured using serum samples to test for antibodies to eptinezumab; any samples positive for anti-eptinezumab antibodies required additional testing to characterize the anti-eptinezumab antibody potential for neutralizing antibody (NAb) activity on eptinezumab.
Sampling timepoints for outcome measures in phase 1 clinical trials (Study 1 and Study 2).

PK: pharmacokinetic; PD: pharmacodynamic.
a PK specimens on day 1 were obtained pre-dose, 10 minutes, 4 hours (± 15 minutes), and 8 hours (± 15 minutes) post-dose; PK specimens on day 84 were obtained pre-dose only. Specimens taken days 15, 28, 56, 84, 98, 112, and 140 ± 2 sampling days, whereas day 168 ± 5 sampling days.
b Skin blood flow obtained pre-dose on second dosing day (day 84). Specimens taken days 15, 28, 56, 84, 98, 112, and 140 ± 2 sampling days, whereas day 168 ± 5 sampling days.
Statistical analysis
Results for Studies 1 and 2 were described using summary statistics. Formal statistical testing was not performed as outlined a priori in the statistical analytical plans. The safety population was defined as all randomized subjects who received at least 1 dose of eptinezumab. Subjects who experienced the same AE more than once were counted only once for that event. For Study 2, the incidence of injection site reactions, the number of subjects with reaction/total number of subjects in the analysis population, the maximum area of the reaction, reaction symptoms, time to onset from eptinezumab dosing, and the duration of the reaction were summarized for each dose and overall by treatment group using frequency tabulations.
The PK population included all randomized subjects who received eptinezumab for whom at least one measurable concentration was available. Subjects for whom the PK profile could not be adequately characterized or who exhibited protocol deviations affecting PK evaluation were flagged and excluded. All PK parameters were calculated according to standard noncompartmental methods. The individual free eptinezumab concentration versus time profiles were used to derive the PK parameters. These included exposure metrics: area under the eptinezumab concentration versus time curve (AUC) from time zero to the time after dosing at which the last quantifiable concentration of free eptinezumab was observed (AUC0–t
), maximum concentration (C
max), and time to peak plasma concentration (t
max) were calculated post eptinezumab doses. The absolute bioavailability (F) for eptinezumab was determined by comparing AUC0–∞ for eptinezumab after SC, IV, and IM administration. Absolute average bioavailability of IM and SC administered free eptinezumab was calculated as
Results
Study population
In Study 1, 139 subjects were screened for participation in the trial, where 104 subjects were randomized and received eptinezumab (n = 67) and placebo (n = 37); 86 subjects completed the trial per protocol as planned (Figure 1). The mean age of participants was 26.9 years and ranged from 21.8 to 36.8 across treatment groups. In Study 2, 123 subjects were screened for participation in the trial, with 61 subjects randomized and 60 subjects allocated to intervention. A total of 55 subjects (90.2%) received all doses of eptinezumab per study protocol and 53 (87.0%) completed the trial as planned (Figure 1). The mean age of subjects was 27.6 years, ranging from 24.8 to 30.5 across treatment groups. For both trials, the majority of the subjects enrolled were non-Hispanic/Latino (96.7%) and white (80.3%); treatment groups were well matched regarding age, sex, weight, height, and BMI.
