
Abstract
Membrane contact sites are molecular bridges between organelles that are sustained by tethering proteins and enable organelle communication. The endoplasmic reticulum (ER) membrane harbors many distinct families of tether proteins that enable the formation of contacts with all other organelles. One such example is the LAM (Lipid transfer protein Anchored at Membrane contact sites) family in yeast, which is composed of six members, each containing a putative lipid binding and transfer domain and an ER-embedded transmembrane segment. The family is divided into three homologous pairs each unique in their molecular architecture and localization to different ER subdomains. However, what determines the distinct localization of the different LAMs and which specific roles they carry out in each contact are still open questions. To address these, we utilized a labeling approach to profile the proximal protein landscape of the entire family. Focusing on unique, candidate interactors we could support that Lam5 resides at the ER-mitochondria contact site and demonstrate a role for it in sustaining mitochondrial activity. Capturing shared, putative interactors of multiple LAMs, we show how the Lam1/3 and Lam2/4 paralogous pairs could be associated specifically with the plasma membrane. Overall, our work provides new insights into the regulation and function of the LAM family members. More globally it demonstrates how proximity labeling can help identify the shared or unique functions of paralogous proteins.
Keywords
Introduction
Eukaryotic complexity is afforded by cell compartmentalization into membrane-bound organelles, each of which is defined by a unique protein and lipid landscape organized into specific substructures. However, these organelles must communicate with one another to coordinate cellular processes and output. This can be achieved through membrane contact sites – areas of close apposition (typically ∼30 nm or less) between specialized membrane subdomains of different organelles, which are tethered to each other via protein-protein or protein-lipid interactions (Scorrano et al., 2019).
All organelles form contacts with each other (Shai et al., 2018) and this has been well documented for the endoplasmic reticulum (ER), which forms multiple and expansive contacts with different organellar membranes (Valm et al., 2017). Lipid transfer is an indispensable function of ER contact sites since the majority of lipid biosynthetic machinery is resident in the ER membrane. One protein domain capable of mediating the transfer of the essential lipid, sterol, is the Steroidogenic Acute Regulatory protein-related lipid Transfer (StART) domain (Clark et al., 1994; Alpy and Tomasetto, 2005). In Saccharomyces cerevisiae (called yeast from here on) there is a family of StART-like domain proteins that resides in the ER and is composed of three homologous pairs: Ysp1 (Lam1) and Sip3 (Lam3); Ysp2 (Ltc4 or Lam2) and Lam4 (Ltc3); and Lam5 (Ltc2) and Lam6 (Ltc1) (Figure 1(A)) (Gatta et al., 2015; Murley et al., 2015). For simplicity and consistency, we will henceforth refer to these proteins only by their LAM nomenclature. Apart from their StART-like domains, these proteins also share an N-terminal Pleckstrin Homology (PH)-like GRAM domain, and a C-terminal transmembrane domain (TMD) which anchors them in the ER membrane (Gatta et al., 2015). The presence of these domains is conserved to the family of human ASTER/GRAMD1 proteins; ASTER-A, ASTER-B, ASTER-C, which are also called GRAMD1A, GRAMD1B and GRAMD1C (Gatta et al., 2015; Sandhu et al., 2018; Naito et al., 2019), and also to plants, where the StART-like domain is referred to as VASt (VAD1 analog of StART (Khafif et al., 2014)).

Proximity profiles of LAM family members differentiate between shared and unique features. (A) A schematic of the yeast LAM family, shown as three pairs of homologs: Lam1 (Ysp1) and Lam3 (Sip3); Lam2 (Ysp2/Ltc4) and Lam4 (Ltc3); Lam5 (Ltc2) and Lam6 (Ltc1). Highlighted are: the cytosolic-facing TurboID-HA tags (yellow) at the N-termini; the BAR (Bin-Amphiphysin-Rvs; green), GRAM (purple) and StART-like (blue) domains; and the C-terminal TMDs (orange). (B) Volcano plots showing -log(P-value) against log2(fold-change) for all proteins identified by LC-MS/MS of the appropriate TurboID-HA-LAM sample and Emc6 control, each in biological triplicate. High-confidence proximal proteins (HCPPs) enriched in each TurboID-HA-LAM sample are marked with their names and highlighted in red. Proteins which passed the criteria for P-value and fold-change, but were only identified by one unique peptide, are colored gray. (C) Hierarchical clustering of the TurboID-ID LAM biological triplicates and their HCPPs, shown as a heatmap. The colors are defined by the normalized intensity. The HCPPs highlighted in red in (B) are marked by an asterisk. The data shows clustering according to the biological replicates. The homologous LAM pairs cluster together, with Lam1–4 separate from Lam5 and Lam6.
The LAM proteins were discovered to localize at distinct ER contact sites. Lam1,2,3 and 4 (Lam1–4) are present at ER-plasma membrane (PM) contacts (Gatta et al., 2015; Murley et al., 2017), Lam5 at ER-Golgi (Weill et al., 2018a) and potentially also ER-mitochondria (Gatta et al., 2015; Fujimoto et al., 2023) contacts, and Lam6 at contacts between ER-mitochondria, and nucleus-vacuole junctions (NVJs) which are contacts between the nuclear ER and the vacuole (Elbaz-Alon et al., 2015; Gatta et al., 2015; Murley et al., 2015). Numerous structure-function studies on the Lam2 and Lam4 StART-like domains have mechanistically defined sterol binding, solubilization and retrograde transfer from the PM to the ER (Gatta et al., 2015, 2018; Horenkamp et al., 2018; Jentsch et al., 2018; Tong et al., 2018; Khelashvili et al., 2019). However, the best characterized member of this family is Lam6, and its presence at multiple membrane contact sites makes it essential for their regulation and cross-talk (Elbaz-Alon et al., 2015). Furthermore, as opposed to Lam1–4, Lam6 mediates anterograde sterol transfer (away from the ER) required for stress-induced formation of a vacuolar membrane subdomain (Murley et al., 2015) by forming a tethering interaction with the vacuolar protein, Vac8. At the ER-mitochondria contact, Lam6 tethers both organelles by interacting with the mitochondrial protein, Tom70 (Elbaz-Alon et al., 2015; Murley et al., 2015).
The fact that Lam6 requires protein tethers on the adjacent membranes of either mitochondria (Tom70) or the vacuole (Vac8) is in some ways surprising, given the presence of its N-terminal GRAM domain. These domains are classically associated with binding anionic lipids such as phosphatidylserine (PS) and different phosphoinositide (PIP) species in the adjacent membrane of the contact (Sandhu et al., 2018; Naito et al., 2019; Ercan et al., 2021). However, analysis of the LAM family GRAM domains shows that they expose fewer basic residues relative to typical GRAM domains (Tong et al., 2018). This would result in a lower affinity to anionic lipids and might underly their requirement for proteinaceous tethering molecules.
Short of Lam6, there is much less information on how other LAMs bind adjacent membranes. Two proteins, Laf1 and Dgr2, were identified as interactors of Lam1–4 (Murley et al., 2015; Topolska et al., 2020). However, neither of these are integral membrane proteins and their deletion did not affect the localization of Lam2 or Lam4 at ER-PM contacts (Topolska et al., 2020). Further still, Lam5 has not been well characterized, neither in terms of interactors, nor function. To better understand the functional significance of the LAM proteins, and how their positioning at specific ER subdomains is determined, we applied our recently-developed enhanced proximity-labeling approach (Fenech et al., 2023) to uncover proximally residing proteins. Through this approach, we were able to shed light on a bioenergetic role for the lesser-characterized Lam5 protein, and also identify a positive regulator of Lam1–4-based ER-PM tethering, which we propose reconciles protein- and lipid-mediated binding to membranes.
Results
Proximity Profiles of LAM Family Members Differentiate Between Shared and Unique Features
The family of yeast LAM proteins act as contact site tethers. LAMs are anchored to the ER membrane by C-terminal TMDs and have long N-terminal domains that extend away from the ER to make contact with different membranes (Gatta et al., 2015). How these ER-resident proteins are directed to different contacts, and which cellular functions they support at these locales, is still not fully understood. We hypothesized that clues to this could come from mapping their precise protein environment at these points of contact.
To map this landscape, we turned to TurboID (Branon et al., 2018), a highly-efficient proximity-labeling enzyme, which conjugates biotin onto available lysine (K) residues in proteins within ∼10 nm, allowing them to be effectively captured by streptavidin and later detected by mass spectrometry (MS). We previously showed that the ABOLISH (Auxin-induced BiOtin LIgase diminiSHing) system enhances the detection of TurboID-labelled proximal proteins by specifically reducing the levels of endogenously biotinylated proteins (Fenech et al., 2023). Therefore, we utilized strains that were part of the whole-proteome yeast TurboID/ABOLISH library (Fenech et al., 2023) which expressed TurboID-HA N-terminal fusions for each of the six LAM family members (Figure 1(A), Supplementary Figure 1(A)). Our reason for using N-terminal TurboID fusions was that the tags would least interfere with the topology of the single TMD at the extreme C-terminus and, moreover, would be poised to label interactors on the adjacent contact site membranes. To ensure that these fusions did not interfere with LAM functionality, we carried out growth assays on media containing Amphotericin B, an anti-fungal drug that affects PM sterols (Anderson et al., 2014). It was previously shown that strains lacking Lam1, Lam2 or Lam3 activity are inhibited in their growth on this drug (Gatta et al., 2015). However, strains expressing either TurboID-HA-Lam1, -Lam2 or -Lam3 were not affected compared to Δlam1, Δlam2 or Δlam3 strains (Supplementary Figure 1(B)) suggesting that N-terminal tagging does not hamper the activity of LAM proteins.
