
Abstract
Aims
Gut microbiota are reported to be associated with the incidence and prognosis of Esophageal cancer (EC) but their genetic association is unclear. We carried out a bidirectional MR analysis to assess the causal relationship between EC and gut microbiota from fecal samples.
Methods
The microbiome genome-wide association studies (GWAS) data of 18,340 individuals provided by MiBioGen consortium and the EC GWAS data (740 esophageal cancers cases and 372 016 controls) provided by UK Biobank were respectively utilized as exposure and/or outcome data. Reliable single nucleotide polymorphisms (SNPs) were obtained after rigorous screening. A bidirectional Mendelian randomization (MR) analysis was conducted using the inverse-variance weighted (IVW) method. The sensitivity analyses including the MR-Egger method, weighted median, weighed mode and leave-one-out method were performed to examine the stability, heterogeneity and pleiotropy of the results.
Results
Forward MR analysis revealed the increase in abundance of the microbial trait by each standard deviation was associated with a higher risk of EC (Coprobacter (OR = 1.001,95%CI = 1.000-1.002, P = .0281, FDR = 0.0424); Ruminococcus1(OR = 1.001,95%CI = 1.000-1.002, P = .0318, FDR = 0.0424); Senegalimassilia (OR = 1.002,95%CI = 1.000-1.003, P = .0062, FDR = 0.0372); Veillonella (OR = 1.001,95%CI = 1.000-1.002, P = .0182, FDR = 0.0372)) or a lower risk of EC (Eubacterium oxidoreducens (OR = 0.999, 95%CI = 0.998-1.000, P = .0379, FDR = 00 433); Lachnospira (OR = 0.998,95%CI = 0.996-1.000, P = .0186, FDR = 0.0372); Romboutsia (OR = 0.999,95%CI = 0.998-1.000, P = .0482, FDR = 0.0482); Turicibacter (OR = 0.999,95%CI = 0.998-1.000, P = .0133, FDR = 0.0372)). Reverse MR analysis showed that genetic liability to EC was also causally linked toincreased susceptibility of changes in the gut microbiome (genera Eggerthella (Beta = 37.63,95%CI = 4.76-70.50, P = .0248, FDR = 0.0331); Coprococcus 2 (Beta = 23.90,95%CI = 1.65-46.15, P = .0353, FDR = 0.0353); Christensenellaceae R.7 (Beta = 22.75,95%CI = 4.22-41.28, P = .0161, FDR = 0.0322); Intestinimonas (Beta = −33.24,95%CI = −54.90-11.58, P = .0026, FDR = 0.0104)).
Conclusions
Our findings supported a bidirectionally causal relationship between gut microbiota and EC, implying the potential role of gut microbiota in preventing the occurrence and development of EC.
Introduction
Esophageal cancer (EC) is one of the most common fatal cancers, imposing significant worldwide health burdens. The global incidence and mortality of EC ranked tenth and sixth, respectively, in 2020. 1 The majority of cases (85% [512 500 cases]) were Esophageal squamous cell carcinoma (ESCC) and Esophageal adenocarcinoma (EAC) accounted for 14% (85 700 cases). The distribution varies from regions and adenocarcinoma is the main type in European and American countries. Moreover, the incidence of EAC has been rising at an alarming rate in the Western world. 2 It is well recognized that host and environmental risk factors, such as obesity, gastroesophageal reflux disease and Barrett's esophagus, contribute to EAC.3–5 The symptoms of early esophageal cancer are often atypical and easy to ignore, thus primary prevention and early screening play key roles in improving clinical outcome and survival of patients. 6 Despite researchers have done a lot of exploring, the exact pathogenic mechanism of esophageal cancer remains unknown to this day, making it difficult to find potential noninvasive biomarkers and predictors.
