
Abstract
To maintain cellular homeostasis and to coordinate the proper response to a specific stimulus, information must be integrated throughout the cell in a well-organized network, in which organelles are the crucial nodes and membrane contact sites are the main edges. Membrane contact sites are the cellular subdomains where two or more organelles come into close apposition and interact with each other. Even though many inter-organelle contacts have been identified, most of them are still not fully characterized, therefore their study is an appealing and expanding field of research. Thanks to significant technological progress, many tools are now available or are in rapid development, making it difficult to choose which one is the most suitable for answering a specific biological question. Here we distinguish two different experimental approaches for studying inter-organelle contact sites. The first one aims to morphologically characterize the sites of membrane contact and to identify the molecular players involved, relying mainly on the application of biochemical and electron microscopy (EM)-related methods. The second approach aims to understand the functional importance of a specific contact, focusing on spatio-temporal details. For this purpose, proximity-driven fluorescent probes are the experimental tools of choice, since they allow the monitoring and quantification of membrane contact sites and their dynamics in living cells under different cellular conditions or upon different stimuli. In this review, we focus on these tools with the purpose of highlighting their great versatility and how they can be applied in the study of membrane contacts. We will extensively describe all the different types of proximity-driven fluorescent tools, discussing their benefits and drawbacks, ultimately providing some suggestions to choose and apply the appropriate methods on a case-to-case basis and to obtain the best experimental outcomes.
Introduction
Cellular organelles in eukaryotic cells are membrane-bound compartments characterized by a particular microenvironment where specific biochemical reactions occur. Nowadays, the idea that organelles are not only single units, but part of a fine-tuned network is well established (Scorrano et al., 2019), and represents an intriguing field of research. The correct inter-organelle crosstalk and the ability to adapt this dynamic equilibrium to different stimuli are now acknowledged as essential to coordinate any cellular activity (Phillips & Voeltz, 2016; Prinz et al., 2020). The inter-organelle communication depends on both vesicular trafficking (Bonifacino, 2014) and sites of contact between the membranes of two opposing organelles, the so-called membrane contact sites. These specific cellular subdomains are enriched in specialized proteins, called tethers, able to keep the two organelles in close proximity, in a range from 10 nm to 80–100 nm, without any fusion event between the two involved membranes (Scorrano et al., 2019). Also, proteins playing functional roles and allowing the exchange of metabolites, such as calcium (Rizzuto et al., 1998) or lipids (Muallem et al., 2017; Vance, 1990; Wong et al., 2017) are present. Sites of membrane contact are crucial hubs to convey and integrate information carrying out a specific signaling pathway, for example, apoptosis (Giamogante et al., 2021), organelle remodeling and trafficking (Henne, 2016), or autophagy (Kohler et al., 2020). The principal membrane contact sites are graphically summarized in Figure 1.
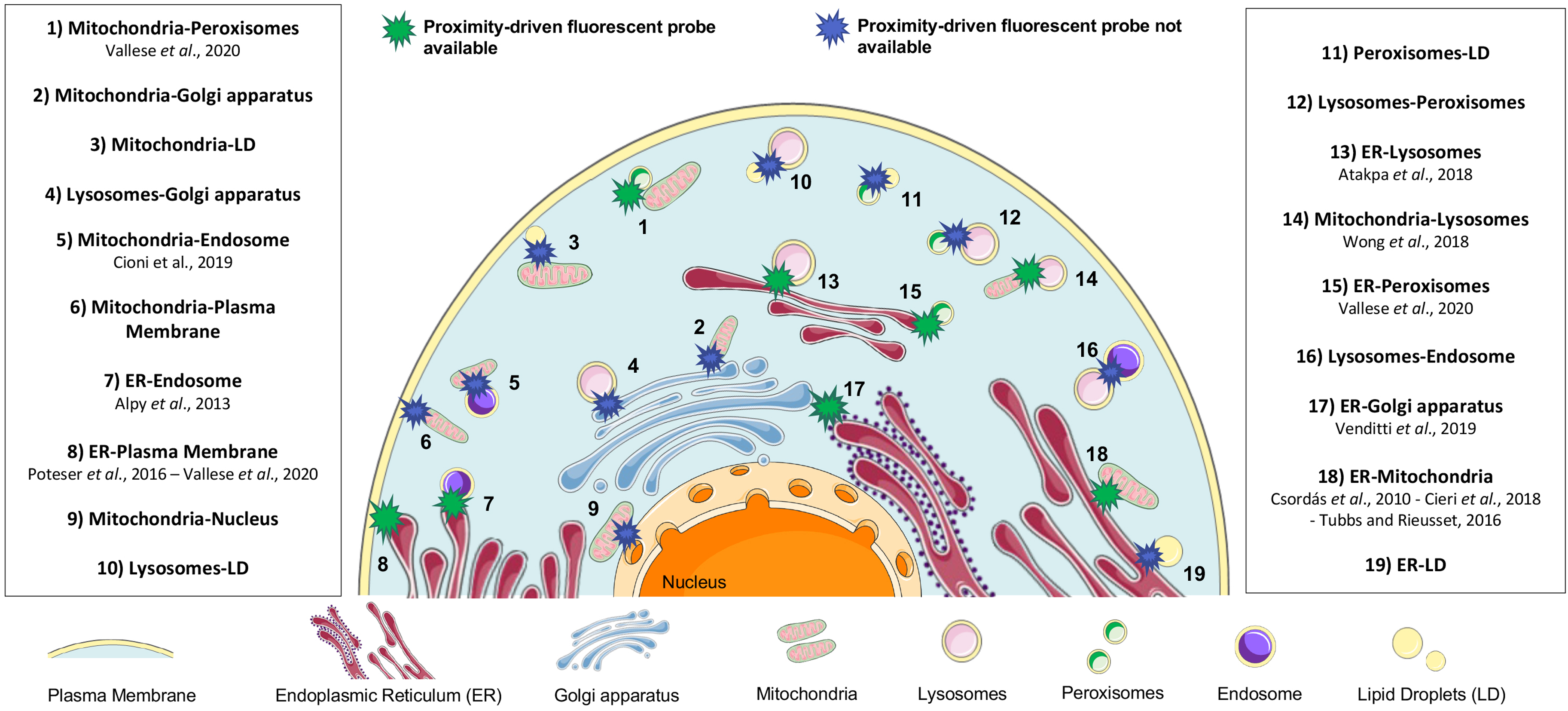
Cellular Landscape of Inter-Organelle Contact Sites. In particular, for those contacts represented with a green star cartoon, a specific proximity-driven fluorescent probe is already available and validated by the reference works cited in the box. Instead, for the contacts represented in the blue star cartoon, no specific proximity-driven fluorescent tools are yet available. Mitochondria-nucleus SPLICS is described in Wu et al., 2022.