These strains, together with a control strain expressing TurboID-HA-Emc6 (an ER-resident protein not known to interact with the LAM family) on the background of ABOLISH, were subject to streptavidin affinity purification (AP) and analysed by MS. To determine high-confidence proximal proteins (HCPPs) for each of the LAMs, their MS profiles were compared to that of the Emc6 sample and enriched proteins were determined as having: a P-value ≤ 0.05; a fold-change ≥ 2; and at least two unique peptides (Figure 1(B), Supplementary Table 1).
The first striking observation was the strong overlap between the HCPPs of Lam1–4, driving the clustering of these four LAMs together (Figure 1(C)). This was not surprising since these proteins are all known to mediate ER-PM contacts (Gatta et al., 2015; Murley et al., 2017). Furthermore, the paralog pairs Lam1/3 and Lam2/4 formed their own subclusters, with a very high degree of overlap shared between the Lam2 and Lam4 HCPPs (Figure 1(B) and (C)). While Lam1 and Lam3 did share HCPPs, Lam1 enriched more unique, cytosolic/nuclear proteins (Hsp26, Yhb1, Dog1/2 and Sam4) relative to Lam3 (and indeed relative to Lam2 and Lam4). Encouragingly, many shared HCPPs of Lam1–4 are known to be PM-localized (Sso1/2, Nbr9, Ras2, and Aqr1) and Lam2 was identified as an HCPP of Lam1, Lam3 and Lam4; the latter being reciprocally found as a Lam2 HCPP (Figure 1(B) and (C)). The interconnectivity between these LAM family members has been previously reported in independent proteomic experiments performed on C-terminally tagged LAM proteins (Murley et al., 2017). Furthermore, this feature extends to the human GRAMD orthologs which were shown to form homo- and hetero-oligomeric complexes (Naito et al., 2019) and we could also reproduce this by immunoprecipitating FLAG-tagged GRAMD1A expressed in HEK293T cells (Supplementary Figure 1(C), Supplementary Table 2) (Naito et al., 2019).
The Lam5/6 paralog pair formed their own separate clusters (Figure 1(C)). As expected, mitochondrial proteins were enriched in the Lam6 sample, including: the outer membrane voltage-dependent anion channel, Por1; and Tom20, a component of the mitochondrial TOM (translocase of the outer membrane) complex. Por1 was previously isolated together with Lam6 (Murley et al., 2015), whereas Tom20 forms a complex together with Tom70; the known tethering partner for Lam6 at ER-mitochondria contact sites (Elbaz-Alon et al., 2015; Murley et al., 2015). Interestingly, Lam5 was also associated with HCPPs linked to different aspects of mitochondrial biology and these are discussed below. Lastly, the vacuolar protein, Vac8, and the ζ-subunit of the coatomer complex, Ret3, were identified as unique HCPPs of Lam6 and Lam5, respectively (Figure 1(B) and (C)). This is consistent with what is already known: Lam6-Vac8 is a known tethering pair at the ER-vacuole contact site (Elbaz-Alon et al., 2015; Murley et al., 2015); and Lam5 localizes to the ER-Golgi interface (Weill et al., 2018a), where the coatomer machinery resides. GRAMD1A-FLAG analysis also recovered a coatomer component (Supplementary Figure 1(C), Supplementary Table 2), increasing our confidence in this putative interaction.
Altogether, our TurboID/ABOLISH approach uncovered known LAM family interactors, as well as many novel putative proximal proteins that could provide clues to the function, regulation and tethering properties of these contact site proteins.
Lam5 Is Localized to ER-Mitochondria Contact Sites and Modulates Mitochondrial Activity
While Lam6 is known to play a role in contact site cross-talk (Elbaz-Alon et al., 2015) and vacuole membrane domain formation (Murley et al., 2015), its close paralog, Lam5, remains uncharacterized in terms of function. The observation that stress-induced domain formation in the vacuolar membrane is perturbed in a Δlam6 strain where Lam5 is still present (Murley et al., 2015) suggests that their roles are at least partially non-redundant. Indeed, the HCPPs of these two LAM family members were largely unique, with only Vps35 being shared (Figure 2(A)).

Lam5 is localized to ER-mitochondria contact sites and modulates mitochondrial activity. (A) An illustration of unique and shared HCPPs of Lam5 and Lam6. HCPP fold-change is marked by a thick (fold-change ≥ 5) or thin (2 ≤ fold-change < 5) arrow, and HCPP localization/pattern (mitochondria – mito; vacuole; or puncta) is annotated according to images of the GFP SWAT libraries (Yofe et al., 2016; Meurer et al., 2018; Weill et al., 2018b) collected on Yeast RGB (Dubreuil et al., 2018). (B) Enhanced resolution confocal microscopy images of strains expressing Lam5 or Lam6 tagged on their N-termini with GFP, stained with vacuolar (FM™4-64) and mitochondrial (MitoView™405) dyes. While it was known that Lam6 localizes to contact sites with both mitochondria and vacuoles, we could also visualize Lam5 colocalization with both the vacuole and mitochondria, as highlighted by solid and dashed arrows, respectively. Scale bars are 5 μm. (C) Confocal microscopy images of strains expressing Lam5 or Lam6 tagged on their N-termini with mCherry and under expression of the strong TEF2 promoter, together with a split-Venus ER-mitochondria contact site (CS) reporter. Overexpressed Lam6 strongly colocalizes with the reporter and even enhances the extent of the contact. Overexpressed Lam5 partially colocalizes with the reporter, as indicated by white arrows. Analysis indicates that 65% of Lam6 signal overlaps with the CS reporter puncta, compared to a 36% overlap for Lam5. Background overlap (from the control cells where no protein was tagged with mCherry) was quantified at 12%. Scale bars are 5 μm. (D) Confocal microscopy images of strains expressing Lam5 or Lam6 tagged on their N-termini with GFP, together with Aim21, that was identified as a putative Lam5 interactor, tagged on its C-terminus with mScarlet. Indeed, Lam5, but not Lam6, shows partial colocalization with Aim21, especially in/around the bud, as indicated by white arrows. Quantitation demonstrates that 31% of signal from Aim21 puncta overlaps with that of Lam5, whereas only 2% overlaps with Lam6. Scale bars are 5 μm. (E) Graphs showing the fold-change in basal and maximal oxygen consumption rate (OCR) of Δlam5 (dark green) and Δlam6 (light green) strains relative to control (black), grown in the non-fermentable carbon source, galactose, measured by Seahorse assay (Agilent). Only the lam6 knockout shows a significant increase in both basal and maximal OCR. The data are from six biological replicates and significance was calculated using two-way ANOVA with Tukey's multiple comparison where: * is p ≤ 0.05; ** is p ≤ 0.01; *** is p ≤ 0.001; and ns is not significant. Error bars show the standard error of the mean (SEM). (F) As in (E) however strains were grown in glucose, where now both knockout strains show significantly elevated basal and maximal OCR.
To begin exploring any overlapping and/or unique functions of Lam5 and Lam6, we first visualized GFP-tagged versions of these proteins together with vacuolar and mitochondrial markers (Figure 2(B)). It was immediately clear that while both proteins displayed a punctate pattern, Lam5, but not Lam6, also showed a more typical ER signature (defined as having a ‘ring’ around the nucleus and another around the cell periphery). Furthermore, there was a high degree of colocalization between the signal from the stained organelles and GFP-Lam6, as expected (Elbaz-Alon et al., 2015; Gatta et al., 2015; Murley et al., 2015). We did, however, find some colocalization of GFP-Lam5 with both vacuole and mitochondria, the latter supporting the previously observed colocalization between Lam5 and Tom6 (Gatta et al., 2015) and recent work exploring proteins proximal to the ER-mitochondria contact (Fujimoto et al., 2023).
To examine whether Lam5 could be a bona fide resident of the ER-mitochondria contact site, we assessed its colocalization with an ER-mitochondria contact site reporter based on the split-Venus probe (Shai et al., 2018). Indeed, overexpressed mCherry-Lam5 signal colocalized with the reporter at discrete puncta (Figure 2(C)). Lam6 tagged with mCherry also colocalized with the ER-mitochondria contact site reporter and its over-expression increased the area of the reporter's signal; a phenomenon associated with some tethering proteins (Eisenberg-Bord et al., 2016), which had been previously observed (Shai et al., 2018). While Lam6 forms a contact site tether with components of the TOM complex (Elbaz-Alon et al., 2015; Murley et al., 2015) we did not identify mitochondrial outer membrane proteins as Lam5 HCPPs (Figures 1(B), (C) and 2(A)). We did, however, identify Aim21 (Altered Inheritance rate of Mitochondria), an actin-associated protein needed for correct mitochondrial motility (Hess et al., 2009; Shin et al., 2018). Strikingly, GFP-Lam5 colocalized with Aim21-mScarlet in the bud and bud neck region, unlike GFP-Lam6 which showed no colocalization with this protein (Figure 2(D)).