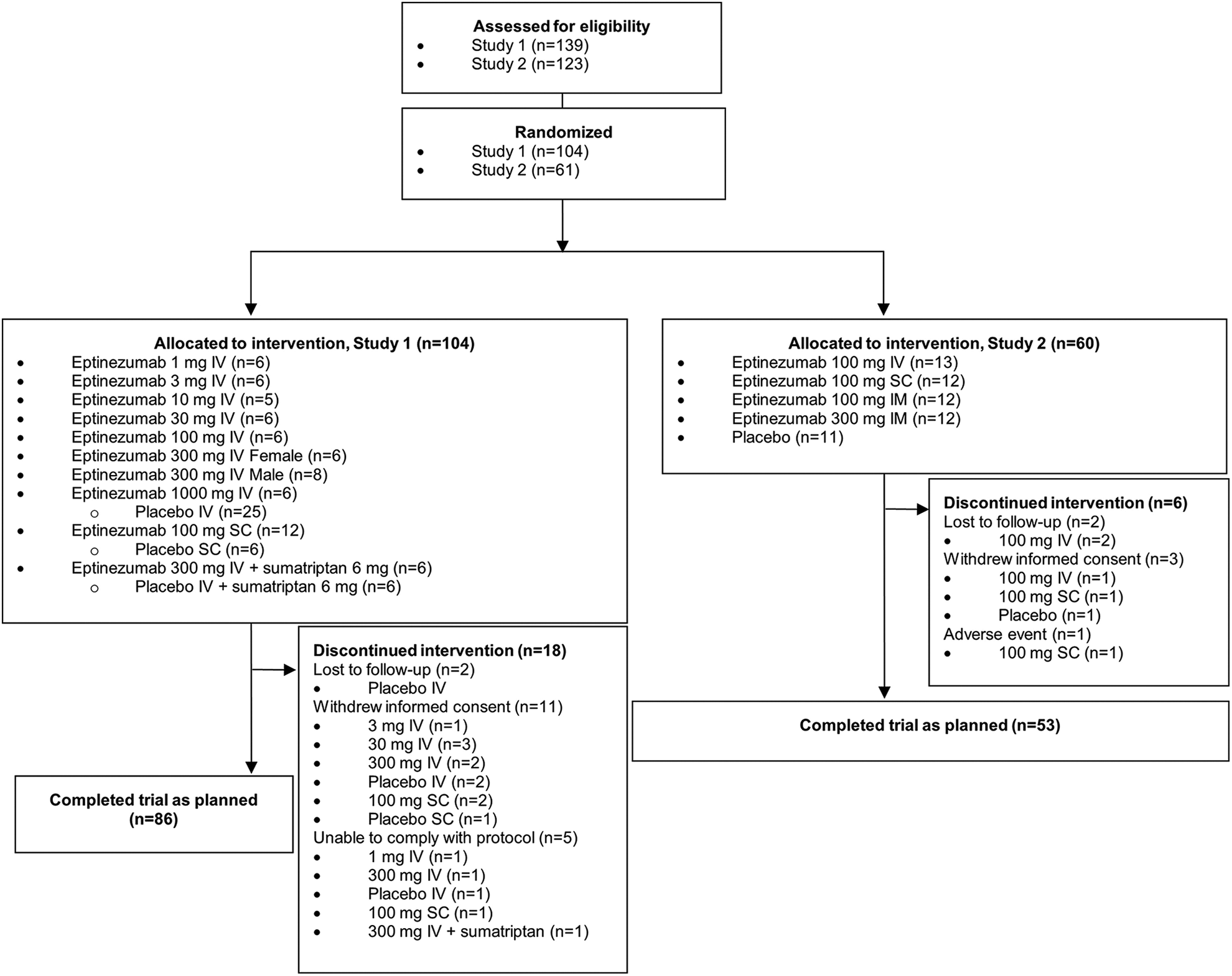
Patient disposition.
Treatment-emergent adverse events
For both studies, a detailed comparison of TEAEs is presented in Table 3. In Study 1, a total of 179 TEAEs were reported during the trial period by 73 subjects (70.2% of total patients). No deaths, withdrawals from the trial, treatment discontinuations due to TEAEs, or TEAEs that were severe or life-threatening/disabling were reported. No differences were observed between IV eptinezumab at any dose cohort or placebo in terms of incidence, type, severity, relationship, or frequency of TEAEs. TEAEs related to dosing with IV eptinezumab included: headache (n (%) number of events = 6 (12.2%) 6), dizziness (2 (18.4%) 2), abdominal pain (1 (2.0%) 1), infusion site pruritus (1 (2.0%) 1), lethargy (1 (2.0%) 1), rhinitis allergic (1(2.0%) 1), and dermatitis allergic (1 (2.0%) 1). Eptinezumab was well tolerated at 100 mg SC; however, the frequency of TEAEs reported for subjects dosed with SC eptinezumab was higher than that for subjects dosed with SC placebo. Specifically, the system organ class with the highest number of TEAEs reported following SC eptinezumab dosing was general disorders and administration site conditions (12 events reported by 9 subjects, 75.0%); these events were all injection site erythema and were considered related to eptinezumab administration. In addition, IV infusion of eptinezumab 300 mg + SC injection of 6 mg sumatriptan were well tolerated; the frequency of TEAEs reported was similar to that for subjects dosed with IV placebo + sumatriptan 6 mg (SC). TEAEs related to dosing with 300 mg IV eptinezumab and 6 mg sumatriptan included headache (n (%) number of events: 2 (33.3%) 3), flushing (3 (50.0%) 3), lethargy (2 (33.3%) 2), dizziness (1 (16.7%) 1), somnolence (1 (16.7%) 1), and throat tightness (1 (16.7%) 1).