The existence of an inter-organelle contact site was first assessed in the 1950s thanks to the observation by EM of juxtapositions between the membranes of the mitochondria and the endoplasmic reticulum (ER), in rat liver cells. The strength of this pioneering observation lay in proposing that the contact between these two organelles is not casual, but due to a precise functional role, even though not yet identified at the time (Bernhard & Rouiller, 1956). Also because of the lack of tools to properly study inter-organelle contact sites, for several years this observation was not fully appreciated. Only 35 years later, the so-called mitochondria-associated membranes of the ER or “MAMs” were isolated and characterized in the course of studies designed to evaluate the import of phospholipids from the ER into mitochondria (Rusinol et al., 1994; Vance, 1990, 2014).
The main challenge in this field is not only being able to visualize precise cellular subdomains, but also to fully understand their functional role. Luckily, nowadays, thanks to a remarkable technical breakthrough, many different experimental methods are available for researchers who want to approach the study of membrane proximity (Huang et al., 2020; Jing et al., 2019). We define two main experimental approaches, each taking advantage of different techniques. The first approach aims to molecularly characterize the architecture of a specific contact site, identifying the proteins involved in the interaction with either a functional or a tethering role. In this regard, biochemical and EM-related methods are the gold standard techniques, and they are briefly discussed in the biochemical methods and microscopy-related techniques sections, respectively. The second approach focuses on the analysis of the overall inter-organelle network from a functional perspective with spatial and temporal details. In this case, fluorescent microscopy coupled with genetically encoded fluorescent proteins targeted to the membrane contact site of interest is the best choice. In this review, we will focus the attention on the experimental methods based on the proximity-driven generation and amplification of a fluorescence signal since, in our opinion, they are very powerful tools. Indeed, not only they are user-friendly and easily accessible, but also consistent and efficient in their functional context. Briefly, two different organelle-targeting sequences or organelle-specific enriched proteins are fused with either fragments or full-length fluorophores with no or different signal on their own. These two moieties reconstitute a strong fluorescent signal only at sites of membrane-membrane juxtaposition where they are allowed to interact. Different experimental methods take advantage of this system: proximity ligation assay (PLA), fluorescence resonance energy transfer (FRET), bimolecular fluorescence complementation (BiFC) and dimerization-dependent fluorescent proteins (ddFP). We thoroughly describe each of them in the proximity-driven fluorescent probes section, also discussing their strengths and weaknesses. Since we believe that the proper application of these proximity-driven fluorescent probes could significantly support the advancement in the study of membrane contact sites, in the last section we highlight some key points to be considered not only while choosing and applying the right tool in a specific model system, but also while interpreting the experimental outcomes.
The Toolkit to Study Membrane Contact Sites
Biochemical Methods
Biochemical methods allow to enrich a protein of interest from cellular extracts and to define its localization or interactions with other molecular partners. We briefly describe a few of them focusing only on those useful to the study of membrane contact sites. In this regard, cell fractionation certainly deserves to be mentioned. It allows to isolate specific subcellular domains or organelles from tissue or cell homogenates, thanks to successive rounds of ultracentrifugation during which the different cell components sediment according to their size and density. For example, the ER-mitochondria contact sites were originally isolated by cell fractionation and sucrose gradient centrifugation and described as an ER “contamination” within the mitochondrial fraction (Lever & Chappell, 1958). Later studies highlighted that these cellular fractions were also enriched in specific enzymes involved in the synthesis and transport of lipids, allowing the first functional characterization of this membrane contact site (Sauner & Levy, 1971). Currently, experimental protocols are not available for all contact sites since the isolation of a membrane contact region is particularly challenging. Several attempts are often needed to find the right cell lysis method and centrifugation workflow in order to obtain a high purification yield. Moreover, it is crucial not only to obtain a cellular fraction not contaminated by other organelles, but also to avoid possible protein modifications or alterations resulting in the destabilization of the contact under investigation. We recommend the protocol optimized by Wieckowski et al. (2009) to study ER-mitochondria contacts, and the one optimized by Grasso et al. (1978) for isolating ER-plasma membrane contact sites. In theory, the isolated fractions retain their original structure and biochemical properties. Thus, they are important substrates for further analysis, as they allow the study of biological processes that would be difficult to investigate otherwise. For example, western blot analyses with specific subcellular markers are usually performed not only to validate which contact site has been isolated, but also to eventually demonstrate the presence or enrichment of an unknown protein. Co-immunoprecipitation experiments are also largely employed to validate the contribution of a novel player in the formation of specific tethering complexes (de Brito & Scorrano, 2008a; Filadi et al., 2016). To explore and identify new contact-localized proteins, affinity chromatography and mass spectrometry can be used, often tagging proteins already known to be involved in that specific contact (Rees & Lilley, 2011; Sala-Vila et al., 2016). Another experimental approach is the proximity enzymatic biotinylation, in which a biotinylating enzyme is targeted to the site of interest. Upon addition of biotin phenol and hydrogen peroxide, all the proteins within a 10–20 nm radius are modified. Then, the rapid isolation of the resulting biotinylated proteins allows their fast identification, for example, via mass spectrometry. To successfully map different membrane contacts (Cho et al., 2017; Hua et al., 2017; Hung et al., 2017), different biotinylating enzymes have been used, such as APEX2, an engineered version of the ascorbate peroxidase (Lam et al., 2015), and BirA*, a nonspecific biotin ligase (Cronan, 2005) in the so-called BioID method (Roux et al., 2012).