Our observations suggested that losing either Lam5 or Lam6 would have a functional consequence at ER-mitochondria contacts, potentially affecting mitochondrial activity. To measure this, we used real-time respirometry to measure the oxygen consumption rate (OCR) in cells lacking either Lam5 or Lam6 and compared them to control cells. When grown on a non-fermentable carbon source (galactose), only the loss of Lam6 significantly affected respiration (Figure 2(E)). However, when grown on glucose as the carbon source (the condition in which we performed our protein landscape profiling and microscopic analyses) we observed that both Δlam5 and Δlam6 had a higher basal and maximal OCR (Figure 2(F)). This demonstrates that these mutations cause elevated levels of respiration and electron transport chain (ETC) activity. Interestingly, loss of the Lam5 HCPP, Aim21, as well as the shared Lam5/6 HCPP, Vps35, also resulted in an increased OCR in glucose (Supplementary Figure 2). Collectively, our data uncover a previously unappreciated role for Lam5 at the ER-mitochondria contact site and suggest that the Lam5/6 paralogs differentially influence mitochondrial function, likely through their association with different proximal proteins.
Proteins Proximal to Lam1–4 Reveal a Novel Mode of Association to the PM
In addition to the Lam5/6 paralogs, there are two other homologous protein pairs; Lam1/3 and Lam2/4, which reside at ER-PM contacts (Gatta et al., 2015; Murley et al., 2017). As tethers they should have binding partners on the PM, yet their identity to date has not been uncovered. Encouragingly, we observed an enrichment of PM-resident proteins as HCPPs (Figure 1(B) and (C)). Furthermore, it seemed that the Lam1/3 paralog pair was less strongly associated with these proteins relative to the Lam2/4 paralogs (Figure 3(A)). This aligned closely with what was observed by imaging. GFP-tagged Lam1/3 display a more typical ER pattern, with both perinuclear and cell peripheral domains visible (Figure 3(B)), as opposed to GFP-Lam2/4, which appear exclusively as peripheral ER, indicating an increased association to the PM. Imaging of both mitochondria and vacuoles showed essentially no overlap with these members of the LAM family (Supplementary Figure 3(A)), highlighting features of Lam1–4 that are non-overlapping with Lam5/6.
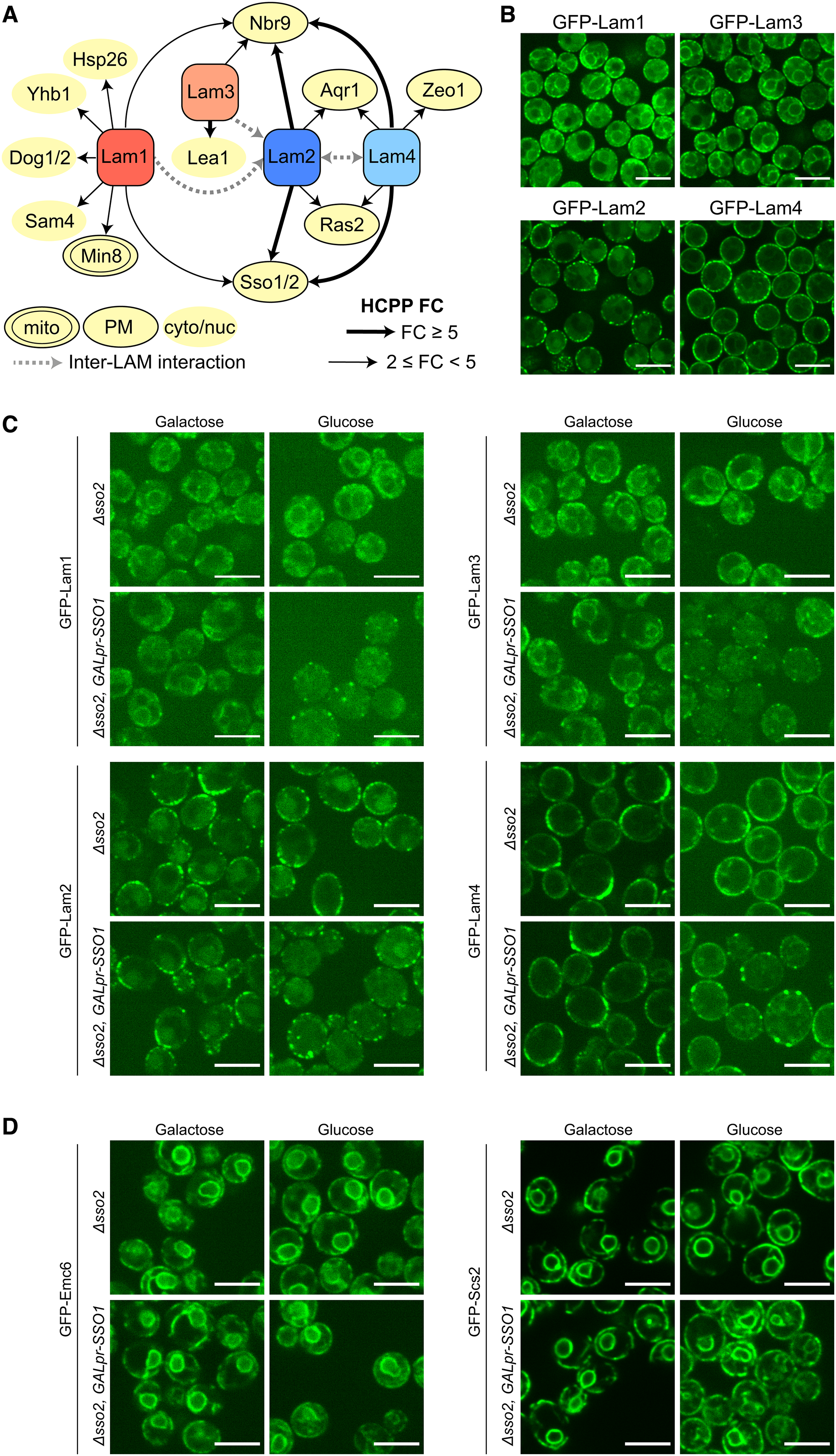
Proteins proximal to Lam1–4 reveal a novel mode of association to the PM. (A) An illustration of unique and shared HCPPs of Lam1–4. HCPP fold-change is marked by a thick (fold-change ≥ 5) or thin (2 ≤ fold-change < 5) arrow, with interactions between different LAM proteins marked by a grey, dashed line. HCPP localization (mitochondria – mito; plasma membrane - PM; cytosolic/nucleoplasmic – cyto/nuc) is annotated according to images of the GFP SWAT libraries (Yofe et al., 2016; Meurer et al., 2018; Weill et al., 2018b) collected on Yeast RGB (Dubreuil et al., 2018). (B) Enhanced resolution confocal microscopy images of strains expressing Lam1–4 tagged on their N-termini with GFP. All display at least some peripheral localization (as reported) with the paralog pair Lam2 and Lam4 situated exclusively at the periphery relative to the Lam1 and Lam3 paralog pair. Scale bars are 5 μm. (C) Enhanced resolution confocal microscopy images of strains grown in either galactose or glucose, expressing Lam1 and Lam3 (top panels), and Lam2 and Lam4 (bottom panels) tagged on their N-termini with GFP, on the background of either Δsso2 or Δsso2 with an inducible/repressible promoter (GALpr) driving the expression of its homolog SSO1 (Δsso2, GALpr-SSO1). When the latter cells are grown in glucose, SSO1 is repressed to create a double mutant of SSO1/2, and each of the tested LAM proteins loses their normal ER localization pattern, instead localizing to a few, dispersed bright puncta, mainly around the periphery. Comparing GFP-tagged Lam1, Lam2, Lam3 and Lam4 grown in glucose on either the single-mutant (Δsso2) background or the double-mutant SSO background, the fold-change increase in this punctate pattern was: 6.0, 1.8, 4.6 and 3.4, respectively. Scale bars are 5 μm. (D) As in (C) however GFP-tagged proteins are Emc6 and Scs2 as negative controls. In glucose, the peripheral ER localization of both persists and their signal does not become punctate. Scale bars are 5 μm.
