
Abstract
In 2020, the pandemic interrupted the series of biannual International Neuroacanthocytosis Meetings that brought together clinicians, scientists, and patient groups to share research into a small group of devastating genetic diseases that combine both acanthocytosis (deformed red blood cells) and neurodegeneration with movement disorders. This Meeting Report describes talks at the 5th VPS13 Forum in January 2022, one of a series of online meetings held to fill the gap. The meeting addressed the basic biology of two key proteins implicated in chorea-acanthocytosis (mutations in VPS13A) and McLeod syndrome (mutations in XK). In a remarkable confluence of ideas, the speakers described different aspects of a single functional unit that comprises of VPS13A and XK proteins working together. Conditions caused by VPS13 (A-D) gene family mutations and related genes, such as XK, previously footnote knowledge, seem to turn central for a novel disease paradigm: bulk lipid transfer disorders.
Keywords
Introduction to the VPS13 Forum
Research into the four members of the VPS13 protein family (VPS13A–D) has intensified in the last 5 years following discoveries from many different approaches revealing them to be conduits for bulk lipid transfer located at sites of membrane contact between distinct organelles (Bean et al., 2018; Dziurdzik & Conibear, 2021; Kumar et al., 2018; Leonzino et al., 2021; Yeshaw et al., 2019).
Community building in this field started in 2002 with an international symposium (Danek, 2003), the first in a biannual series made possible by the Advocacy for Neuroacanthocytosis patients. Meetings continued until 2018 (Peikert & Hermann, 2018), while the 10th symposium had to turn virtual (Masana et al., 2021). Now, the bimonthly, virtual VPS13 Forum (Table 1) attracts from 50 to 150 participants from all continents and maintains communication until the next in-person conference (Homburg, Germany, September 2023). Here, we summarize the talks of the 5th VPS13 Forum and focus on their interconnections, their experimental details being available in full elsewhere (Guillén-Samander et al., 2022; Park et al., 2022; Ryoden et al., 2022).
VPS13 Forum Virtual Meetings Initiated in 2021 by the Global Community of Clinicians, Scientists, and Families Faced With VPS13A or XK Diseases and Related Disorders (Inclusive of January 2023).

Clinical Background
In humans, the four genes of the VPS13 family are linked to inherited diseases mainly of the nervous system. These are chorea-acanthocytosis (VPS13A disease) (Velayos Baeza et al., 2019), Cohen syndrome (VPS13B disease) (Kolehmainen et al., 2003), one form of familial parkinsonism (VPS13C disease) (Lesage et al., 2016; Smolders et al., 2021) and various ataxic conditions (SCAR4/SCA24/SCASI, now VPS13D disease) (Gauthier et al., 2018; Seong et al., 2018).
McLeod syndrome (XK disease) (Jung et al., 2021) shares a variety of features with chorea-acanthocytosis (VPS13A disease), such as degeneration of the basal ganglia and presence of acanthocytes in blood (Peikert et al., 2022). Both diseases have been historically referred to as “neuroacanthocytosis syndromes” (Walker & Danek, 2021). Knowledge on XK has hardly advanced since gene identification (Ho et al., 1994). The molecule first was regarded as a transmembrane protein linked to the Kell protein by a disulfide bond (see Figure 1, left) (Ho et al., 1994) in erythrocytes. In contrast, XK and Kell seem not linked in both brain and skeletal muscle (Clapéron et al., 2007; Jung et al., 2001; Russo et al., 2000) and XK might associate with other proteins in these tissues. Delineation of a family of XK-related proteins (XKr), after discovery of members “XPLAC” and “XTES” (Calenda et al., 2006), initially added no functional insight. XKr8, however, was shown to function as a lipid scramblase by Nagata’s group in 2013 (Suzuki et al., 2013) and its role in apoptotic phosphatidylserine exposure was subsequently identified (Suzuki et al., 2014). Interestingly, reduced levels of phosphatidylserine (PtdSer) subspecies in the inner membrane leaflet of McLeod erythrocytes (with absent XK protein) had been noted in the late 1980s (Redman et al., 1989).

(A) Original model of the XK-Kell protein complex in comparison to the revised model in (B). AlphaFold predicts that two stretches originally regarded as transmembrane (original domains 3 and 8) are intramembrane reentrant loops. This results in inversion of the transmembrane stretches (4 through 7) and relocates the third (extramembrane) loop intracellularly (arrows). As discussed in the VPS13 Forum, this is the predicted site of the VPS13A-XK interaction.
In the clinic, VPSA13 disease and XK disease are often mistaken for each other: because of this similarity some form of interaction between the VPS13A and XK proteins was long suspected (Danek, 2004) and later substantiated by Urata et al. (2019). Proof for the interaction came from Jae-Sook Park in Neiman’s lab (Park & Neiman, 2020) and in 2019 won her the newly established Glenn Irvine prize (see www.naadvocacy.org: NA News Issue 33).