Microscopy-Related Techniques
The first critical point in the study of membrane contact sites is how to visualize inter-organelle contacts with sufficient resolution. In this regard, EM is a powerful technique to image and morphologically study subcellular compartments and, therefore, also inter-organelle membrane tethering. To gain high spatial resolution, up to 0.2 nm, samples are illuminated by an electron beam, used as a source of light. In transmission EM (TEM), electrons are transmitted through ultrathin cell slices leading to an accurate 2D visualization of the structures of interest (Csordas et al., 2006). An evolution of TEM is electron tomography (ET), in which the sample is tilted along an axis and multiple images are acquired allowing a 3D reconstruction (Hönscher et al., 2014). It is also possible to obtain 3D reconstructions of cellular subdomains in scanning EM (SEM), collecting the backscattered electrons obtained from a few nanometers below the surface of the irradiated sample. A development of this technique is the focused ion beam-SEM (FIB-SEM), where a highly focused gallium ion beam ablates a thin layer of the sample between one acquisition and the following one (Drobne, 2013). These methods have been successfully applied in the study of membrane contact sites leading to the high-resolution structure of ER-mitochondria contacts by ET (de Brito & Scorrano, 2008a), or to the reconstruction of the ER-plasma membrane (PM) and the ER-mitochondria contacts in mice neurons (Wu et al., 2017). However, EM and all its related techniques have some significant drawbacks: they are time- and cost-consuming with a low through-put efficiency. Moreover, the samples need to be heavily chemically processed during fixation and staining steps, possibly leading to artifacts or alterations of the organelle structure. Also, the main limitation is the impossibility of imaging living cells and, thus, to study the dynamics of inter-organelle contacts. For this latter purpose, fluorescent microscopy is the best choice. Instead of performing immunofluorescence analyses, that also require cell fixation, it is possible to use genetically encoded fluorescent proteins targeted to the organelles of interest. With this experimental approach, the membrane proximity can be detected in living cells and described as the level of colocalization between the fluorescent signals from the two involved organelles. This parameter is quantified as the level of pixel-pixel overlap or correlation with Pearson's and Mander's coefficients (Dunn et al., 2011). However, epifluorescence and confocal microscopy have low spatial resolution (∼200 nm on the x-y axis, and ∼500 nm on the z-axis using a confocal microscope) due to the diffraction limit of the incident light. Total internal reflection fluorescence microscopy (TIRFM) partially overcomes this problem inducing an evanescent wave able to specifically excite fluorophores at the surface of the specimen up to a maximum distance of 100 nm. For example, TIRFM has been successfully applied to study ER-PM proximity and its functional importance in calcium signaling (Wu et al., 2006). Although to better observe sites of membrane contact with a high axial resolution, super-resolution microscopy (SRM) methods are now rapidly evolving, such as structured illumination microscopy (Modi et al., 2019), stimulated emission depletion (Galiani et al., 2016), photoactivation localization microscopy (Sengupta et al., 2014), and stochastic optical reconstruction microscopy (Shim et al., 2012). These methods overcome the spatial resolution limit of conventional microscopy allowing the excitation of only a small subset of fluorophores, that can be precisely localized with computer-fitting algorithms. However, to generate the final image, thousands of frames of a restricted region of the specimen need to be acquired losing temporal resolution. Furthermore, these techniques require expensive equipment, experienced personnel, time-consuming data processing and computing, making them not readily available for researchers that want to easily approach the study of contacts. Over the years, the use of fluorescent proteins targeted to a specific organelle has been overcome by the application of specific probes, such as the PLA-, FRET-, BiFC-, and ddFP-based probes, in which the fluorescence signal is generated and amplified locally, thanks to the membrane proximity within the site of contact. As mentioned above, since these probes are very powerful and versatile, we will focus our attention on them, discussing their properties and application in the following sections.
Proximity-Driven Fluorescent Probes
Proximity Ligation Assay
PLA is primarily employed to detect the interaction between two proteins at a maximal distance of 40 nm. More precisely, the proteins of interest are targeted by two primary antibodies raised in different species, while the PLA probes are two different species-specific secondary antibodies conjugated with single-stranded oligonucleotides. Recently, the applicability of this technique has been increased by conjugating the PLA oligonucleotide strands directly to the primary antibodies. If the two targeted proteins are in proximity, the oligonucleotide strands will react with hybridizing connectors acting as primers for the synthesis of rolling-circle DNA molecules, a process called rolling-circle amplification (RCA) and carried out by a specific DNA polymerase. Then, labeled oligonucleotides hybridizing the amplified DNA are added to visualize a discrete fluorescent signal where the two proteins of interest are in contact. Even though PLA has been mainly used to visualize interacting proteins within macromolecular complexes and their precise localization in situ (Fredriksson et al., 2002), recently this technique has been also employed to identify sites of membrane contact (Benhammouda et al., 2021). It is possible to target the two PLA probes to two tethering proteins of the contact site of interest. Figure 2 depicts schematically the experimental workflow. Tubbs and Rieusset (2016) first visualized and quantified ER-mitochondria contact sites by targeting the voltage-dependent anion channel 1 (VDAC1) at the outer mitochondrial membrane and the inositol 1,4,5-triphosphate receptor (IP3R) localized on the ER membranes. Other examples are summarized in Table 1, indicating which specific proteins were chosen as the target for the PLA probes. One strength of this assay is the possibility to easily quantify the contacts under investigation. Furthermore, PLA does not require any protein overexpression allowing the direct visualization of the endogenous players in situ. However, this method also presents a few drawbacks. First, the primary antibodies must have high affinity and specificity to their targets to prevent nonspecific signals and fluorescence background. The reactivity of the paired antibodies should also be comparable to avoid over- or under-estimation of the contact number. Thus, the obtained results need to be validated by the appropriate negative controls or by the colocalization with the organelles of interest Also, since variations in the number of fluorescent dots revealed by this technique could reflect not only increased/decreased inter-organelle juxtaposition but also the expression/downregulation/post-translational modifications of the targeted proteins, it is important to always check the expression level of the proteins under investigation. In addition, the PLA technique requires the fixation of the sample, that could lead to artifacts and limits its applicability to living cells.

Schematic Representation of a PLA Workflow for Detecting Sites of Contact Between Two Generic Organelles (organelle 1 and 2). Two different tethers or proteins specifically enriched within the organelles of interest (protein red and blue) should be identified. In step 1, the selected proteins are targeted with two primary antibodies raised in different species. Then, species-specific secondary antibodies conjugated with an oligonucleotide strand (PLA probes) bind to the primary antibodies. In step 2, the hybridizing connectors can anneal with the PLA probes, only if the two proteins of interest are in close proximity (distant less than 40 nm). In step 3, thanks to the ligase activity, a closed and circular DNA molecule is formed. In step 4, this DNA molecule acts as a primer for the rolling-circle amplification (RCA) process, mediated by a DNA polymerase. Then, in the last step, fluorescently labeled nucleotides can hybridize to the obtained amplicon allowing the detection of inter-organelle contact sites as discrete puncta with a fluorescent microscope. PLA = proximity ligation assay.