Of the potential PM-resident binding proteins, we focused on the SSOs (paralogs Sso1 and Sso2, orthologous to human Syntaxin1A, STX1A). These proteins were found as HCPPs of Lam1, Lam2, and Lam4 and have already been implicated in ER-PM contact site formation in both yeast and humans (Petkovic et al., 2014). Importantly, none of the other known ER-PM tethers (Manford et al., 2012; Filseck et al., 2015), including the SSO proteins, had been physically associated to the LAM proteins before. Since the SSOs must be in areas of close apposition between the ER and PM, we hypothesized that they may play a role in the Lam1–4-mediated ER-PM contacts. To test this, we generated GFP-tagged Lam1–4 strains on the background of an SSO mutant. Since the double knockout of SSO1/2 is lethal, we created either Δsso2 strains or Δsso2 strains which also harbored SSO1 under regulation of a galactose-inducible/glucose-repressible promoter (GALpr-SSO1). When the latter strain is cultured in glucose-containing media, SSO1 expression is shut-off, enabling the generation of cells with minimal levels of SSO proteins. While we were unable to assay these double mutant strains for sensitivity to Amphotericin B (as done for LAM knockout strains (Gatta et al., 2015), Supplementary Figure 1(B)) we could see that the single Δsso2 mutant challenged by incubation at 37 °C, grew slower on media containing the drug compared to the control (Supplementary Figure 3(B)). More striking however, was that when the Δsso2/GALpr-SSO1 double mutant cells were grown in glucose (Figure 3(C), bottom right image in each of the four panels), the localization of the GFP-Lam1–4 proteins changed dramatically. Specifically, the perinuclear signal from Lam1/3 was largely lost, and the majority of the peripheral signal from Lam1–4, attributed to the ER-PM contact, disappeared, leaving only few individual and more isolated puncta which were mainly situated around the cell periphery. This supports the hypothesis that the SSO proteins play a role in the tethering of Lam1–4 at the PM. To control for nonspecific effects on either the ER membrane as a whole, or the general ER-PM contact site structure, we imaged, respectively, either a GFP-tagged ER membrane protein (GFP-Emc6) or the yeast ortholog of VAMP-associated protein (VAP); an ER-PM contact site protein not associated with the LAM family members (GFP-Scs2). These tagged proteins, which were imaged on the same genetic background as the GFP-Lam1–4 strains, were not affected by SSO protein loss (Figure 3(D)), and these data suggest that SSO proteins are specifically important for Lam1–4 binding the PM at ER-PM contact sites.
Interestingly, it has been shown that human STX1A can cluster the anionic PIP, PI(4,5)P2, in the PM (Honigmann et al., 2013). Consistently, we noticed that PI(4,5)P2 is partially mislocalized in cells depleted for both SSO proteins (Supplementary Figure 4). Based on this information, we hypothesized that the SSO proteins may help promote Lam1–4 PM tethering by enhancing PI(4,5)P2-GRAM domain interaction. To test this, we visualized these LAMs following the depletion of PI(4,5)P2 using an alternative method. Specifically, we GFP-tagged Lam1–4, Emc6 and Scs2 on the background of a mutant of Mss4; the kinase which generates PI(4,5)P2 from PI4P. Since Mss4 is an essential gene, we fuzed it to an auxin-inducible degron (AID* (Morawska and Ulrich, 2013)), which enables its degradation in the presence of the mutated TIR1(F74G) adaptor protein and the modified auxin compound, 5-Ph-IAA (Yesbolatova et al., 2020). Upon treatment with 5-Ph-IAA, Lam1–4 localization was altered and large, internal accumulations were observed (Figure 4(A)). Again, loss of the perinuclear signal from Lam1/3 was more pronounced than the changes in the peripheral signal from Lam1–4, and both control proteins, Emc6 and Scs2, were unaffected. Based on these collective results, we propose a hypothetical model in which SSO-mediated PI(4,5)P2 clustering supports Lam1–4 tethering (Figure 4(B)). Upon depletion of the SSO proteins (or Mss4), PI(4,5)P2 is no longer adequately clustered, leading to a reduction in the number of Lam1–4-contacts, which correspond to the punctate pattern observed in Figure 3(C). In summary, our data suggest that both PI(4,5)P2 and the SSO proteins play an important role in determining the correct localization of Lam1–4 at ER-PM contact sites.
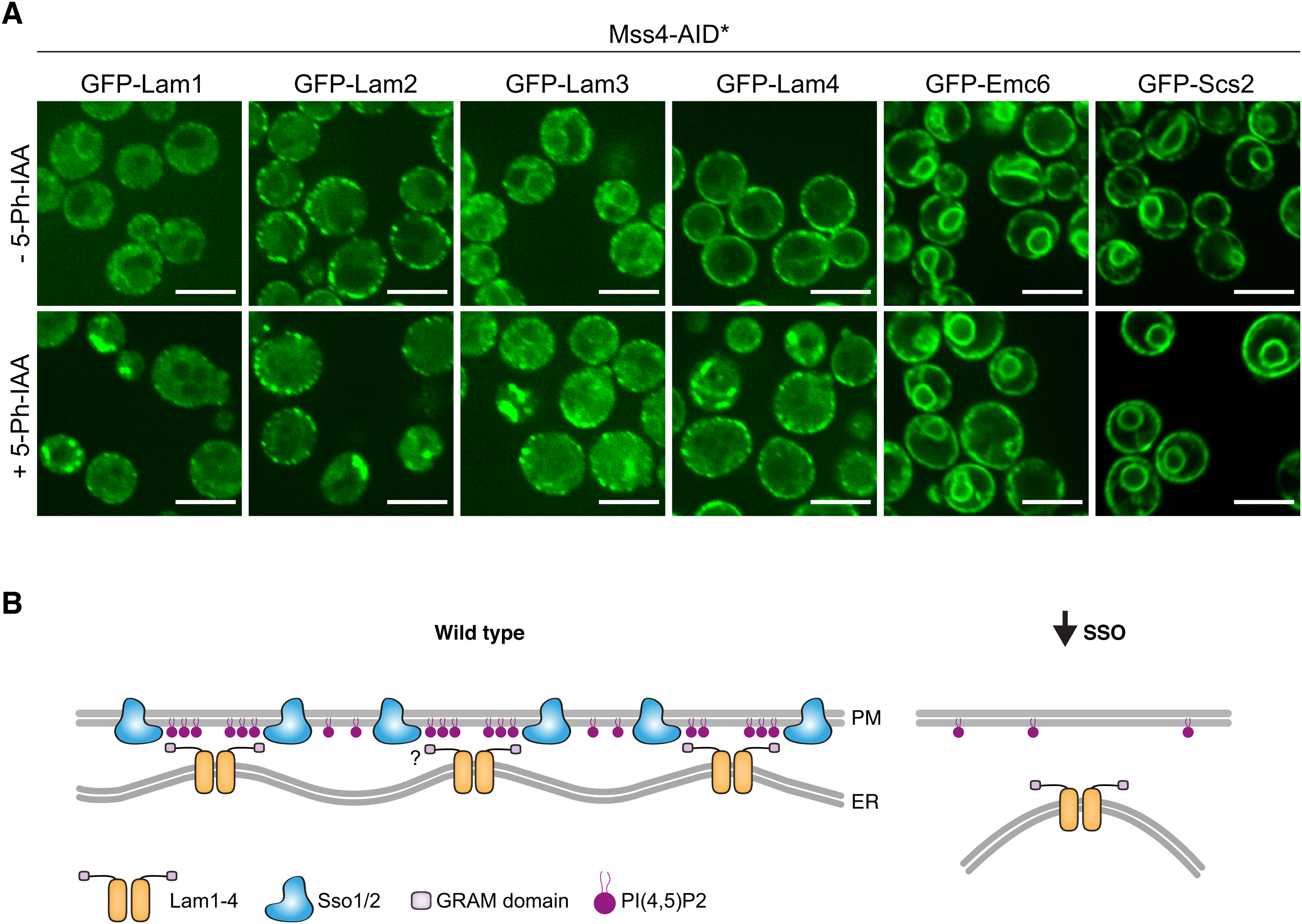
SSO proteins are important for Lam1–4 association at the ER-PM contact potentially via PI(4,5)P2 clustering. (A) Enhanced resolution confocal microscopy images of strains grown either with or without auxin, expressing either Lam1, Lam2, Lam3, Lam4, Emc6, or Scs2 tagged on their N-termini with GFP, on the background of TIR1(F74G) expression and Mss4 tagged on its C-terminus with AID*-9myc. In cells grown in auxin, where Mss4 is depleted, Lam1–4 are no longer correctly localized, however no effect is observed for Emc6 and Scs2. Scale bars are 5 μm. (B) A hypothetical model for how SSO loss could affect LAM association with the PM: ER localized Lam1–4 may oligomerize, potentially through Lam2, bringing together their N-terminal GRAM and StART-like domains (for simplicity, only the GRAM domain is depicted). The GRAM domain can interact with PI(4,5)P2 on the PM, which has been shown to be corralled by the human SSO orthologs (Honigmann et al., 2013). These local concentrations of PI(4,5)P2, potentially together with Lam1–4-SSO protein-interaction (shown as a question mark), may act to increase the avidity of binding between Lam1–4 and the PM. On the contrary, when SSO protein levels are limiting, PI(4,5)P2 is randomly diffused within the PM, reducing its effective concentration at contact sites and decreasing the binding capacity for the LAMs, hence reducing the amount of contacts that they form.