Proceedings of the 5th VPS13 Forum
Pietro De Camilli (Yale University, New Haven, Connecticut, USA) spoke about
De Camilli indeed showed that in a small population of COS7 cells VPS13A is localized not only at contacts between the ER and mitochondria but also at contacts between the ER and the PM. His lab reproduced the observation of the Neiman lab (Park & Neiman, 2020) that XK overexpression leads to relocalization of VPS13A from the mitochondria to the ER (where newly synthesized XK accumulates), but not to the PM, where the bulk of XK resides. Under these conditions, VPS13A co-localized with XK at the ER suggesting that VPS13A competitively binds to XK and mitochondrial membranes. Binding of VPS13A at the C-terminus - to XK or the mitochondrial membrane - requires its PH-domain and ATG2-C region. De Camilli showed that a beta-sheet of the PH domain of VPS13A is predicted to bind to a beta-sheet of the second cytoplasmic loop of XK. This prediction relies on two separate findings from AlphaFold. AlphaFold in 2021 had brought about a revolution in structural biology (Thornton et al., 2021): it reliably predicts three-dimensional protein structures from amino acid sequences. With respect to XK, AlphaFold first led to a revision of XK topology, changing the location of some key conserved sequence from extracellular to cytoplasmic (Figure 1); and second, elements of both VPS13A's PH domain and XK's cytoplasmic loop were recognized to form single extended beta-sheets. Expression of only the PH domain of VPS13A led to its binding to mitochondria, but when co-expressed with XK it completely relocalized both to the ER and PM along with XK. This is in contrast to the behavior of full length VPS13A by itself.
In summary, De Camilli introduced a new site of action of VPS13A: the ER-PM contact site where, under certain conditions, it interacts with the putative scramblase XK. This interaction competes with the tethering function of VPS13A at intracellular membrane contact sites. One possible role of the VPS13A-XK complex might be delivery of lipids from the ER to the PM and their exposure at the cell surface. In this context, De Camilli referred to the Drosophila model of the Sibon lab that had shown Vps13-dependent membranous structures in close association with the PM, possibly representing such ER-PM contact sites, which may be relevant for the removal of nurse cells from drosophila ovaries undergoing degradation in the late stage of oogenesis (Faber et al., 2020). The role of such interaction in neurons and erythrocytes has yet to be studied in detail. However, the exposure of PtdSer at the PM by scramblases has long been implicated in removal of dead cells and in synaptic pruning as an “eat-me” signal (Damisah et al., 2020; Scott-Hewitt et al., 2020). With VPS13A or XK absent, this signal may be affected.
Aaron M Neiman (Stony Brook University, New York, USA) described work from his lab in his talk
With respect to the corresponding XK binding site, Neiman referred to its novel topology (Figure 1) and noted that its second intracellular loop is highly conserved in XK and XKr2, the latter known to interact with VPS13A (Huttlin et al., 2015; Suzuki et al., 2014). He proposed that six conserved residues (EEPYVS, amino acids 97–102) are necessary for interaction with VPS13A by showing that mutation of this sequence disrupted interaction with VPS13.
In conclusion, Neiman emphasized the importance of this region for regulation of competitive partitioning of VPS13A within the cell, along with additional binding sites (VAB domain, FFAT motif, amphipathic helices in the ATG-C domain) and their respective adaptor/partner proteins.
Shikegazu Nagata (Osaka University, Osaka, Japan) dealt with
XK and VPS13A came into the picture as the Nagata lab screened for genes responsible for another mechanism for PtdSer exposure, the P2X7-mediated pathway. P2X7 is a receptor for extracellular ATP and initiates cell lysis upon high extracellular ATP concentrations, such as in an inflammatory or a tumor environment. It is expressed in immune and some tumor cells. Knockout of Xk or Vps13a in mouse T-cell lines resulted in a reduction of ATP-induced PtdSer exposure and subsequent cell death. This indicates a scramblase activity of XK-VPS13A triggered by an as yet unidentified P2X7-dependent signal. As mice deficient in P2X7 do not exhibit a neuronal phenotype, Nagata hypothesized that other systems might be responsible for XK-VPS13A activation in neurons. He concluded from additional experiments that XK recruits VPS13A to the PM where both form a protein complex, which is in line with the data presented by the two other speakers of this session.
Conclusions
The data presented at the 5th VPS13 forum clearly underpin interactions of XK and VPS13A via defined binding regions as well as a bulk lipid transfer and scramblase activity of their complex at sites of contact between endoplasmic reticulum and PM.