Summary of TEAEs by treatment and eptinezumab dose for Study 1 and Study 2.a

Epti: eptinezumab; IM: intramuscular; IV: intravenous; NR: not reported; SAE: serious adverse event; SC: subcutaneous; SMT: sumatriptan; TEAE: treatment-emergent adverse event; TRAE: treatment-related (drug) adverse event.
a n is the number of subjects, % is the percentage of subjects, E is the number of events.
In Study 2, a total of 126 TEAEs were reported by 47 subjects (78.3%). No important differences were observed between the groups in grade of severity of the events, with only one event graded as severe (worsening of migraine symptoms). No TEAEs were fatal or life-threatening/disabling, and 41 TEAEs from 39 subjects (65%) were considered related to study drug, where the most frequently reported TEAE was headache, followed by injection site reactions. Headaches were reported in 100 mg IV (n (%) number of events: 3 (23.1%) 3), 100 mg SC (2 (16.7%) 2), 100 mg IM (1 (8.3%) 1), and 300 mg IM (3 (25.0%) 3) compared to placebo (2 (18.2%) 2). Injection site reactions were reported in 100 mg SC (2 (16.7%) 3) and 100 mg IM (1 (8.3%) 1) compared to (2 (18.2%) 3) in placebo. All remaining related TEAEs were reported in three or fewer subjects. In addition, there were no study drug–related clinically significant findings based on laboratory parameters, vital signs, or electrocardiogram parameters following any eptinezumab dosing. With the exception of one injection site reaction, all events started and stopped on the same day as the dose administration and were graded mild in intensity.
Pharmacokinetics
For both studies, a detailed list of the PK parameter results is given in Table 4; for Study 2, Figure 2 displays the relationship between free eptinezumab by weeks after dosing with either 100 mg SC or 100 mg IV eptinezumab. Briefly, in Study 1, the 1-mg to 1000-mg doses were associated with a plasma half-life between 12.1 days and 34.2 days between 1-mg and 1000-mg doses and exhibited a linear PK (from 1-mg to 1000-mg doses), with no observable sex differences following IV eptinezumab administration. Specifically for the 100-mg and 300-mg IV doses, in Study 1, the plasma half-life for 100 mg IV and 300 mg IV were 22.9 days and 29.5 days, respectively, and 31.5 days for 100 mg IV in Study 2. In addition, the mean C max for 100 mg IV and 300 mg IV were 25.33 µg mL−1 and 93.16 µg mL−1, respectively, and 36.0 for 100 mg IV in Study 2. AUC0–t values for 100 mg IV were 13,655 h·µg mL−1 for Study 1, 18,270 h·µg mL−1 for Study 2, and 39,967 h·µg mL−1 for 300 mg in Study 1. Co-administration with sumatriptan did not alter single-dose PK of IV eptinezumab, and the PK of sumatriptan was not impacted by the co-administration of eptinezumab. 16 In Study 2, the IM injections of eptinezumab resulted in 79% bioavailability of free eptinezumab at 100 mg and 85% at 300 mg compared to the SC route of administration (73% at 100 mg); bioavailability generally increased from 100 mg to 300 mg. 17 The accumulation ratio (accumulation of a drug after repeated administration compared to a single dose) of free eptinezumab was 1.4 for IM injections compared to IV (1.3) and SC (1.0) administration. 17 The apparent higher absolute bioavailability of free eptinezumab combined with a higher accumulation ratio resulted in an increase of overall exposure to eptinezumab after IM administration (partial AUC after second dose: 1.4) relative to the IV or SC route (both AUC 1.1). 17
Pharmacokinetic parameters of eptinezumab following first dose of administration from Study 1 and Study 2.