Discussion
Using the newly-developed TurboID/ABOLISH system (Fenech et al., 2023) we set out to uncover novel proximal proteins for the entire ER-resident LAM family of contact site proteins, to gain a deeper understanding of their unique and shared features, and how these define their localization or regulation. Since the LAM proteins are anchored in the ER membrane by C-terminal TMDs, and their N-termini extend towards adjacent membranes to form a contact, we tagged these proteins N-terminally (Figure 1(A)) to capture putative interactors which may be involved in membrane tethering. While proximity-labeling methods are distinct from classical immunoprecipitation (IP), we could still recapitulate previously identified interactors including the Lam6-Vac8 interaction (Elbaz-Alon et al., 2015; Murley et al., 2015) and the association between ER-PM LAM members (Murley et al., 2017). As well as verifying known interactions, we also identified new putative proximal proteins, which we explored further.
From the proximity profiling, it was evident that the paralog pair Lam5 and Lam6 were the most distinct – both from the rest of the LAM family, and from each other (Figure 1(C)). Nevertheless, inspection of their localization revealed some overlap in terms of the organelles they are adjacent to (Figure 2(B)). Intriguingly, Lam5 showed partial colocalization with mitochondria, and encouragingly also with an ER-mitochondria contact site reporter (Figure 2(C)). In addition, Aim21, a soluble actin-binding protein, was identified as one of its HCPPs (Figures 1(B), (C), and 2(A)), and since this protein was shown to be required for proper mitochondrial movement (Hess et al., 2009), we reasoned this may be at least part of the missing link between Lam5 and mitochondria. Indeed, we observed a unique colocalization between these proteins (Figure 2(D)). However, since we were not able to find evidence for Aim21 being present at ER-mitochondria contacts, more work is required to understand its relationship with Lam5 at this contact site. An intriguing possibility is that the function of Lam5 at the ER-mitochondrial interface may be influenced by an Aim21-Lam5 interaction at a distinct cellular locale. Alternatively, other factors may be involved, such as Vps35, a protein identified as an HCPP of both Lam5 and Lam6. Despite it being a highly-conserved component of the retromer complex in both yeast and humans, it is also known to regulate mitochondrial dynamics (Cutillo et al., 2020). One way it does this is via the removal of inactive human DNM1L (the ortholog of yeast Dnm1), which promotes mitochondrial fission (Wang et al., 2016). This would be interesting to explore in the future, especially within the functional context of Lam5 and Lam6.
To investigate the functionality of this LAM paralog pair and their HCPPs at ER-mitochondria contacts, we used Seahorse technology to assay bioenergetics. Here, we found that the loss of Lam5 led to increased respiration when the cells were grown in glucose (Figure 2(F)), as opposed to Lam6, which appears to be required for maintaining normal respiration levels independent of the carbon source (Figure 2(E) and (F)). Since LAM HCPPs were identified in glucose-rich media, we assayed respiration in glucose upon deletion of either Aim21 or Vps35, and also found it to be increased (Supplementary Figure 2). Evidence suggests these changes are at least in part conserved, since loss of human VPS35 (Tang et al., 2015) and one of the human Lam5/6 orthologs, GRAMD1C, both lead to changes in oxidative phosphorylation in media containing glucose (Ng et al., 2022).
Interestingly, another Lam5/6 ortholog, GRAMD1B has been proposed to transfer cholesterol between the trans-Golgi network (TGN) and the ER (Naito et al., 2023). This is in line with localization data from yeast (Weill et al., 2018a) and our identification of Ret3, a COPI component, as a Lam5 HCPP (Figures 1(B), (C), and 2(A)). Other GRAMD1 orthologs have been shown to localize to various membrane contact sites, including: ER-PM contacts (in the case of GRAMD1A/B/C (Sandhu et al., 2018; Naito et al., 2019)); ER-lysosome contacts (in the case of GRAMD1B (Höglinger et al., 2019)), and ER-mitochondria contacts (in the case of GRAMD1C (Ng et al., 2022)). The role of the GRAMD1 proteins and Lam5/6 at multiple contacts is particularly intriguing, and, for Lam5, exactly how these different pools are maintained and regulated remains an open question. It does seem, however, that while there is some overlap in some of the locations and functions of the Lam5 and Lam6 paralogs, they each clearly have distinct characteristics. This is corroborated by our data (Figure 1(B) and (C)) and previous work on Lam6 interactors (Elbaz-Alon et al., 2015; Murley et al., 2015), which would indicate they do not interact with each other to form a complex. This is in contrast to the Lam1–4 family members, which appear to be in close proximity to each other (Figures 1(B), (C), and 3(A) (Murley et al., 2017)).
In a similar way to Lam1–4, the three human GRAMD1 proteins also interact with one another (Naito et al., 2019), despite being structurally more similar to Lam5/6 (Gatta et al., 2015). The Lam1–4 family members however, have recently-discovered orthologs in Cryptococcus neoformans (CnYsp2, which is most similar to Lam2 (Choy et al., 2023, 2024)) and in Magnaporthe oryzae (MoVast1/2, which are similar to Lam2 and Lam1/3, respectively (Zhu et al., 2021, 2023)). Amphotericin B hypersensitivity has also been observed in these pathogenic fungi upon disruption of either CnYsp2 or MoVast1 (Zhu et al., 2021; Choy et al., 2023), and also in HeLa cells lacking all three GRAMD1 proteins (Ercan et al., 2021; Naito et al., 2023), indicating evolutionarily conserved functions of this protein family in the regulation of PM sterol homeostasis. Despite this, questions still remain about how exactly these proteins are correctly localized. Encouragingly, we found several HCPPs of TurboID-HA-Lam1–4 to be PM-resident proteins (Figure 3(A)), whose enrichment was greater for Lam2/4, relative to Lam1/3 (Figures 1(B), (C), and 3(A)). This matched our microscopy observations, where Lam2/4 seem to be specifically localized to the peripheral ER, whereas Lam1/3 can also be seen at the perinuclear ER (Figure 3(B)). The different structural domains of Lam1/3, including the Bin-Amphiphysin-Rvs (BAR) domain, may explain these differences, however our proximity mapping did not yield any insights into the perinuclear Lam1/3 subdomain, and this would be of interest to resolve in the future.
The PM-resident Sso1/2 paralogs were particularly notable HCPPs. This is because in addition to being t-SNARE proteins for vesicle fusion, Sso1 and its human ortholog, STX1A, are also involved in non-fusogenic ER-PM tethering together with the ER-resident, Sec22 (yeast)/SEC22b (human) (Petkovic et al., 2014). In cells depleted for both SSO proteins, the localization of Lam1–4 was drastically reduced to a few puncta per cell (Figure 3(C)). Interestingly, the loss of SSO2 alone rendered cells sensitive to Amphotericin B (Supplementary Figure 3(B)), an anti-fungal which acts on PM sterols (Anderson et al., 2014). This is in line with previous observations that the loss of either LAM1, LAM2 or LAM3 negatively affects cell growth on this drug (Gatta et al., 2015).
While our data imply the SSOs are required to correctly localize Lam1–4, further investigation is needed to determine whether Sso1/2 are bona fide tethering partners or whether they affect the locale of these LAM proteins indirectly. Indeed, it was previously suggested that Lam1–4 may form contacts through protein interactions (Tong et al., 2018), in a manner similar to how Lam6 is tethered at contacts (Elbaz-Alon et al., 2015; Murley et al., 2015). This suggestion was corroborated by the fact that LAM GRAM domains expose fewer basic residues compared to similar PH domains (Tong et al., 2018), and would therefore bind to anionic phospholipids less effectively. It is possible, however, that both protein-protein and protein-lipid interactions are required for these ER-PM contacts to form. A role for lipid-based interaction is supported by our observation that perturbing the PI(4,5)P2-generating enzyme, Mss4, also leads to Lam1–4 mislocalization (Figure 4(A)). This raises the possibility that PI(4,5)P2 clustering, which is promoted by the human SSO ortholog STX1A (Honigmann et al., 2013), may help overcome the binding limitations of these GRAM domains.
In line with what would be expected from STX1A orthologs, we observed that PI(4,5)P2 is partially mislocalized in yeast cells where both SSO proteins are depleted (Supplementary Figure 4). While this shows that the SSOs play a role in PI(4,5)P2 distribution, this observation may be, at least in part, due to their capacity as SNARE proteins. This idea is supported by previous work showing that PI(4,5)P2 is synthesized on human exocytic vesicles by PIP5K1C (a human ortholog of the yeast Mss4 kinase) (Maib and Murray, 2022) .The presence of PI(4,5)P2 is what allows exocyst complex binding and subsequent tethering to the PM, prior to SNARE-mediated fusion. Therefore, the internal puncta we see in our SSO mutant cells may represent an accumulation of untethered vesicles. It is attractive to speculate that fusion of these vesicles may contribute to local pools of PI(4,5)P2 on the PM in proximity to the SSOs.