Proteins Chosen as Target for the PLA Probes.

ER = endoplasmic reticulum; PLA = proximity ligation assay.
Fluorescence Resonance Energy Transfer
The FRET process was first theorized by Theodor Förster in the late 1940s. The underlying principle is that, when a fluorescent molecule, called the donor, is excited, the electron energy can be redistributed not only intramolecularly, but also intermolecularly to a different fluorophore, called the acceptor, as long as these two molecules are less than 10 nm distant from each other (Förster, 1948). If the two fluorophores are fused to two proteins of interest, this technique allows detecting their interaction. Only when the two targeted proteins are close to each other and the donor is excited, the non-radiative (or dipole-dipole) transfer of energy results in the excitation of the acceptor molecule and, subsequently, its emission light is detectable. The FRET technique allows monitoring protein–protein interactions with a higher spatial resolution compared to the other standard fluorescence microscopy methods. To better understand why, it is worth mentioning the definition of the efficiency of the energy transfer (E):
However, frequently the FRET event results in a signal of low intensity since the efficiency of the energy transfer is limited to only a few conditions. The donor and the acceptor molecules must be in a precise orientation relative to each other, and the excitation spectrum of the acceptor must overlap with the emission spectrum of the donor molecule. In the last decade, to improve the FRET dynamic range and the application of this system in living cell imaging, many fluorescent molecules have been engineered to increase their brightness, photostability, maturation, and folding processes. For a more comprehensive discussion, we refer to Müller et al. (2013) and Bajar et al. (2016). The most widely used fluorophores are the cyan fluorescent protein (CFP) and the yellow fluorescent protein (YFP). In particular, the improved CFP variant mTurquoise2 (Goedhart et al., 2012), the fluorescent molecule with the highest quantum yield (0.93), has been successfully paired, as donor partner, with mVenus, a YFP derivative, as acceptor, leading to an R0 of 5.8 nm. Moreover, compared to other YFPs, mVenus has better folding and maturation properties at 37°C, it is less sensitive to pH and more photostable (Kremers et al., 2006; Nagai et al., 2002).
Monitoring the development of the FRET signal not only provides information with high spatial resolution but also allows to observe the formation and dissociation of protein complexes in real-time. Thus, all these aspects make very appealing the application of FRET in the study of membrane contacts. A few works summarized in Table 2 have already successfully targeted FRET pairs to inter-organelle contact sites, as schematically shown in Figure 3. As already mentioned, one of the main drawbacks of the FRET system is the low output signal, principally due to high background fluorescence. For this reason, quite often, to strengthen the membrane juxtaposition and increase the signal-to-noise ratio, rapamycin-induced dimerization domains are fused to the donor and acceptor molecules. In this way, the initial increase of the FRET signal, obtained right after the addition of rapamycin, most likely represents the real dynamics of the contact, while the following maximal signal, reached after minutes, is due to a forced expansion of the contact area. Thus, the results obtained with this experimental approach have to be carefully considered and contextualized (Csordas et al., 2010). However, except for genetically encoded artificial tethers (Kornmann et al., 2009; Shai et al., 2018), this is the only available method when it is necessary to experimentally force contacts between organelles (Csordas et al., 2006, 2010). Other factors need to be considered before performing FRET experiments: specific equipment and specialized personnel are required making this technique not often easily accessible in every laboratory.
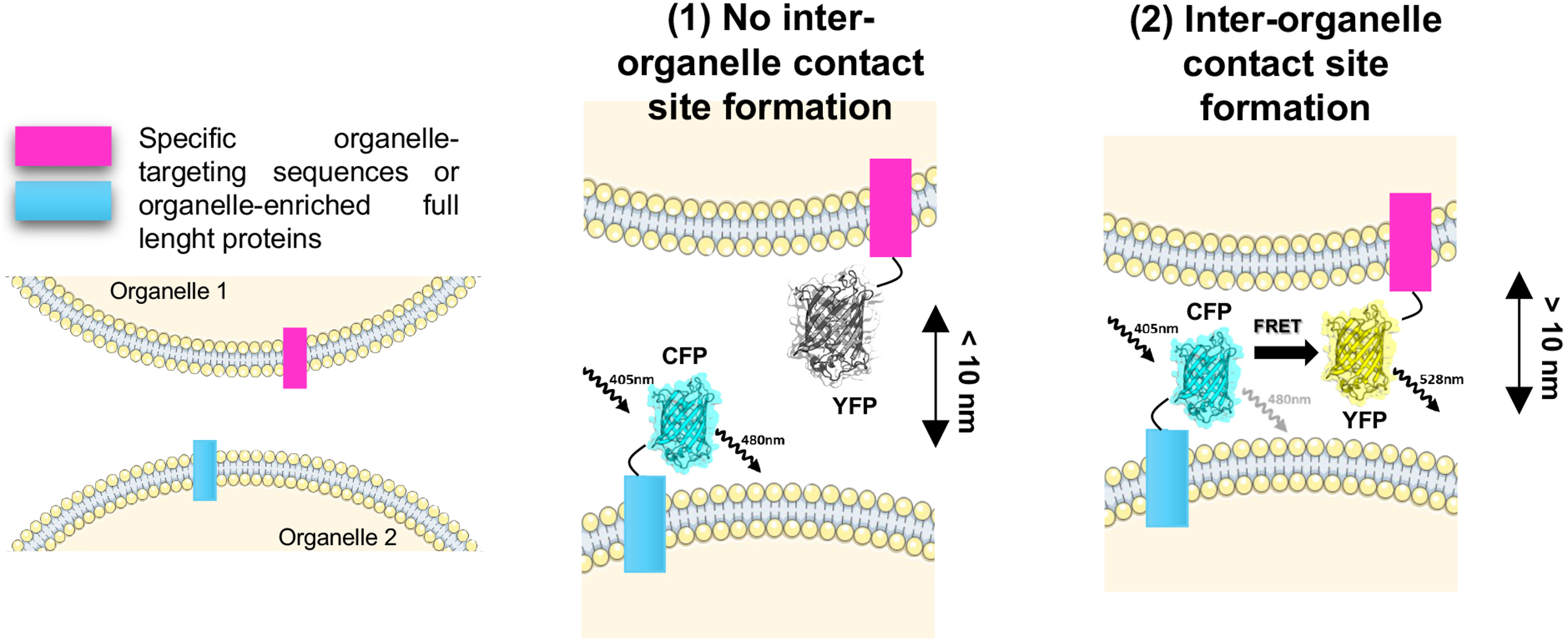
Schematic Representation of a FRET-based Sensor to Monitor the Formation of a Site of Contact Between Two Generic Organelles (organelle 1 and 2). As an exemplificative FRET-pair, we selected the cyan fluorescent protein (CFP) and the yellow fluorescent protein (YFP), respectively, as donor and acceptor molecules. Each fluorophore is fused either to a protein specifically enriched within the organelle of interest or to a specific organelle-targeting sequence (box magenta and light blue). (1) When there is no interaction between the two organelles, the distance between the two fluorophores is more than 10 nm, preventing the fluorescence energy transfer from the CYP to the YFP. Thus, if the donor molecule is excited at 405 nm, the only emission signal detected is at 480 nm, corresponding to the emission wavelength of the donor molecule itself. (2) Instead, when the two organelles come in proximity, the distance between the CFP and the YFP is less than 10 nm. Thus, if the donor molecule is excited, the FRET event can occur resulting in the excitation of the acceptor molecule. Hence, only in this case, the emission wavelength of the YFP can be detected at 528 nm. FRET = fluorescence resonance energy transfer.