The Gastrointestinal tract (GIT) is colonized by trillions of microorganisms that comprise the gut microbiota. Gut microbiota not only take part in protecting against pathogens and maintaining gut ecosystem homeostasis but also are involved in several cancers. 7 According to a recent study, about 13% of all cancer cases in 2018 can be attributed to infectious etiology. 8 During the past few years, much attention has been paid to the association between esophageal cancers and gut microbes. Gut microbiota have a crucial role in the onset, development, and treatment efficacy of gastrointestinal malignancies, which may be caused by inflammation and immune disorders. 9 Several researches have revealed the different esophageal bacterial strains, from normal esophagus to ESCC and EAC.10–12 Ajayi et al 12 showed that normal esophagus is mainly colonized by Streptococcus while esophagitis and BE are associated with gram-negative bacteria. Furthermore, certain G-bacteria, including Escherichia coli and Fusobacterium nucleatum, are linked to EAC. Yamamura et al 13 revealed that Fusobacterium nucleatum aggravates the survival of ESCC patients. The limitation is, most of the sample were obtained from esophagus by invasive examination. It has been proven that intestinal microbiome is involved in inflammatory bowel diseases, 14 diabetes, 15 colorectal cancer 16 and so on. Recently, Deng et al 17 demonstrated an increased richness of gut microbes and remarkable change of the distribution proportion of some bacteria in EC compared with healthy individuals through 16S rDNA gene sequencing of fresh stool. While it is still unclear of the causal relationship between gut microbiota and esophageal cancer.
Mendelian randomization (MR) is an original approach to exploring the causality between exposure or risk factor and outcome, which has been widely applied to research in various fields. It screens for genetic variants that can represent exposure as an instrument to examine the relationship between instrument and outcome. Compared to traditional observational studies, which are prone to biases such as reverse causality and residual confounding, MR rules out these biases due to the law of independent assortment. 18 In this text, we utilize the genome-wide association study (GWAS) summary statistics from UK Biobank and MiBioGen consortium. A two-sample MR is carried out to evaluate the causal relationship between gut microbiota and esophageal cancer.
Methods
Figure 1 shows the flowchart of the MR analysis. A bidirectional two-sample MR study was designed to explore the causal effect of gut microbiota and esophageal cancer.
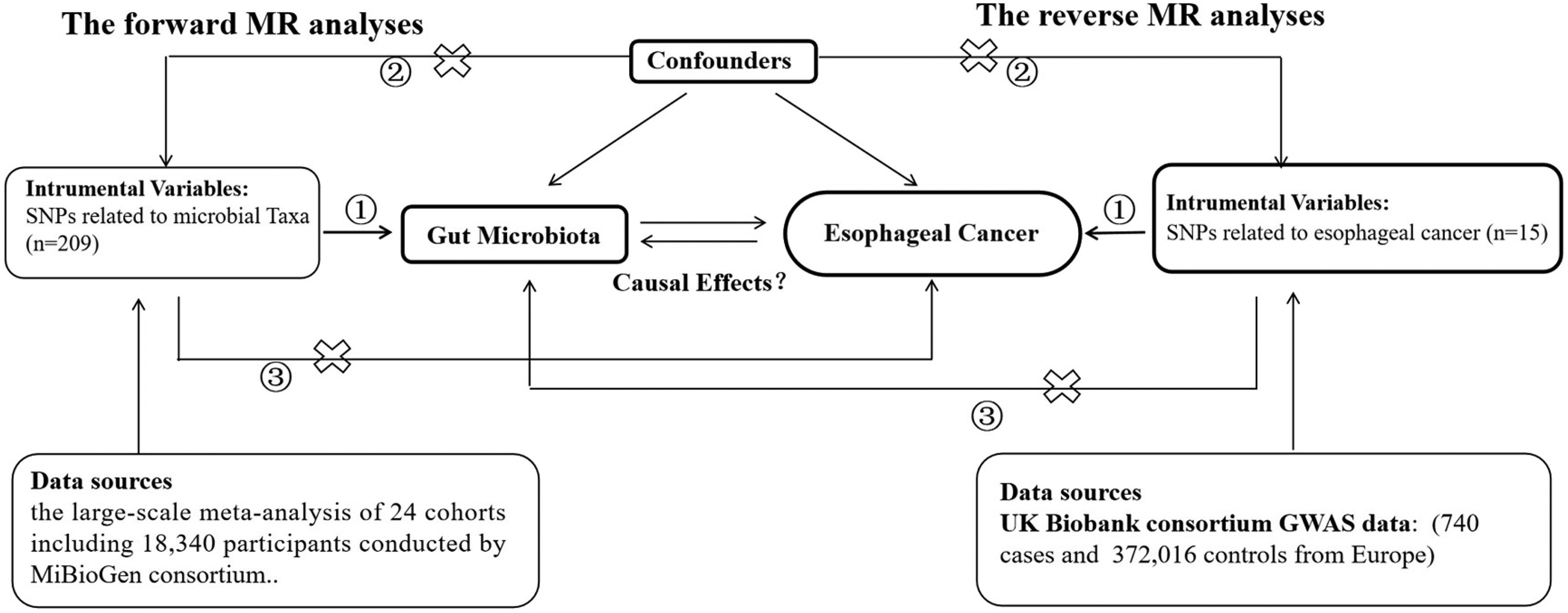