Independent of the exact mechanism of tethering, anionic lipids such as PI(4,5)P2 play an important role, since their presence accelerates sterol transfer for yeast Lam2/4 and human GRAMD1B StART-like domains (Horenkamp et al., 2018; Jentsch et al., 2018). In addition, the GRAM domain of human GRAMD1B was shown to act as a co-incidence detector of anionic lipids and sterols. It was shown that these anionic species enhanced binding of this GRAM domain to liposomes containing cholesterol (Ercan et al., 2021). This aligns with recent work in yeast where the Lam2 GRAM domain was localized to sterol-enriched PM (Alli-Balogun et al., 2024). Furthermore, cholesterol can cluster STX1A (Murray and Tamm, 2009) and therefore it is possible that the orthologous SSO proteins and their associated lipids may create an optimal and complementary environment for Lam1–4 membrane association and lipid transfer.
More generally, by uncovering novel HCPPs for the LAM family we revealed both overlapping and specific properties for its paralogous members. We shed light on the localization and unique function of the relatively uncharacterized protein, Lam5, and discovered the SSO proteins as novel, shared regulators of Lam1–4 ER-PM localization. Future work will tell if the SSO proteins are the direct tethering molecules for Lam1–4, however this discovery highlights the need to explore whether different contact site machineries can cooperate and coordinate with one another. With many different proteins and protein families mediating ER-PM tethering (Manford et al., 2012), we should question if, how, and under which conditions these machineries can work together. There is some evidence to suggest that tethering proteins collaborate in human cells, where GRAMD2 colocalizes with the ER-PM extended-synaptotagmin 2/3 tethers (Besprozvannaya et al., 2018). Lastly, several contact site proteins are known to be mutated in different human disorders. For example, an intellectual disability-linked mutation in GRAMD1B affects a residue conserved to yeast and is known to hinder anionic lipid detection by the protein's PH-like GRAM domain (Ercan et al., 2021). These points emphasize the importance of elucidating the precise molecular mechanisms behind how these proteins work – for understanding both fundamental principles of cellular organization and potential implications for human health.
Materials and Methods
Yeast Strains
The strains used in this work were either picked and verified from libraries, or constructed using the lithium acetate-based transformation protocol (Gietz and Woods, 2002). All strains are listed in Supplementary Table 3, together with references. Plasmids and primers are listed in Supplementary Tables 4 and 5, respectively.
Yeast Growth
Yeast cells were grown on solid media containing 2.2% agar or liquid media. YPD (2% peptone, 1% yeast extract, 2% glucose) was used for cell growth if only antibiotic selections were required, whereas synthetic minimal media (SD/Gal; 0.67% [w/v] yeast nitrogen base (YNB) without amino acids and with ammonium sulfate or 0.17% [w/v] YNB without amino acids and with monosodium glutamate, 2% [w/v] glucose (for SD) or 2% [w/v] galactose (for SGal), supplemented with required amino acid) was used for auxotrophic selection. Antibiotic concentrations were as follows: nourseothricin (NAT, Jena Bioscience) at 0.2 g/l; G418 (Formedium) at 0.5 g/l; and hygromycin (HYG, Formedium) at 0.5 g/l. Yeast grown for transformation or protein extraction was first grown in liquid media with full selections overnight at 30 °C and subsequently back-diluted into YPD/SD media to an OD600 of ∼0.2. Cells were collected after at least one division but before reaching an OD600 of 1 and either immediately used for transformation or snap-frozen for later processing.
For mass spectrometry, strains encoding TurboID-HA-LAM proteins on the background of ABOLISH were grown overnight at 30 °C in SD liquid media supplemented with: an amino acid mix without leucine and histidine; G418; HYG; and 1 mM auxin (Sigma, #13750). The following day, cells were back-diluted to an OD600 of ∼0.15 and collected by centrifugation after ∼5 h (before reaching on OD600 of 1). Media for back-dilution was SD containing a complete amino acid mix, 1 mM auxin and 100 nM biotin (Supelco, #47868), as determined in (Fenech et al., 2023). Cell pellets were washed once in 1 ml LC-MS/MS-grade H2O before being snap-frozen for later processing.
For imaging, the yeast strains were grown overnight in 100 μl SD/SGal media (with appropriate amino acid and antibiotic selections) in round-bottomed 96-well plates at 30 °C with shaking. For the non-SSO experiments, cells grown in SD-based media were back-diluted (5 μl culture into 100 μl fresh media) into SD (with complete amino acids) and grown for ∼4 h at 30 °C with shaking. For the Mss4 experiments, cells were back-diluted with or without 5-Ph-IAA for 7 h, with 2.5 μl and 5 μl culture diluted into 100 μl fresh media for untreated and treated cells, respectively. For the SSO experiments, 5 μl of overnight culture grown in either SD or SGal-based media was back-diluted into 100 μl of the same respective media and grown for another overnight. The following day, the cells were back-diluted (5 μl culture into 100 μl fresh media) into either SD or SGal (with complete amino acids), respectively, and grown for ∼4 h. After the 4 h growth period, cells were processed for imaging (see protocol below).
For the Seahorse assays, cells were initially grown for 24 h on YPD agar plates supplemented with G418 at 30 °C. Cells were then grown in liquid YPD (2% Glucose) or YPGal (1% Galactose) media overnight at 30 °C with slight agitation. After 12 h and on the day of the measurement, cells were back-diluted to an OD600 of 0.5 and grown for an additional 2.5 h at 30 °C with slight agitation, in either YPD or YPGal.
Protein Extraction and SDS-PAGE Analysis
Protein extraction, sample preparation, SDS-PAGE electrophoresis, Western blotting and imaging were performed as described in (Eisenberg-Bord et al., 2021). Briefly, cell pellets were resuspended in 8 M urea-based lysis buffer and subject to glass bead-beating. Lysates were denatured with SDS (final concentration ∼2%) and incubated at 45 °C for 15 min. Denatured lysates were centrifuged to separate cell debris. The resulting supernatants were reduced with sample buffer containing dithiothreitol (DTT) (final concentration ∼25 mM) and incubated at 45 °C for 15 min. Sample was separated on pre-cast 4–20% gradient gels (Bio-Rad) which were transferred onto nitrocellulose membrane using the Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked in SEA BLOCK buffer (Thermo Scientific), incubated overnight at 4 °C with primary antibodies (anti-HA, 1:1000, BioLegend, #901502; anti-myc, 1:3000, Abcam, #ab9106; anti-Histone H3, 1:5000, Abcam, #ab1791), washed, and finally incubated with fluorescent secondary antibodies (goat anti-rabbit IgG 800, 1:10000, Abcam, #ab216773; goat anti-mouse IgG 680, 1:10000, Abcam, #ab216776) for 1 h at room temperature. After washing, the probed membranes were imaged on the LI-COR Odyssey Infrared Scanner.
Affinity Purification and LC-MS/MS Sample Preparation for TurboID-HA-Tagged Protein Samples
Samples were prepared exactly as reported in (Fenech et al., 2023). Briefly, cell pellets were resuspended in 400 μl lysis buffer (150 mM NaCl, 50 mM Tris–HCl pH 8.0, 5% Glycerol, 1% digitonin (Sigma, #D141), 1 mM MgCl2, 1 × protease inhibitors (Merck), benzonase (Sigma, #E1014)) and lysed by 6 × 1 min maximum speed cycles on a FastPrep-24™ cell homogenizer (MP Biomedicals) using 1 mm silica beads (lysing matrix C, MP Biomedicals). Lysates were cleared by centrifugation and subject to affinity purification overnight at 4 °C with streptavidin-conjugated magnetic beads (Cytiva, #28985799). The beads were subsequently washed twice in 2% SDS, twice in 0.1% SDS, and lastly, twice in basic wash buffer (150 mM NaCl, 50 mM Tris–HCl pH 8.0). Elution buffer (2 M urea, 20 mM Tris–HCl pH 8.0, 2 mM DTT and 0.25 μg/μl trypsin/sample) was added to the beads, followed by alkylation, and digestion overnight. The following morning 0.25 μg/μl trypsin was added to each sample and incubated for a further 4 h. Peptides were acidified and desalted using Oasis HLB, μElution format (Waters, Milford, MA, USA). The samples were vacuum dried and stored at −80 °C until further analysis.
LC-MS/MS Settings, Analysis and Raw Data Processing (for TurboID-HA-Tagged Protein Samples)
Settings, analysis and data processing were carried out exactly as described in (Fenech et al., 2023). In short, samples were run on a Q Exactive HF instrument (Thermo Scientific) and data were acquired in data-dependent acquisition (DDA) mode. Raw data were processed using MaxQuant v1.6.6.0 and searched with the Andromeda search engine against the SwissProt S. cerevisiae database (November 2018, 6049 entries). The generated LFQ intensities were used for subsequent analysis on Perseus v1.6.2.3 and Student's t-tests were carried out between appropriate groups to identify significantly enriched proteins. The raw datasets were deposited on the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2021), under the identifier PXD051047. The LAM HCPPs are listed in Supplementary Table 1, together with information on their localization from (Dubreuil et al., 2018).