FRET Pairs to the Inter-Organelle Contact Site of Interest.

ER = endoplasmic reticulum; FRET = fluorescence resonance energy transfer.
A variant of the FRET technique is based on the bioluminescence resonance energy transfer (BRET) process. The BRET pair consists of an enzyme, specifically a luciferase, as the donor partner and a fluorescent molecule as the acceptor. The luciferase catalyzes the oxidation of its substrate, for example, luciferin or coelenterazine, leading to the emission of bioluminescence. The bioluminescent energy is transferred by resonance and excites the acceptor fluorophore. Thus, in the presence of the luciferase substrate, the acceptor is excited and emits light only if the proteins of interest are interacting with each other (Pfleger & Eidne, 2006). Recently, Hertlein et al. (2020) developed a new BRET-based tool to study ER-mitochondria contacts, called mitochondria-ER length indicator nanosensor (MERLIN). The Renilla luciferase 8 is targeted to the ER thanks to the fusion of a truncated form of calnexin, while the acceptor, consisting of a mVenus molecule, is localized at the outer mitochondrial membrane thanks to the C-terminal domain of Bcl-xL. Compared to the FRET system, the BRET event does not cause phototoxicity in living cells, but the output signal could be even lower, mainly because of the luciferase sensitivity to microenvironmental conditions (such as pH and ATP levels) and problems related to the proper availability of its substrate.
Bimolecular Fluorescence Complementation
BiFC is one of the most widely used techniques to visualize protein-protein interactions in living cells. It is based on splitting a fluorophore into two parts, complementary to each other and not fluorescent on their own. These two moieties, fused to the two proteins of interest, will reconstitute the fluorophore and restore the fluorescent signal only upon interaction. The stability of the system allows the detection of low-affinity complexes and transient interactions at very narrow distances. The first split protein was the green fluorescence protein (GFP), which was split between the amino acidic residues 157 and 158, generating the GFPN and GFPC fragments, respectively. The reassembly of the fluorophore was driven by the association of two antiparallel leucine zipper domains, fused to the C-terminus of GFPN and the N-terminus of GFPC (Ghosh et al., 2000; Magliery et al., 2005). After that, many variants have been developed to improve the BiFC system. The main challenge was to increase the solubility and folding efficiency of the two GFP moieties, without increasing the background signal due to spontaneous assemblies, without affecting the folding properties of the fusion protein partners. By expressing libraries of GFP variants, Pédelacq et al. (2006) identified a particularly stable molecule, thus called super-folder GFP (sfGFP). The sfGFP was split between the residues 214 and 215 into two fragments, further mutated, to obtain the so-called GFP1-10 and GFP11 moieties (Cabantous et al., 2005). This split-GFP (spGFP) system is, nowadays, the most widely used BiFC method. Further evolution was the development of a tripartite spGFP, consisting of three fragments, the GFP1-9 (residues 1–193), the GFP10 (residues 194–212), and the GFP11 (residues 213–233) (Cabantous et al., 2013; Liu, 2021). Now, split-YFP (spYFP) and split-CFP (spCFP) variants are also available thanks to the introduction of different point mutations in the GFP1-10 sequence. These mutations not only change the spectral properties of the fluorophore (Kamiyama et al., 2016) but also improve the stability of the molecule enhancing its self-assembly ability and making more efficient its maturation process (Köker et al., 2018). Efforts to engineer fluorescent molecules and expand the BiFC toolbox are always in progress. We refer to Romei and Boxer (2019) for a complete list of all the up-to-now available split fluorescent proteins.