The study design of the bidirectional MR study. The core assumptions of Mendelian Randomization (MR) are: ① Relevance: Genetic variants must be strongly associated with the exposure. ② Independence: There are no confounders between the genetic variants and the outcome. ③ Exclusion Restriction: Genetic variants affect the outcome only through the exposure, without any other pathways (no pleiotropy). Abbreviations: MR, Mendelian Randomization; SNP: Single nucleotide polymorphisms.
Data Sources
We obtained the genetic variants datasets from public-available GWAS. The summary statistics of esophageal cancer were provided by UK Biobank. The project is a population-based cohort study involving in 500 000 middle-aged participants. The study consisted of 740 esophageal cancers cases and 372 016 controls, and almost of them are European. 19 The summary statistics of gut microbes were acquired from the large-scale meta-analysis of 24 cohorts including 18 340 participants conducted by MiBioGen consortium. In this study, fecal microbial composition was analyzed by targeting the V4, V3-V4, and V1-V2 variable regions of the 16S rRNA gene and classified by direct taxonomic classification. To identify host genetic variants that was mapped to the genetic loci impacting the relative abundance or presence of microbial taxa, microbiome quantitative trait loci (mbQTL) mapping analysis was carried out. A total of 211 taxa (131 genera, 35 families, 20 orders, 16 classes, and 9 phyla) were involved. 20
Selection of the Instrumental Variables
Because of the limited number of SNPs reaching the genome-wide statistical significance threshold (P < 5*10−8), we set a P-value threshold of 5*10−6 to filter the SNPs strongly associated with the the gut microbiome in the forward MR analysis. The threshold of 5*10−8 was set for the selection of instrumental variables related to esophageal cancer in the reverse analysis.Then, on the basis of the European ancestral individuals from the 1000 Genomes Project, we assessed the linkage disequilibrium to ensure the independence, and appropriate SNPs within 10 000 kb clumping window with an r2 < 0.001 were kept. 18 Before harmonizing the exposure and outcome datasets, we excluded the palindromic sequences due to the uncertainty of the direction for exposure and outcome. Moreover, F-statistic was calculated to evaluate the strength of IVs by the formula: F = R2(n−1−k)/(1−R2) k, (R2 represents the proportion of variation explained by each IV, k represents the number of eligible SNPs, and n represents the sample size). 21 When F statistic > 10, the IV was considered as strongly-correlated instruments.The F-statistics we calculated in the forward MR analysis were greater than 35.