Vz and CL are reported for vascular (IV) administration; Vz/F and CL/F are reported for extravascular (SC, IM) administration. AUC: area under the curve; CL: clearance; h: hours; IM: intramuscular; IV: intravenous; NR: not reported; SC: subcutaneous; SD: standard deviation; SMT: sumatriptan; Vz : distribution volume in terminal elimination phase for IV administration for dose 1.
a Results from Study 1.
b Results from Study 2.
c Includes results from Study 1 and Study 2, respectively.

Free mean ±SD eptinezumab plasma concentrations (100 mg IV [n=13] vs 100 mg SC [n=12]) by weeks after dosing in Study 2.
In addition, 100-mg IV infusion reported the highest C max in Study 1 and Study 2 relative to 100-mg SC and IM doses (Table 4). Similarly, the 300-mg IV dose reported a higher C max in Study 1 than the 300-mg IM dose in Study 2. Additionally, the shortest t max was found in the 100-mg and 300-mg IV doses (Table 4).
Pharmacodynamics
In Study 1, the higher doses (30–1000 mg IV eptinezumab) reduced the dermal vasodilation induced by topical treatment with capsaicin relative to placebo that was generally dose dependent and persisted through week 12 of treatment. 16 Following dosing with 1, 3, or 10 mg IV eptinezumab, the median capsaicin/vehicle dermal perfusion ratios post-treatment were generally similar to subjects dosed with IV placebo (Supplemental Figures 1A–C). 16 In Study 2, subjects receiving eptinezumab 100 mg administration via IV, SC, or IM, or 300 mg via IM, were observed with marked reductions in capsaicin-induced dermal perfusion relative to placebo patients within 24 hours following the first treatment that persisted for at least 12 weeks following each of two treatments (Supplemental Figure 2). 17
Immunogenicity
In Study 1, four subjects developed anti-eptinezumab antibodies following dosing with 100–1000 mg IV eptinezumab (8.2% of subjects), whereas two subjects developed anti-drug antibodies (ADAs) following dosing with 100 mg SC eptinezumab (16.7%), and one subject developed anti-eptinezumab antibodies following co-administration of 300 mg IV eptinezumab with SC sumatriptan (16.7%). Of these subjects, five displayed increased anti-eptinezumab titers and four of these subjects displayed reduced t 1/2; however, there was no consistent correlation among immunoreactivity, higher titers, and reduced t 1/2. In Study 2, one subject developed ADAs from 100 mg IV (7.7% of subjects), three from 100 mg SC (25.0%), two from 100 mg IM (16.7%), and one from 300 mg: of those subjects with positive ADAs, one subject each in the 100 mg SC, 100 mg IM, and 300 mg IM treatment groups had positive neutralizing antibodies (NAbs). However, there was no apparent impact by the formation of ADAs or NAbs on PK parameters or PD results. The day of ADA onset and the incidence of positive and negative ADAs and NAbs are presented in Table 5.
Summary of anti-eptinezumab antibody and neutralizing antibodies results by treatment for subjects receiving at least one dose of eptinezumab in Study 2.

ADA: anti-drug antibody; IM: intramuscular; IV: intravenous; NAb: neutralizing antibody; SC: subcutaneous.
a Number of subjects with positive ADA results/number of subjects treated.
b Number of subjects with positive ADA results/number of subjects treated; highest titer observed for each subject with positive ADA results.
c Number of ADA-positive subjects with negative ADA results at the end of the trial/number of subjects with positive ADA results.
d Number of subjects with positive NAb results/number of subjects with positive ADA results.
e Number of NAb-positive subjects with negative NAb results at end of trial/number of subjects with positive NAb results.