In the last decade, the BiFC system has been used not only to monitor protein–protein interactions, but also to study sites of membrane contact, as shown in Figure 4. We have generated the first genetically encoded reporter for monitoring ER-mitochondria contact sites (Cieri et al., 2018) and named it split GFP-based contact site (SPLICS) sensor. We have targeted the GFP11 to the ER membranes and the GFP1-10 to the mitochondria, by adding the minimal Sac1 ER targeting sequence (Csordas et al., 2006) and the Tom20 N33 targeting sequence (Giacomello et al., 2010), respectively. Since the non-fluorescent fragments are protruding from the cytosolic side of each organelle, only when membrane juxtaposition occurs at a precise distance, they can come in proximity and re-assemble the original fluorescent protein (Cieri et al., 2018). Thus, only at the site of contact, the fluorescent signal is detectable as discrete puncta. An important advantage of this system is the possibility to identify the spatial range at which the contact site occurs, thanks to the insertion of flexible spacers linked to the GFP11. In particular, two different versions of the SPLICS reporters have been developed: the SPLICS short with a 29 residues-long spacer to specifically detect contacts sites occurring within a range of 8–10 nm, and the SPLICS long with a 146 residues-long spacer, to monitor those contacts occurring at 40–50 nm. Thus, this system overcomes one of the main drawbacks of the BiFC technology in fluorescent and confocal microscopy, that is the low spatial resolution. The two spGFP portions could be fused to different proteins, that could be either already known as involved in a specific contact site or just enriched at the site of interest. For example, to monitor ER–mitochondria interactions, Yang et al. (2018) fused the GFP1-10 moiety to the N-terminus of an ER-resident and highly conserved protein, called UBE2J2, and the GFP11 part to the C-terminus of the first 59 residues of TOM70. Notably, since the overexpression of certain proteins could affect cell homeostasis, we suggest fusing the split fluorescent proteins to a minimal targeting sequence for the organelle of interest, rather than to an entire membrane-anchored protein (Cali & Brini, 2021). The first SPLICS was generated to monitor ER-mitochondria tethering (Cieri et al., 2018), but the family is constantly growing: in addition to a palette of single vector-based SPLICS sensors designed to ensure the equimolar production of the organelle-targeted spGFP, we have also engineered a bivalent SPLICS probe to simultaneously detect and quantify two different inter-organelle interactions (Vallese et al., 2020). All the SPLICS sensors and the other BiFC methods used in mammalian cells to investigate membrane contact sites are summarized in Table 3. BiFC mVenus-based probes are also widely used and have been successfully applied to investigate contacts in Saccharomyces cerevisiae (Lahiri et al., 2014; Shai et al., 2018; Toulmay & Prinz, 2012).

Schematic Representation of a BiFC-Based Sensor to Monitor the Formation of a Site of Contact Between Two Generic Organelles (organelle 1 and 2). As an exemplifying probe, we selected the split-green fluorescent protein (spGFP) system. The two non-fluorescent moieties, in particular, the first ten β-strands of the GFP (GFP1.10) and the eleventh β-strand (GFP11) are each fused either to a protein specifically enriched within the organelle of interest or to a specific organelle-targeting sequence (box green water and violet). (1) When the organelles of interest do not interact with each other, the distance between the GFP1.10 and the GFP11 does not allow the reassembly of the spGFP. Thus, even if the sample is excited at 488 nm, the emission light of the GFP is not detectable. (2) On the contrary, with the formation of an inter-organelle contact site, the distance between the GFP1.10 and the GFP11 decreases allowing their interaction and the reassembly of the fluorophore. Thus, upon excitation, the emission signal of the GFP is detectable at 488 nm. BiFC = bimolecular fluorescence complementation.
SPLICS Sensors and Other BiFC Methods (ddFP) to Investigate Inter-Organelle Contacts Sites.

ER = endoplasmic reticulum; SPLICS = split GFP-based contact site; BiFC = bimolecular fluorescence complementation; ddFP = dimerization-dependent fluorescent protein.
The BiFC system has many points of strength: it is less cost- and time-consuming, easier to perform compared to the other techniques, and results in a straightforward experimental outcome, the dotted fluorescent pattern, that is easy to quantify. However, a few drawbacks must be considered. It is always necessary to validate the chosen targeting sequence to ensure the specificity of the fluorescent signals, often simply performing colocalization analyses with the organelles of interest. This step could be useful to verify also whether the protein overexpression causes alteration in the organelle morphology. Then, the expression of the BiFC probes needs to be carefully titrated not only to avoid mistargeted or aspecific fluorescent signals but also to prevent fluorescent protein aggregation and accumulation, which can result in the forced induction of membrane proximity (e.g., by artificially “zippering” opposing membranes). It should be considered, however, that all methods used to study contact sites have their limitations and thus all results should be validated using more than one method.
Dimerization-Dependent Fluorescent Proteins
The ddFP method consists of two dark fluorescent proteins, weakly fluorescent in their monomeric form, able to interact with each other in a fluorescent heterodimeric complex (Ding et al., 2015; Mitchell et al., 2018). Engineered versions of fluorescent proteins are required to limit their natural oligomerization tendency (Shaner et al., 2004) and to increase their fluorescent signal only upon dimerization (Alford et al., 2012). The ddFPs derive from a homodimeric variant of a red fluorescent protein from Discosoma species (DsRed), but nowadays many ddGFPs and ddYFPs are available. Usually, these molecules have a modest reciprocal binding affinity making this system reversible and, thus, suitable for monitoring protein–protein interactions in living cells with temporal resolution (Tchekanda et al., 2014). ddFP tools have been mainly applied to study protein–protein interactions. However, not surprisingly, they have been successfully used also to monitor sites of membrane contact, as indicated in Figure 5, by targeting the two dark molecules to the different organelles of interest (Table 3). The applicability of this technique is mainly limited because of the intrinsic lower fluorescence of ddFPs compared with other systems including BiFC probes.

Schematic Representation of a ddFP-Based Sensor to Monitor the Formation of a Site of Contact Between Two Generic Organelles (organelle 1 and 2). As exemplifying probes, we selected two dimerization-dependent green fluorescent proteins (ddGFPs). These two proteins are non-fluorescent on their own. Each one is fused either to a protein specifically enriched within the organelle of interest or to a specific organelle-targeting sequence (box yellow and purple). (1) When the organelles of interest do not form any contact site, the distance between the two ddGFPs prevents their dimerization. Thus, even if the sample is excited, the emission signal is not detectable. (2) Instead, if the inter-organelle contact occurs, the two ddGFPs can form a homodimeric complex, that, upon excitation, emits fluorescence light at a wavelength of 508 nm. ddFP = dimerization-dependent fluorescent protein.
Proximity-Driven Fluorescent Tools for Different Purposes: How to Choose
The tools to investigate inter-organelle crosstalk are constantly growing and improving. For those who approach the study of membrane contacts for the first time, the choice of the most suitable method could be tricky. First, we would like to stress that none of the above-mentioned methods should be used as their own. Since each tool is imperfect and has drawbacks, the results obtained with one technique must be validated also by applying another system. However, according to the specific research purpose, it is possible to define a “tool of choice” that should be applied at first to obtain preliminary, yet insightful, results with as little effort as possible in terms of time and costs. In this section, we will discuss how, thanks to their great versatility, we believe that the proximity-driven fluorescent tools perfectly fit this description on several occasions.