Statistical Analysis
In this study, we performed the analysis in R 4.1.0 using TwoSampleMR 22 and MR-PRESSO packages. To assess whether there is a causal relationship between esophageal cancer and gut microbes,we applied the Inverse variance weighted (IVW) method predominantly. The IVW method calculated a weighted average of Wald ratio estimates of each SNP. If all of the SNPs are valid instrumental variables, there will be no bias in the results. 23 Since MR analysis may encounter potential pleiotropy, where the IVs influence the outcome through multiple pathways, multiple methods were applied for sensitivity test: MR Egger regression, Weighted median and Weighted mode. Moreover, the Leave-one-out analysis demonstrated the meta effects of the remaining SNPs when a certain SNP was removed in turn. If the results changed greatly after the elimination, it indicated the heterogeneity of the removed SNP. Considering the increased likelihood of Type I errors due to multiple comparisons, we have applied the FDR(False Discovery Rate) method. An adjusted P-value < .05 was considered statistically significant. In reverse Mendelian Randomization (MR) analysis, the Inverse Variance Weighted (IVW) method was still used as the primary approach for estimating causal relationships. MR Egger regression, weighted median, weighted mode and Leave-One-Out Analysis were utilized as sensitivity analyses to ensure robustness.
Results
Genetic Association Between Gut microbiota and EC
A total of 209 SNPs were screened out to be valid instrumental variables for 211 bacterial genera according to the selection principles. Figure 2 and Figure S1 are the forest plot and the scatter plots of filtered microbial taxa with risksof esophageal cancer. In the forward MR analyses, eight significant microbial taxa were found to be closely associated with esophageal cancer: the genera Eubacterium oxidoreducens, Coprobacter, Lachnospira, Romboutsia, Ruminococcus1, Senegalimassilia, Turicibacter, and Veillonella. Our analyses revealed that specific gut bacteria may play a critical role in reducing EC risk. For instance, each standard deviation increase in the abundance of Eubacterium oxidoreducens results in a 0.001% decreased incidence of EC (OR = 0.999, 95%CI = 0.998-1.000, P = .0379, FDR = 0.0433). Similarly, Lachnospira, Romboutsia and Turicibacter show reductions of 0.002% and 0.001%, respectively, with comparable confidence intervals and P-values, indicating robust statistical results. In reverse MR analysis,each standard deviation increases in the abundance of Coprobacter, Ruminococcus1, and Veillonella was each associated with a 0.001% rise in EC (OR = 1.001, 95%CI = 1.000-1.002, P-values ranging from 0.0182 to 0.0318, FDR = 0.0372,0.0424). A more substantial increase was observed with Senegalimassilia, linked to a 0.002% rise in EC (OR = 1.002, 95%CI = 1.000-1.003, P = .0062,FDR = 0.0372). The results of the forward MR analyses were comprehensively presented in Table S1. Figure S2 shows the results of MR leave-one-out sensitivity analyses, indicating the stability of the IVs.Moreover, based on the results of MR Egger regression, no evidence of pleiotropy was identified.The causal effect estimate does not change significantly after removing each SNP in the leave-one-out analysis, and the confidence intervals of most points overlap, indicating that the results are robust(Figure S2).

Associations of genetically predicted gut microbiota with risk of esophageal cancer.
Genetic Association Between EC and Gut microbiota
We extracted 14 strongly-associated and independent SNPs of esophageal cancer after excluding a palindromic SNP (rs3802909). The reverse MR analysis was carried out as prescribed. The forest plot (Figure 3) and scatter plot (Figure S3) clearly demonstrated the effect of esophageal cancer on gut microbes. We found esophageal cancer has causal relationship with the change in genera Intestinimonas, Eggerthella, Coprococcus2 and Christensenellaceae R.7. The occurrence of Esophageal carcinoma is consistent with the increased enrichment of the genera Eggerthella (Beta = 37.63, 95%CI = 4.76-70.50,P = .0248,FDR = 0.0331),Coprococcus2(Beta= 23.90,95%CI = 1.65-46.15,P = .0353,FDR = 0.0353) and Chri-stensenellaceaeR.7 (Beta = 22.75, 95%CI = 4.22-41.28, P = .0161, FDR = 0.0322). Nevertheless,the abundance of the genera Intestinimonas (Beta = −33.24, 95%CI = −54.90-11.58, P = .0026, FDR = 0.0104) tends to decrease under the influence of EC. The results of all five statistical methods applied in the forward MR analyses have been displayed in Table S2. The MR Egger regression intercept which is close to zero also proved that there no pleiotropy existed. Moreover, no single SNP has an overly large influence on the results according to the leave-one-out analysis (Figure S4).