Discussion
In this report of two phase 1 clinical studies evaluating the safety and tolerability of eptinezumab, there were no withdrawals from the trial or treatment discontinuations due to TEAEs, and no life-threatening/disabling or fatal TEAEs. Overall, IV infusions were well tolerated at all dose levels of eptinezumab and those co-administered with SC sumatriptan, where TEAEs and drug or TRAEs were similar to placebo in both studies, with the exception of 300 mg IV plus 6 mg sumatriptan dose which had slightly higher TRAEs than placebo (Table 3). In contrast, the frequency of TRAEs in subjects dosed with SC eptinezumab was higher than in those receiving SC placebo in Study 1. Further, more subjects experienced injection site reactions after receiving SC and IM injections than with IV infusion, where only one injection site reaction was reported. Finally, the only TEAE that led to study discontinuation (worsening migraine) was found in the 100 mg SC treatment group. As expected, these data suggest that IV eptinezumab administration may have a more advantageous tolerability profile relative to SC and IM routes of administration. These data are further supported in the phase 3 studies (the placebo-controlled NCT01772524, NCT02275117, PROMISE-1, and PROMISE-2, and open-labeled PREVAIL), which pooled five clinical trials of IV eptinezumab (10 mg, 30 mg, 100 mg, 300 mg, and 1000 mg) compared to placebo and found that IV administration of eptinezumab every 12 weeks has a favorable safety and tolerability profile. 11,18
Overall, the observed PK profile was consistent with previous population-level analysis that included four phase 1, one phase 2, and three phase 3 trials and indicated that eptinezumab doses of 100 mg produce mean C max and AUC0–12wk values of >37 µg mL−1 and >17,000 h·µg mL−1, respectively, with a terminal elimination half-life of 27 days. 19
Comparatively, in Study 1, the plasma half-life for 100 mg IV and 300 mg IV were 22.9 days and 29.5 days, respectively, and 31.5 days for 100 mg IV in Study 2. The mean C max for 100 mg IV and 300 mg IV was 25.33 µg mL−1 and 93.16 µg mL−1, respectively, and 36.0 for 100 mg IV in Study 2. AUC0–t values for 100 mg IV were 13,655 h·µg mL−1 for Study 1 and 18,270 h·µg mL−1 for Study 2 and 39,967 h·µg mL−1 for 300 mg in Study 1. In addition, co-administration with sumatriptan did not appear to affect the single-dose PK of IV eptinezumab. In general, eptinezumab reduced the dermal vasodilation induced by topical treatment with capsaicin relative to placebo that was generally dose dependent.
The observed PK profile was consistent with reported PK values for other mAbs given differences in routes of administration. For example, after single SC administration of 225 mg, 675 mg, and 900 mg fremanezumab, the t max was 5–7 days and reported a t 1/2 of approximately 31 days. 20 After SC administration of 70 mg and 140 mg once monthly dose of erenumab, the mean (SD) C max was reported as 6.1 (2.1) mcg/mL and 15.8 (4.8) mcg/mL for the 70 mg and 140 mg doses, respectively, with the t 1/2 reported as 28 days. 21 Finally, the loading dose of 240 mg SC galcanezumab reached a t max at 5 days and reports a t 1/2 of 27 days. 22
In Study 1, seven subjects in total (10.4%) developed anti-eptinezumab antibodies; however, immunoreactivity did not appear to be correlated with shorter t 1/2 or higher doses. Similarly in Study 2, seven subjects developed ADAs, but the presence of ADAs and NAbs had no apparent impact on PK or PD parameters, and a significant portion of the ADA and NAb observations were resolved by the end of the trial. The frequency of ADAs appeared to be lower with IV administration, which had slightly lower incidence compared to other routes of administration (Study 1 IV: 8.2% vs. 16.7 SC; Study 2 IV: 7.7% vs. 25.0% SC, 16.7% IM) in this study. Due to the differences in the ADA assay methodologies, the results for different mAb products are not directly comparable. For galcanezumab, the reported incidence of ADAs was 4.8% (33/688) in patients receiving SC galcanezumab once monthly in a controlled 6-month study; 6.2% (48/778) in patients receiving 70 mg erenumab once monthly in controlled studies; and 1.6% (30/1888) of patients receiving 225 mg fremanezumab once monthly in a long-term, open-label study. 20 –22 Overall, the data of ADA development on the efficacy or safety of any of these mAbs are too limited to make definitive conclusions.
Overall these results are consistent with the rapid onset of action demonstrated in the analyses of the PROMISE-1 and PROMISE-2 data, which demonstrated that the migraine preventive effect of IV infusion of eptinezumab in patients with chronic or episodic migraine is observed on the day following the initial dose, 14 and the phase 3 RELIEF study that suggested treatment with IV eptinezumab shortened the time to 2 hours’ resolution of headache and MBS. 15
Limitations
These safety, PK, PD, and immunogenicity data are based on a limited population of younger, white adults who may not be representative of the total population affected by migraine. In addition, for the comparison of AEs between the different routes of administration, there was a limited number of subjects on IM and SC relative to those on IV or placebo, as well as a low overall sample size within groups and a high number of subgroups.