Generation and Culture of GRAMD1AFLAG Cell Lines
Constructs enabling expression of GRAMD1A with a C-terminal FLAG tag were generated by amplification of gene-specific PCR fragments using HEK293T cDNA. Primers were designed based on the NCBI (National Center for Biotechnology Information) sequence (NM_020895.5). Amplicons and pcDNA5/FRT/TO (Thermo Fisher Scientific) were digested with appropriate enzymes, ligated and the final constructs confirmed by sequencing. Human embryonic kidney (HEK) cell lines that inducibly express GRAMD1AFLAG using the HEK293T-Flp-In™ T-Rex™ system were generated according to manufacturer's instructions (Thermo Fisher Scientific). Single clones were selected and confirmed. Expression of the construct was induced using 1 μg/ml doxycycline (Sigma-Aldrich, D9881) at 14 h prior to harvest. For interactomic experiments, a stable-isotope labeling of amino acids in cell culture (SILAC) approach was taken whereby cells were grown for five passages in DMEM medium lacking arginine and lysine, supplemented with 10% (v/v) dialyzed fetal bovine serum, 600 mg/l proline, 42 mg/l arginine hydrochloride (or 13C6, 15N4-arginine in ‘heavy’ media) (Cambridge Isotope Laboratories) and 146 mg/l lysine hydrochloride (or 13C6, 15N2-lysine in ‘heavy’ media) (Cambridge Isotope Laboratories).
Crosslinking, Immunoprecipitation and LC-MS/MS Sample Preparation of GRAMD1AFLAG
To stabilize any potential GRAMD1A interactions, cells were washed twice in PBS with 0.1 mM CaCl2 and 1 mM MgCl2 (PBS++) and then crosslinked with 0.5 mM ethylene glycol bis(succinimidyl succinate) (EGS) in PBS++ for 30 min at 37 °C. Cells were then washed with PBS++ and the reaction was quenched using 10 mM NH4Cl in PBS++ for 10 min at 37 °C. Cells were harvested, washed in PBS and resuspended in solubilization buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 10% (v/v) glycerol, 1 mM EDTA, 1% (v/v) digitonin, 1 mM PMSF, 1 x protease inhibitors (Roche)) at a protein/buffer ratio of 2 mg/ml. Cells were solubilized for 30 min at 4 °C with mild agitation. Unsolubilized cells were sedimented by centrifugation at 12,000 g for 15 min at 4 °C. Solubilized protein was added to pre-equilibrated anti-FLAG agarose affinity resin (Sigma, A2220) and incubated for 90 min at 4 °C on a rotating wheel. Affinity resins were washed extensively with buffer containing 0.3% (v/v) digitonin. Bound proteins were eluted with 5 μg/ml FLAG peptide (Sigma, F3290) in buffer and subsequently separated on 4–12% NuPAGE Novex Bis-Tris Minigels (Invitrogen). Gels were stained with Coomassie Blue for visualization purposes, and each lane sliced into 21 equidistant slices regardless of staining. After washing, gel slices were reduced with DTT, alkylated with 2-iodoacetamide and digested with Endopeptidase Trypsin (sequencing grade, Promega) overnight. The resulting peptide mixtures were then extracted, dried in a SpeedVac, reconstituted in 2% acetonitrile/0.1% formic acid (v/v) and prepared for nanoLC-MS/MS as described previously (Atanassov and Urlaub, 2013).
LC-MS/MS Settings, Analysis and Raw Data Processing (for GRAMD1AFLAG Samples)
For mass spectrometric analysis, samples were enriched on a self-packed reversed phase-C18 precolumn (0.15 mm ID × 20 mm, Reprosil-Pur 120 C18-AQ, 5 µm, Dr. Maisch) and separated on an analytical reversed phase-C18 column (0.075 mm ID × 200 mm, Reprosil-Pur 120 C18-AQ, 3 µm, Dr. Maisch) using a 30 min linear gradient of 5–35% acetonitrile/0.1% formic acid (v/v) at 300nl/min. The eluent was analysed on a Q Exactive hybrid quadrupole/orbitrap mass spectrometer (ThermoFisher Scientific) equipped with a FlexIon nanoSpray source and operated under Excalibur 2.4 software using a DDA method. Each experimental cycle was of the following form: one full MS scan across the 350–1600 m/z range was acquired at a resolution setting of 70,000 FWHM, an AGC target of 1 × 106 and a maximum fill time of 60 ms. Up to the 12 most abundant peptide precursors of charge states 2 to 5 above a 2 × 104 intensity threshold were then sequentially isolated at 2.0FWHM isolation width, fragmented with nitrogen at a normalized collision energy setting of 25%, and the resulting product ion spectra recorded at a resolution setting of 17,500 FWHM, an AGC target of 2 × 105 and a maximum fill time of 60 ms. Selected precursor m/z values were then excluded for the following 15 s. All gel fractions were acquired with two technical injection replicates.
Raw MS files were processed using MaxQuant (version 1.5.7.4) and MS/MS spectra were searched against the UniProtKB database human reference proteome (February 2017) using default SILAC settings for K8/R10 heavy channels, and the ‘Requantify’ option enabled for improved quantitation. Statistical analysis was performed in Perseus software (version 1.6.15.0); the Significance B test was used to establish significant abundance changes on the protein group level. The raw datasets were deposited on the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2021), under the identifier PXD051014. Significantly enriched/depleted proteins are listed in Supplementary Table 2.
Imaging and Organelle Staining
After back-dilution of strains for imaging (see Yeast Growth methods above), 50 μl of cell culture was transferred to a 384-well glass-bottomed plate (Azenta Life Sciences) coated with Concanavalin A (ConA, Sigma, 0.25 mg/ml) and incubated at RT for ∼20 min. Cells were then washed twice in SD (or SGal for Figure 3(C), (D) and Supplementary Figure 4) supplemented with a complete amino acid mix and imaged in the same media. For the Mss4 experiments, 5-Ph-IAA was included in the wash and imaging media, for the appropriate samples. If MitoView™405 (Biotium, #70070) / FM™4-64 (Invitrogen, #T13320) dyes were being used for mitochondrial/vacuolar staining, then prior to washing, the media was removed and 50 μl of SD supplemented with a complete amino acid mix containing the required dye/s was added and incubated at RT for 15 min. Dye concentrations were 500 nM and 16 μM, respectively.
For Figure 2(C), (D) and Supplementary Figure 4, images were obtained using a VisiScope Confocal Cell Explorer system composed of a Yokogawa spinning disk scanning unit (CSU-W1) coupled with an inverted Olympus IX83 microscope. Single focal plane images were acquired with a 60x oil lens and were captured using a PCO-Edge sCMOS camera, controlled by VisiView software (V3.2.0, Visitron Systems; GFP/Venus at 488 nm and mCherry at 561 nm). Images were transferred to ImageJ (Schindelin et al., 2012) for slight contrast and brightness adjustments. The contrast and brightness settings for the ER-mitochondria contact site (CS) reporter images (Figure 2(C)) and for Aim21-mScarlet images (Figure 2(D)) were set relative to the mCherry-/GFP-Lam5 strains to enable direct comparison with the mCherry-/GFP-Lam6 strains.
For all other microscopy-based figures, images were obtained using an automated inverted fluorescence microscope system (Olympus) containing a spinning disk high-resolution module (Yokogawa CSU-W1 SoRa confocal scanner with double micro lenses and 50 μm pinholes, with a Hamamatsu ORCA-Flash 4.0 camera). Several planes were recorded using a 60x oil lens (magnification 3.2x, NA 1.42). Fluorophores were excited by a laser and images were recorded in three channels: GFP (excitation wavelength 488 nm, emission filter 525/50 nm), mCherry / mScarlet / FM™4-64 (excitation wavelength 561 nm, emission filter 617/73 nm) and MitoView™405 (excitation wavelength 405 nm, emission filter 447/60). Image acquisition was performed using scanR Olympus soft imaging solutions version 3.2. Images were transferred to ImageJ (Schindelin et al., 2012), for slight contrast and brightness adjustments to each individual panel.
Split-Venus Assay
Generation and imaging of strains expressing TEF2pr-mCherry-tagged Lam5 or Lam6 together with an ER-mitochondria CS reporter (Pho88-VC in the ER membrane and Tom70-VN in the outer mitochondrial membrane) was carried out exactly as detailed in (Castro et al., 2022). Briefly, a strain expressing Pho88-VC and Tom70-VN was crossed against strains picked from the TEF2pr-mCherry library (Yofe et al., 2016; Weill et al., 2018b) and then sporulated and selected for haploids containing both traits using automated methods (Tong and Boone, 2006; Cohen and Schuldiner, 2011). The resulting haploid strains were imaged on a Yokogawa spinning disk scanning unit (see under ‘Imaging and organelle staining’).