Associations of genetically predicted esophageal cancer with gut microbiota dysbiosis.
Discussion
The Two-Sample MR study we conducted clearly showed the bidirectional causal relationship between esophageal cancer and gut microbes without the influence of confounding factors. The forward MR revealed that genera Eubacterium oxidoreducens, Lachnospira, Romboutsia and Turicibacter had a positive effect on esophageal cancer, while Coprobacter, Ruminococcus1, Senegalimassilia and Veillonella might be the risk factors for the development of esophageal malignant neoplasms. Meanwhile, in the reverse MR, Esophageal cancer enriched the abundance of the genera Eggerthella, Coprococcus2 and Christensenellaceae R.7, and the abundance of the genera Intestinimonas was found to be decreased.
In recent years, there has been an increasing focus on the correlation between microorganisms and digestive tract diseases in academic research. For example, Helicobacter pylori has been the well-recognized key causative factor for gastric cancer. 24 Previous researches have clarified that virus infection such as human papillomavirus (HPV) and Epstein-Barr virus, as well as the changes in gut microbial composition contribute to the carcinogenesis of the esophagus. 25 In a recent study, the tumor development in LFD-fed GF mice, which received the fecal microbiota from HFD-fed L2-IL1B mice, indicated the crucial role of intestinal microbes in esophageal cancer development and verified its linkage with the IL8/CXCL1 chemokine family. 26 In an observational study, which recruited 23 EC patient and 23 healthy individuals from Huai'an First People's Hospital (Huai'an, China), the abundance of Lachnospira was observed to be reduced in the EC patients, and Lachnospira, of which the AUC = 1, showed a great discrimination ability between EC patients and the healthy. 17 Reinforcing our analyses, it is plausible that Lachnospira plays a protective role in the pathogenesis of esophageal cancer. The genera Lachnospira, belonging to the phylum Firmicutes and the family Lachnospiraceae, generates acetic acid and butyric acid. It has been recognized that SCFAs not only maintain the stability of Gastrointestinal homeostasis but also function through immunoregulation. 27 Studies have shown that Lachnospira might connect with nonalcoholic fatty liver disease, 28 Asthma, 29 gastric cancer, 30 colorectal cancer, 31 depression and anxiety in patients with active ulcerative colitis. 32 However, most of them are association studies, lacking in exploration of mechanisms. Veillonella, one of the major oral microorganisms, promoted the cancer development according to our analyses. The genera contain Veillonella parvum and Veillonella alkalinogens. Oral microbial colonization of the intestine is concerned with many diseases, such as inflammatory bowel disease (IBD) 33 and colorectal cancer. 34 A recent study illustrated that the Veillonella ectopically colonize the intestine by taking advantage of inflammatory nitrates derived from inducible nitric oxide synthase (iNOS) activity in host cells. 35 As a member of Type II bacteria (Gram-negative and anaerobic/microaerophilic bacteria) of the Esophageal Flora, the genera Veillonella may serve as collaborative mechanism to facilitate gastric reflux through activating the iNOS pathway. It is considered that the alteration of Veillonella promotes the progression of gastroesophageal reflux to Barrett's esophagus, which eventually leads to adenocarcinoma. 36 The studies mentioned above all further validated our analysis. Further studies are urgently needed to determine the mechanism of intestinal bacteria in the pathogenesis of esophageal cancer.