The phenotypes of the corresponding diseases (VPS13A and XK disease, respectively) suggest the complex function in membrane lipid homeostasis and survival of erythrocytes as well as central and peripheral neuronal cells, myocytes and spermatocytes, resulting perhaps from deficient PtdSer exposure at the PM. Alternative mechanisms might be at play depending on the exact sites (tissues) and possible additional partners of the XK-VPS13A interaction. We speculate that impaired function of bulk lipid transfer proteins represents a novel disease mechanism shared by various neurodegenerative and neurodevelopmental disorders in addition to the VPS13 proteins. The disease spectrum possibly involves conditions such as the ALS subtype related to VAPB mutations (Borgese et al., 2021), diseases related to anoctamins (Benarroch, 2017; Lenoir et al., 2021), and some of the Neurodegeneration with Brain Iron Accumulation disorders (e.g., BPAN due to WDR45/WIPI4 mutations [Melia & Reinisch, 2022]).
Open questions for future investigation include the following:
XK has been shown to form complexes with two different proteins, with VPS13A (Guillén-Samander et al., 2022; Park et al., 2022; Park & Neiman, 2020; Ryoden et al., 2022; Urata et al., 2019) and with Kell (as part of the 4.1R protein complex) (Ho et al., 1994; Roulis et al., 2018) (Figure 2). Absence of co-expression of XK and Kell in both central nervous system and skeletal muscle (Clapéron et al., 2007; Jung et al., 2001; Russo et al., 2000) suggests tissue-specific roles of XK and of the complexes that XK is part of. Does a “Kell-equivalent” exist for brain or skeletal muscle XK? As XKr8 is in a complex with Basigin, could there be a “Basigin-equivalent” also for XK? Roles of the XK-VPS13A complex in neurodegeneration and for the survival of nerve cells must be elaborated further. Are the neuronal populations with specific vulnerability in XK and VPS13A disease (mainly within the basal ganglia) those most dependent on proper functioning of this complex? Why is XK disease less severe than VPS13A disease? How about compensatory pathways that would explain the delayed consequences of mutations present from birth? Particularly in erythrocytes, the relations of XK-VPS13A and XK-Kell complexes appear enigmatic. VPS13A is clearly present in red cell membranes, as obvious from the routine utilization of the respective Western blot in the clinic (Dobson-Stone et al., 2004; Niemelä et al., 2020). XK is reduced in VPS13A disease erythrocyte membranes (Urata et al., 2019) indeed suggesting the existence of a VPS13A-XK complex in mature erythrocytes. As Kell is an integral membrane protein of erythrocytes, are larger XK-Kell-VPS13A complexes conceivable in these cells? Is there a function for lipid transferring VPS13A in mature red cells, these empty bags devoid of organelles (thus without ER and without ER-PM contact sites), or is its presence just reminiscent of earlier stages of erythropoiesis, that is, progenitor maturation towards red cells? Are the respective interactions, if at all occurring, of these three molecules competitively regulated? Will further (tissue-specific) interaction partners of XK be identified and, if so, will they bind to XK at already known or novel sites? How does VPS13A or XK dysfunction lead to formation of acanthocytes, the bizarre deformation of red cell membranes that appears to derive from leaflet asymmetry, either with the outer leaflet “overgrowing” or the inner leaflet “contracting”? Do complexes with XK stabilize the shapes of normal erythrocytes by distributing specific lipids between the two leaflets? Here, the role of XKr proteins for membrane deformation through phospholipid scrambling seems an important clue (Shiomi et al., 2021). Defects in maturation might also contribute to acanthocyte formation in absence of XK or VPS13A as acanthocytes from VPS13A disease patients and Vps13a-/- mice, in contrast to normal mature erythrocytes, retain double membrane remnants and vesicles (Lupo et al., 2016; Peikert et al., 2021). These remnants might be related to a disturbed degradation process possibly requiring lipid transfer at membrane contact sites. While XK appears to be the scramblase at one end of the VPS13A lipid transfer mechanism, there might be a scramblase also at the channel’s opposite end. We speculate that this molecule (perhaps, according to tissue environment, a variety of them) as well as other putative partners within an extended XK-VPS13A complex may be found implicated in as yet to be identified diseases that share features with the currently known two diseases due to VPS13A and XK mutations, respectively.