Image Analysis
For Figure 2(C), three independent images of either control, mCherry-Lam5 or mCherry-Lam6 on the background of the split-Venus CS reporter were used for quantifying colocalization using ImageJ. Using the green reporter channel only, the contrast was auto-adjusted and a crop was taken from the center of the image, measuring 800 × 800 pixels. From this crop, around 100 CS puncta in the green channel were randomly selected and marked (in the absence of the red channel). Colocalization was then measured by performing the ‘coloc2’ analysis function in ImageJ, where the marked CS puncta in the green channel were set as the regions of interest (ROIs). The average thresholded Mander's coefficient (tM1) is reported in the figure legend and this value indicates how much red signal (from control (background)/Lam5/Lam6) colocalizes with the signal of the green CS reporter channel in the ROIs. For Figure 2(D), three independent images of either GFP-Lam5 or GFP-Lam6 together with Aim21-mScarlet were quantified similarly. For these images, the brightfield channel was used as a reference for cropping. Around 300 Aim21 puncta in the red channel were randomly selected and marked (in the absence of the green channel). These were then defined as ROIs when running the ‘coloc2’ function, and the average tM2 reported in the legend indicates how much red Aim21 signal colocalizes with the green Lam5/6 signal in the ROIs.
For Figure 3(C), two independent, confocal microscopy images (equivalent to the enhanced-resolution confocal images shown in the main figure) of either GFP-Lam1, -Lam2, -Lam3 or -Lam4 on the background of either the single or double SSO mutants, all grown in glucose, were used for quantifying the localization change. Using the brightfield images only, 50 cells were selected at random from each image (meaning 100 cells were analysed for each of the eight different strains). These cells were then assessed in the green channel, and the number of cells with clear, discernible, individual puncta were counted. The fold-change reported in the figure legend represents the number of cells with this punctate pattern in the double SSO mutant, relative to the single mutant.
Seahorse Metabolic Analysis
One day prior to measurement, a Seahorse XFe96/XF Pro Cell Culture microplate (Agilent) was coated with 0.1 mg/ml Poly-D-Lysine (Sigma Aldrich) and incubated at 4 °C overnight. Seahorse XF Calibrant (Agilent) was added to the Seahorse XFe96/XF Pro Sensor Cartridge plate (Agilent) and incubated overnight in a non-CO2 incubator at 37 °C. The Seahorse XFe96 Analyzer (Agilent) was set to 30 °C. On the day of measurement, cells were pelleted by centrifugation at 500 g for 5 min to remove growth media, after which they were resuspended in assay medium (0.67% yeast nitrogen base, 2% potassium acetate, and 2% ethanol) as described in (Zhang et al., 2022), to an OD600 of 0.1. 180 μl of cell suspension per well was added to the Seahorse XFe96/XF Pro Cell Culture microplate followed by incubation at 30 °C for 30 min. Measurements were taken under basal conditions (from 0 min) and upon the addition of 40 mM CCCP (at 18 min; Sigma, #C2759), and 2.5 mM Antimycin A (at 36 min; Sigma #A8674) in the Seahorse Analyzer. Three 6 min cycles of mixing (for 3 min) and measuring (for 3 min) time were allotted to each condition. Data analysis was done using Graph Pad Prism (V10.2.0) and the data presented is the average of six independent experiments. Graphs are plotted showing the standard error of the mean (SEM). Statistical significance was determined using two-way ANOVA with Tukey's multiple comparison. P-values ≤0.05, ≤0.01, ≤0.001, or ≤0.0001 are shown as *, **, ***, or ****, respectively. P-values >0.05 are not significant (ns).
Amphotericin B Serial Dilution Growth Assay
Cells were grown overnight in liquid YPD media supplemented with either, NAT, G418 or HYG. The following day, cells were back-diluted in YPD to an OD600 of 0.2 and grown for ∼3 h at 30 °C. Serial dilutions were prepared exactly as described in (Castro et al., 2022) and cells were plated on SD with complete amino acids agar plates with or without 1 μg/ml Amphotericin B (Sigma, #A2932). For Supplementary Figure 1(B), cells were incubated at for three days at 30 °C and then imaged. For Supplementary Figure 3(B), cells were incubated for two days at 37 °C and then imaged.
Supplemental Material
sj-xlsx-1-ctc-10.1177_25152564251321770 - Supplemental material for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function
Supplemental material, sj-xlsx-1-ctc-10.1177_25152564251321770 for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function by Emma J. Fenech, Meital Kupervaser, Angela Boshnakovska, Shani Ravid, Inês Gomes Castro, Yeynit Asraf, Sylvie Callegari, Christof Lenz, Henning Urlaub, Peter Rehling and Maya Schuldiner in Contact
Supplemental Material
sj-xlsx-2-ctc-10.1177_25152564251321770 - Supplemental material for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function
Supplemental material, sj-xlsx-2-ctc-10.1177_25152564251321770 for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function by Emma J. Fenech, Meital Kupervaser, Angela Boshnakovska, Shani Ravid, Inês Gomes Castro, Yeynit Asraf, Sylvie Callegari, Christof Lenz, Henning Urlaub, Peter Rehling and Maya Schuldiner in Contact
Supplemental Material
sj-xlsx-3-ctc-10.1177_25152564251321770 - Supplemental material for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function
Supplemental material, sj-xlsx-3-ctc-10.1177_25152564251321770 for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function by Emma J. Fenech, Meital Kupervaser, Angela Boshnakovska, Shani Ravid, Inês Gomes Castro, Yeynit Asraf, Sylvie Callegari, Christof Lenz, Henning Urlaub, Peter Rehling and Maya Schuldiner in Contact
Supplemental Material
sj-xlsx-4-ctc-10.1177_25152564251321770 - Supplemental material for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function
Supplemental material, sj-xlsx-4-ctc-10.1177_25152564251321770 for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function by Emma J. Fenech, Meital Kupervaser, Angela Boshnakovska, Shani Ravid, Inês Gomes Castro, Yeynit Asraf, Sylvie Callegari, Christof Lenz, Henning Urlaub, Peter Rehling and Maya Schuldiner in Contact
Supplemental Material
sj-xlsx-5-ctc-10.1177_25152564251321770 - Supplemental material for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function
Supplemental material, sj-xlsx-5-ctc-10.1177_25152564251321770 for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function by Emma J. Fenech, Meital Kupervaser, Angela Boshnakovska, Shani Ravid, Inês Gomes Castro, Yeynit Asraf, Sylvie Callegari, Christof Lenz, Henning Urlaub, Peter Rehling and Maya Schuldiner in Contact
Supplemental Material
sj-zip-6-ctc-10.1177_25152564251321770 - Supplemental material for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function
Supplemental material, sj-zip-6-ctc-10.1177_25152564251321770 for Profiling the LAM Family of Contact Site Tethers Provides Insights into Their Regulation and Function by Emma J. Fenech, Meital Kupervaser, Angela Boshnakovska, Shani Ravid, Inês Gomes Castro, Yeynit Asraf, Sylvie Callegari, Christof Lenz, Henning Urlaub, Peter Rehling and Maya Schuldiner in Contact
Footnotes
Acknowledgements
We thank Dr. Ehud Sass, Sivan Arad and Noga Preminger from the Schuldiner lab for critical reading of this manuscript, and Reut Ester Avraham for making all the yeast media. We also thank: Dr Ofir Klein, Rosario Valenti and Noga Preminger for their technical support with experiments; Dr. Yury Bykov and Dr. Katja Hansen for their help and discussions on image analysis; Tanja Gall for her assistance with setting-up Seahorse assays; Corine Katina for helping with LC-MS/MS sample preparation; and Dr. Mike Tyler for his help with plotting heatmaps. GRAMD1A mass spectrometric experiments were supported by the University Medical Center Göttingen (UMG) Core Facility Proteomics. This project was supported by the Deutsche Forschungsgemeinschaft through the SFB1190 (P11, MS; P13, PR; Z02, HU). Emma Fenech was supported by a senior postdoctoral award from the Weizmann Institute of Science. Inês Gomes Castro is a recipient of an EMBO Long-term Fellowship (ALTF-580–2017). Maya Schuldiner is an incumbent of Dr. Gilbert Omenn and Martha Darling Professorial Chair in Molecular Genetics.
Author Contributions
Emma J Fenech conceptualized the project, designed and performed experiments, analyzed data, and wrote the original manuscript draft. Meital Kupervaser ran TurboID-HA-LAM samples and processed the raw LC-MS/MS data. Angela Boshnakovska performed and analyzed Seahorse experiments. Shani Ravid performed microscopy and drop assays. Inês Gomes Castro performed the split-Venus assay. Yeynit Asraf facilitated enhanced-resolution microscopy. Sylvie Callegari performed GRAMD1A-FLAG pulldowns and the LC-MS/MS was performed by Christof Lenz. Henning Urlaub supervised GRAMD1A LC-MS/MS analyses and provided analytical resources. Peter Rehling supervised the project as well as acquired resources and funding. Maya Schuldiner conceptualized and supervised the project, acquired resources and funding, and wrote the original manuscript draft. All authors contributed to reviewing and editing the manuscript.
Data Availability
Yeast TurboID-HA-tagged proximity labeling LC-MS/MS data is available via ProteomeXchange with identifier PXD051047. Human GRAMD1A-FLAG IP-LC-MS/MS data is available via ProteomeXchange with identifier PXD051014.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Deutsche Forschungsgemeinschaft, (grant number SFB1190).
ORCID iDs
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.