Currently, only a few contact sites are entirely unexplored, while for most of them we have already identified at least one tethering complex and/or hypothesized a functional role. Table 4 is intended to group this set of information. In particular, the tethering complexes involved in mitochondria-lysosomes, mitochondria-peroxisomes, and mitochondria-lipid droplet contact sites are still largely unknown. Thus, for example, if the research focus is the structural and morphological characterization of these inter-organelle interfaces, the most suitable tools would be the biochemical and EM-related methods, as discussed in the biochemical methods and microscopy-related techniques section. However, proximity-driven fluorescent methods could be also used as ancillary techniques, after the identification of a specific targeting sequence or a particularly enriched protein for each of the organelles involved. This prerequisite allows, for example, the proper targeting of a spGFP at the sites of contact. Then, it is possible to perform a screening for tethering players amongst a series of candidate proteins by genetically modulating their expression (overexpression or silencing) and evaluating whether the number of contacts varies accordingly. Even though this trial-and-error strategy is time-consuming, it has been proven successful in the past: for example, it led to the identification of the key role of OSBP1, ORP9, and ORP10 in the ER-trans Golgi network interaction (Venditti et al., 2019). Moreover, while applying this strategy, it is necessary to consider that the modulation of only one tether could not directly result in an overall modulation of the contact sites, often due to the presence of other compensatory tethering complexes. Thus, in this specific case, we do not recommend the use of proximity-driven fluorescent tools as the first choice, or at least not exclusively.
Tethering Complexes and/or Hypothesized Functional Roles of Specific Membrane Contact Site.

ER = endoplasmic reticulum.
In contrast, if the research aim is to identify a novel tethering player within an already well-characterized contact site, both the BiFC and the PLA methods could be easily applied. It is possible to modulate the expression (overexpression or silencing) of the candidate protein and to monitor how the number of contacts changes either by applying an already available BiFC probe (see Table 3) or targeting the PLA probes to proteins already selected for that particular inter-organelle interface (see Table 1). For instance, thanks to PLA and coimmunoprecipitation experiments, a novel tethering function at the ER–mitochondria interface has been described for the Parkinson's disease (PD)-related protein DJ1, as part of the already identified IP3R-VDCA1-GRP75 complex (Liu et al., 2019).
On the other side, if the research focus is the functional characterization of a specific site of membrane contact, proximity-driven fluorescent methods are undoubtedly the “tools of choice,” principally because they allow to monitor contact sites in living cells either in physiological conditions or upon specific stimuli. As already mentioned, many BiFC-based probes are now available for several contacts, but it is possible to easily create new ones by selecting a proper targeting sequence and carefully assessing the specificity of the detected fluorescent signal. In this regard, we suggest referring to Cali and Brini (2021) as a guideline to develop other SPLICS sensors.
If the research aims to unravel spatial details of a particular site of contact, FRET-based tools can be a good strategy. Since the data obtained with this system have such a high spatial resolution, it is possible to detect changes in the contacts within a specific organelle subdomain. This level of detail makes it possible to appreciate small, but important membrane dynamics in a functional context, for example, upon a specific stimulus. In particular, the FRET technique was used to better characterize the ER-PM junction, unraveling its molecular nature and role in cellular calcium handling (Zhou et al., 2018). A FRET-based tool was also applied to better define the role of the protein mitofusin 2 (MFN2) at the ER-mitochondria interface (Naon et al., 2016), although its precise role is still under deep investigation because of its pleiotropic functions at different contact sites (Zervopoulos et al., 2022). As described in the fluorescence resonance energy transfer section, many different factors can affect the efficiency of the energy transfer, especially within sites of membrane contact, which are narrow spaces enriched in tethering complexes with significant steric hindrance. Thus, to avoid misinterpretation of the results, our suggestion is to apply a FRET-based strategy only if the laboratory already has high expertise in using these tools. Otherwise, the PLA- and BiFC-based probes are a valid alternative since they allow to directly visualize and quantify membrane contact sites in situ and, for the latter, in living cells. Additionally, with this approach, it is also possible to observe not only whether the number of contacts varies in response to a specific stimulus, but also how contacts are redistributed throughout the cell. For example, considering organelles with a widespread network such as ER and mitochondria, it is possible to study if some specific inter-organelle contacts are preferentially located either in the perinuclear or in the peripheral cell area, defining potential organellar subdomains specialized in different cellular functions (Bravo-Sagua et al., 2016; Cieri et al., 2018). All these interesting aspects are still under investigation, but, in our opinion, the use of BiFC tools could accelerate their understanding. We recommend, while applying this kind of strategy, to monitor not only the redistribution of sites of membrane contact but also the potential morphological changes of the organelles of interest taking advantage of the numerous available in vivo organelle-targeting fluorescent dyes, such as MitoTracker™, LysoTracker™, etc.
If the research aim is studying the dynamics of contact with time-resolved details, suitable tools are mainly lacking. Further studies are needed to fill this technical and experimental gap, especially considering their importance for this field. Up to now, one available technique is the FRET method. It is possible to obtain time-resolved measurements detecting the variations in the donor fluorescence lifetime. During the FRET event, the energy transfer corresponds to a quenching process that ultimately results in the shortening of the donor’s lifetime. The fluorescent lifetime, in the range of nanoseconds (ns), is specific for each fluorophore and can be affected by many factors in the surrounding microenvironment, such as pH and ionic strength. Obviously, fluorescence-lifetime imaging microscopy (FLIM) must be coupled with the use of FRET-based probes to observe in living cells the donor fluorescence lifetime, as performed by Poteser et al. (2016) to study ER-PM contact sites remodeling in response to store-operated calcium-entry. Once again, we suggest to apply these tools with a high expertise.
Regarding the application of BiFC-based tools in studying contact dynamics in real time, the limiting characteristic is the reversibility of the fluorescent probes. Whether a split fluorescent protein may or may not dissociate again after re-complementation is still under discussion. Data in vitro suggest that the re-assembly of the fluorophore is an irreversible process (Magliery et al., 2005). However, a few studies observed in live imaging the formation and dissociation of spGFP-based signals, demonstrating that the re-complementation could be reversible in living cells. For example, Yang and colleagues monitored ER-mitochondria contacts sites, in real-time, with a spinning-disk confocal microscope with a spGFP probe. They observed that approximately 27% of the contacts last less than 60 s from their appearance, while 22% of the contacts lasts between 60 s and 250 s. The remaining 51% of the contacts is stable, lasting more than 250 s (Yang et al., 2018). We also have observed ER–mitochondria contact sites tracking their lifetime and movement speed in Hela cells and mouse hippocampal neurons (Vallese et al., 2020). All these data suggest that the reversibility of a BiFC-based probe depends not only on the affinity between the two moieties of the fluorescent molecule, but also on the affinities between the endogenous tethers and the dynamics of the organelles involved. Further studies are needed to fully clarify this aspect.