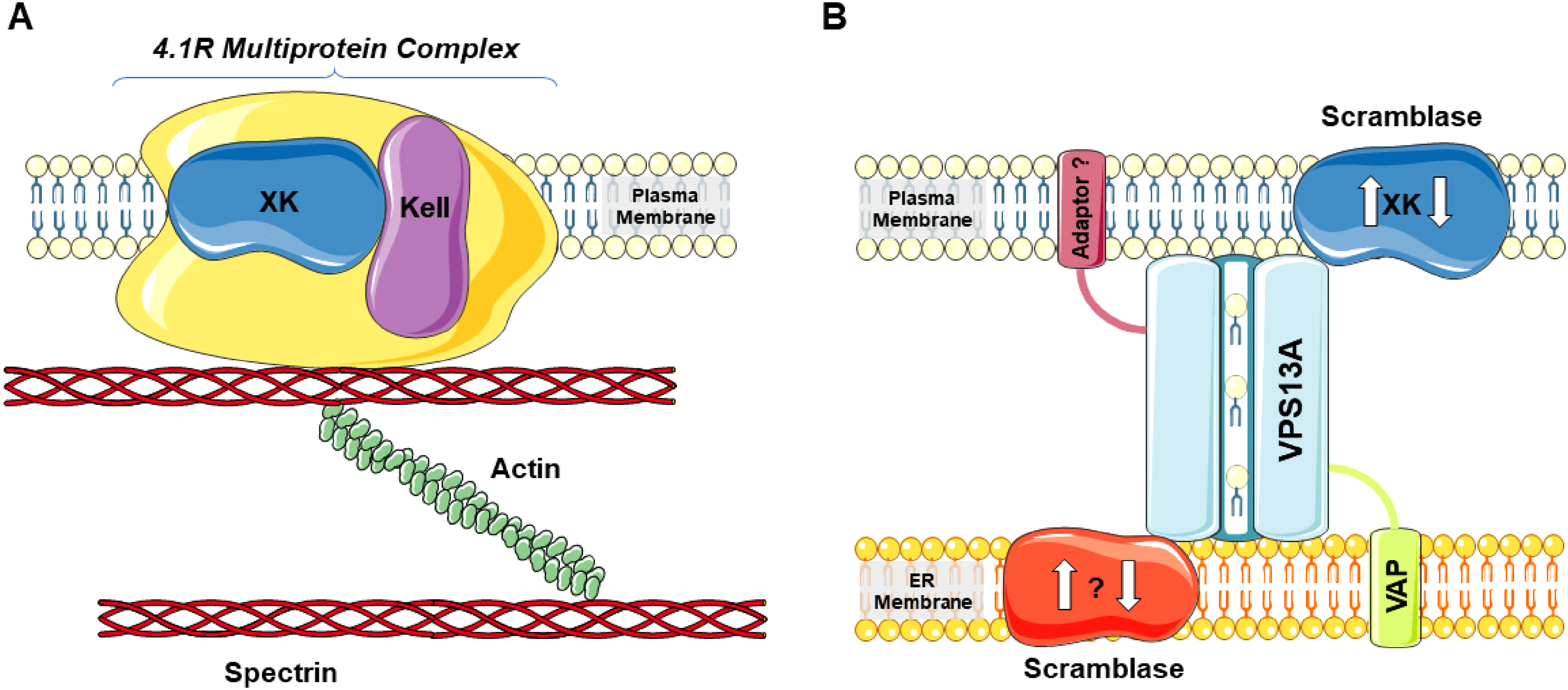
The XK protein in two types of interactions: (A) Together with Kell in the 4.1R protein complex of the red cell membrane (drawn simplistically) and (B) in interaction with the “intermembrane bridge” of VPS13A that tethers the inner leaflet of the plasma membrane (PM) and the endoplasmic reticulum (ER) membrane as a membrane contact site. VAP is the ER-sided adaptor protein of VPS13A, while the opposite adaptor is as yet undefined as is a putative ER-sided scramblase (working in synchrony with XK?). Presence of a PM-sided adaptor protein still is also speculative [This figure contains modified images from Servier Medical Art (https://smart.servier.com) licensed by a Creative Commons Attribution 3.0 Unported License.].
Footnotes
Acknowledgments
We thank all speakers of the VPS13 forums, in particular the three speakers of the 5th forum: Pietro De Camilli (Yale University, New Haven, Connecticut, USA), Aaron M Neiman (Stony Brook University, New York, USA), and Shikegazu Nagata (Osaka University, Osaka, Japan). These three have approved of the report given here. We are grateful to Glenn (†) and Ginger Irvine, founders of the Advocacy for Neuroacanthocytosis Patients (www.naadvocacy.org), and to Susan Wagner and Joy Willard-Williford as representatives of NA Advocacy USA (![]() ). K.P. is supported by the Rostock Academy for Clinician Scientists (RACS) and the FORUN program (University of Rostock, Germany). We thank Tim Levine for his comments on an earlier version of this text.
). K.P. is supported by the Rostock Academy for Clinician Scientists (RACS) and the FORUN program (University of Rostock, Germany). We thank Tim Levine for his comments on an earlier version of this text.
Authors’ Note
For invitations to future VPS13 forum sessions, please contact kevin.peikert@med.uni-rostock.de.
Author Contributions
Conceptualization: AD and KP. Writing of the first draft of the manuscript: AD and KP. Critical review and editing of the manuscript: AD and KP.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.