In our reverse MR analyses, we revealed that the alteration of gut microbes after suffering from esophageal cancer. Eggerthella is among the top five most abundant genera of the phylum Actinobacteria in the human gut. A study found that the colonization of Eggerthella activates intestinal Th17 by removing from inhibition of the Th17 transcription factor Rorγt, exacerbating the severity of colitis. 37 Moreover, with proinflammatory properties, Eggerthella is involved in the synthesis of key neurotransmitters in depression, such as glutamate, butyrate, serotonin, and γ-aminobutyric acid (GABA). 38 In another study, Eggerthella lenta was found to promotes the accumulation of uremic toxin in serum and exacerbate the progression of kidney disease in a CKD rat model. 39 However, there is little research on the association of Eggerthella and EC, let alone the mechanism. Our analyses showed the decreased abundance of Intestinimonas under the exposure of EC. Intestinimonas produces butyrate from both sugars and amino acids, protecting the host from harmful metabolites. The enrichment of the genus Intestinimonas in gastric intraepithelial neoplasia 40 and the reduced abundance of this genera in the obesity 41 has been observed. Besides, there's already evidence that Intestinimonas showed a good distinction ability between the patients with chronic renal diseases (CKD) and the healthy 42 and correlated with the total functional ability score and the IL-4 level in patients with Huntington's disease. 43 Currently, there is limited research on the intestinal microbiota identified in our reverse study. Further investigation is necessary to determine whether alterations in gut microbiota following esophageal cancer have a positive or negative impact, which could enhance the prognosis and adjuvant therapy of EC.
Our study has several strengths. Previous studies are more focused on esophageal cancer and esophageal microbiota. Moreover, changes in the microbiota in observational studies are merely clinical manifestations. We analyzed GWAS data by MR to confirm the possible interaction and causal relationship between EC and intestinal microbiota. Bidirectional analyses guarantee causality inferred from both directions. The selected bacteria lay the foundation for further exploration in the future. This also has contributed to the screening of biomarkers for esophageal cancer and the improvement of prognosis. In the procedure of the MR analyses, we strictly screened for strong-associated SNPs as instrumental variables (F statistics >10). MR Egger regression was conducted to exclude SNPS with pleiotropy. Despite our exploration is meaningful, there are several limitations. The population we obtained from the UK Biobank and MiBioGen consortium were restricted to the European, in which the most common type of esophageal cancer is adenocarcinoma.We did not get the original information on the patients with EC, making it difficult for us to perform subgroup analysis. In addition, there may be overlap between the two groups, which could lead to bias.Besides, genus was the lowest taxonomic level in the gut microbe datasets, further exploration at the species level was restricted. Furthermore,we used a relativey lenient P-value threshold of 5*10−6 to select the genetic variants strongly-associated with gut microbiota in the forward MR analysis, inevitably violating the MR assumptaton to some extent. We chose the IVW as the major method to estimate the causal relationship, while as presented in the forest plot there was contradictory results in other methods. Moreover, the OR value we obtained in the forward MR is close to 1. The interpretation of our results requires caution, and further in-depth clinical and scientific research is necessary to provide more robust evidence.
In conclusion, this two sample MR found that the bilateral causality between esophageal cancer and intestinal microbes. Several genera such as Lachnospira and Veillonella had a positive or carcinogenic effect on esophageal cancer. Moreover, the alteration of the genera Eggerthella and Intestinimonas demonstrated the influence of EC on the gut microbiome. Further RCT study and animal experiments are in an urgent need to figure out the mechanisms involved, thus making the candidate intestinal microbes useful for clinical precaution and screening.
Footnotes
Acknowledgements
The authors thank all investigators and participants from GWAS databases for sharing these data.
Author Contributions (Roles)
S.W. and C.H. drafted the manuscript. C.H. and H.D. were responsible for conception and design the study. Z.G.X. and Y.B.X. performed data analysis and interpretation. S.X.M. and Z.W.F. reviewed the data analysis. Z.X.Y. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The present MR analysis was based on summary data from previous studies that had gained written informed consent and ethics approval. No ethical permit is required for the secondary analysis of summary data.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Natural Science Foundation of China (No. 82100594), China Postdoctoral Science Foundation (No. 2023M731417), Spring Sunshine Program from the Ministry of Education of China (No.202201552) and Jiangsu province Hospital (the First Affiliated Hospital with Nanjing Medical University) Clinical Capacity Enhancement Project (JSPH-MC-2022-29).