Finally, a growing research field is represented by the study of pathological conditions and how they affect membrane proximity, such as in cancer (Gil-Hernandez et al., 2020) or in neurodegenerative diseases (Petkovic et al., 2021). To monitor the overall dynamics of inter-organelle contacts in specific experimental conditions or under the administration of different stimuli, the BiFC tools are undoubtedly the most suitable ones. It is possible to transfect BiFC probes in virtually all types of cells either carrying different pathogenic gene mutations (such as stable cell lines) or transiently expressing mutated proteins associated with specific diseases. For example, we suggested a possible relationship between defective ER-mitochondria communication in model cells for genetic PD by monitoring the effects of the PD-related α-synuclein mutations A53T and A30P on ER-mitochondria proximity with SPLICS sensors (Cali et al., 2019). Moreover, SPLICS sensors have also been used in vivo in Zebrafish (Vallese et al., 2020), opening the possibility of extending their application also in other model organisms to further study the relationship between pathological conditions and the remodeling of contact sites. Dysregulation in ER–mitochondria crosstalk was also demonstrated in NSC34 cells via PLA experiments after silencing of the FUS protein, a well-known condition implicated in the development of familial forms of amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD) (Stoica et al., 2016). In this regard, we would like to highlight that the application of PLA methods is also extended to patient specimens and ex vivo samples.
Another strategy to unravel some pathogenic mechanisms is mimicking different types of cellular stress, such as oxidative or metabolic stress. For instance, by using BiFC-based tools, different groups demonstrated that the number of ER-mitochondria contact sites varies significantly under starvation, apoptosis, and ER stress conditions (Cieri et al., 2018; Vallese et al., 2020; Yang et al., 2018). Since the dysregulation of inter-organelle communication seems to play a crucial role in the onset of pathological conditions, membrane contact sites could also become an interesting and appealing pharmacological target. For example, recently, Coku et al., 2022 demonstrated that reduced ER–mitochondria crosstalk promotes multidrug resistance in neuroblastoma cells. Thus, testing the effect of different chemotherapeutic agents in modulating the contact sites of interest could be insightful. Also in this case, the use of proximity-driven fluorescent probes such as BiFC- or PLA-based methods, could be a key instrument to easily perform drug or target screenings in a very short time.
Conclusions
Studying contact sites and their dynamics is an expanding field of research. Comprehension of the mechanistic details underlying inter-organelle communication could provide new insights into cell homeostasis and metabolism adaptation in response to different stimuli. Also, many recent studies have highlighted that the dysregulation of the inter-organelle crosstalk is a crucial aspect shared in many pathological conditions, such as in cancer and in neurodegenerative diseases. Thus, studying where, when, and how the interaction between organelles occurs is ever-more pressing. In this review, we introduced a wide toolkit to study membrane proximity and mainly focused on proximity-driven fluorescent tools. We consider them not only the most versatile methods but also the tools of choice to apply in this field of research. Principally, this is because FRET- and BiFC-based sensors allow the observation of membrane contact sites in situ and in living cells. With FRET it is possible to resolve organelle interactions up to a few nanometers overcoming the spatial resolution limit of any microscopy technique. Instead, BiFC-based sensors allow to easily quantify contact changes and monitor their rewiring in physiological conditions or in response to different stimuli. Moreover, in this review, we have extensively discussed the advantages and disadvantages of these proximity-driven fluorescent tools, suggesting which one is more suitable for a specific research purpose, as summarized in Figure 6.

Schematic Representation of the Suitability of the Different Experimental Methods (rows) According to the Different Research Purposes (columns). If one tool is highly suitable for one specific research purpose, it is represented with three pink dots meaning that its application leads to more reliable and informative results, compared to other techniques. In particular, we highlighted in green the proximity-driven fluorescent probes, since they are the focus of this review. As discussed in the proximity-driven fluorescent tools for different purposes: how to choose section, for the identification of tethering complexes or molecular players involved in a specific contact site, biochemical methods are the most used techniques, but also some proximity-driven fluorescent probes could be applied. Regarding the morphological characterization of a contact site, all the microscopy-related techniques that allow gaining high spatial resolution compared to light microscopy, such as electron microscopy, are highly recommended. Proximity-driven fluorescent probes can also lead to informative results, but only if applied performing proper staining of the organelles of interest. To study how contact sites change in time, the development of a proper technique is still ongoing. However., currently, high temporal resolution could be obtained thanks to the application of super resolution-microscopy methods and some proximity-driven fluorescent probes. To study how contact sites rearrange in space, all microscopy-related techniques might be useful, but super resolution-microscopy methods and some proximity-driven fluorescent probes are more suitable thanks to the possibility of monitoring living cells. The same techniques are also highly recommended to study pathological conditions, since they could be applied not only in cultured cells, but also in animal models, in particular the proximity-driven fluorescent probes. However, in this specific context, morphological alteration of the contact site structure could be observed also with other microscopy-related and biochemical techniques in clinical samples, for example, tissues from patients. TEM = transmission electron microscopy; SEM = scanning electron microscopy; ET = electron tomography; Cryo-ET = cryogenic electron tomography; FIB-SEM = focused ion beam-scanning electron microscopy; TIRFM = total internal reflection fluorescence microscopy; SRM = super resolution microscopy; PLA = proximity ligation assay; FRET = fluorescence resonance energy transfer; BRET = bioluminescence resonance energy transfer; BiFC = bimolecular fluorescence complementation; ddFP = dimerization-dependent fluorescent protein.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work is supported by grants from the Ministry of University and Research (Bando SIR 2014 no. RBSI14C65Z and PRIN2017 to T.C.) and from the Università degli Studi di Padova (ProgettoGiovani 2012 no. GRIC128SP0 to T.C., Progetto di Ateneo 2016 no. CALI_- SID16_01 to T.C., STARS Consolidator Grant 2019 to T.C. and Local Funds from the University of Padova to M.B.).