Conclusion
These phase 1 safety and tolerability data supported eptinezumab intravenous infusions at 100 and 300 mg; both were eventually approved for migraine prevention, were well tolerated, had low immunogenicity and rapid attainment of high plasma concentrations.
Key findings
Eptinezumab is approved for the preventive treatment of migraine in adults.
Approved dose levels are 100 mg and 300 mg administered via intravenous infusion.
Two phase 1 studies support 100 mg and 300 mg doses administered as intravenous infusions through measures of safety, tolerability, pharmacokinetics, pharmacodynamics, and immunogenicity compared to placebo and other routes of administration (subcutaneous, intramuscular).
Supplemental material
Supplemental Material, sj-docx-1-rep-10.1177_25158163221131326 - Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies
Supplemental Material, sj-docx-1-rep-10.1177_25158163221131326 for Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies by Brian Baker, Vivienne Shen, Roger Cady, Anders Ettrup and Frank Larsen in Cephalalgia Reports
Supplemental material
Supplemental Material, sj-jpg-1-rep-10.1177_25158163221131326 - Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies
Supplemental Material, sj-jpg-1-rep-10.1177_25158163221131326 for Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies by Brian Baker, Vivienne Shen, Roger Cady, Anders Ettrup and Frank Larsen in Cephalalgia Reports
Supplemental material
Supplemental Material, sj-jpg-2-rep-10.1177_25158163221131326 - Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies
Supplemental Material, sj-jpg-2-rep-10.1177_25158163221131326 for Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies by Brian Baker, Vivienne Shen, Roger Cady, Anders Ettrup and Frank Larsen in Cephalalgia Reports
Supplemental material
Supplemental Material, sj-jpg-3-rep-10.1177_25158163221131326 - Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies
Supplemental Material, sj-jpg-3-rep-10.1177_25158163221131326 for Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies by Brian Baker, Vivienne Shen, Roger Cady, Anders Ettrup and Frank Larsen in Cephalalgia Reports
Supplemental material
Supplemental Material, sj-jpg-4-rep-10.1177_25158163221131326 - Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies
Supplemental Material, sj-jpg-4-rep-10.1177_25158163221131326 for Eptinezumab administered intravenously, subcutaneously, or intramuscularly in healthy subjects and/or patients with migraine: Early development studies by Brian Baker, Vivienne Shen, Roger Cady, Anders Ettrup and Frank Larsen in Cephalalgia Reports
Footnotes
Acknowledgments
The authors thank Beth Reichard, PhD, and Nicole Coolbaugh, CMPP, of The Medicine Group, LLC (New Hope, PA, USA) for providing medical writing support in drafting and revising the manuscript, which was funded by H. Lundbeck A/S (Copenhagen, Denmark) in accordance with Good Publication Practice guidelines.
Author contributions
All authors had full access to all study data and take responsibility for all aspects of the work, including the integrity of the data and the accuracy of the data analysis. All authors provided final approval of the manuscript for submission. Substantial contributions to the acquisition, analysis, or interpretation of data for the work: BB, VS, RC, AE, and FL. Drafting of the manuscript and critical review/revision of the manuscript for important intellectual content: BB, VS, RC, AE, and FL.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: BB, AE, VS, and FL are employees of Lundbeck or one of its subsidiary companies. RC was an employee of Lundbeck at the time of manuscript development.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was sponsored and funded by H. Lundbeck A/S. All authors and H. Lundbeck A/S prepared, reviewed, and approved the manuscript and made the decision to submit the manuscript for publication. Writing and editorial support for the development of this manuscript was funded by H. Lundbeck A/S.
Research ethics and patient consent
Both studies included in this analysis were approved by an independent ethics committee. The studies were registered at ClinicalTrials.gov under NCT01579383 and ![]() under ACTRN12615000531516. In each study, patients provided written informed consent to participate.
under ACTRN12615000531516. In each study, patients provided written informed consent to participate.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.