
Abstract
The endoplasmic reticulum (ER) is a hub that coordinates neutral lipid synthesis, storage, and export. To fulfill this role, the ER maintains close contact with lipid droplets (LDs), which are evolutionarily conserved organelles for the storage of neutral lipids. Decades of biochemical evidence points to fatty acid modification and neutral lipid synthesis in the ER. Conceptually, lipid export into extracellular space or lipid retention intracellularly require the subsequent remodeling of an ER membrane leaflet that faces the lumen or cytoplasm, respectively. This is because LDs and very-low-density lipoprotein particles are all structures surrounded by a phospholipid monolayer. While the export of neutral lipids via very-low-density lipoprotein production is well characterized, there has been increasing interest in the mechanisms that underlie neutral lipid retention in LDs. Structural determination, in vitro reconstitution, and localization of key proteins by advanced microscopy techniques collectively enrich models of ER-LD engagement. In this review, we consider current concepts on how LDs emerge from the ER in a directional manner and how sustained ER-LD contacts support LD expansion.
The endoplasmic reticulum (ER) is a single continuous membrane system and its morphology ranges from tubules to sheets (Palade, 1955). The original observations have been progressively enriched and reconciled by technical advancements in electron, confocal, and more recently superresolution microscopy techniques (West et al., 2011; Puhka et al., 2012; Terasaki et al., 2013; Nixon-Abell et al., 2016; Schroeder et al., 2019). The complexity of the ER structure supports its many indispensable functions in protein and lipid turnover and signaling cascades. With the increasing appreciation of interorganelle contacts, perhaps it is not surprising that the ER fulfills its functions via physical and functional coupling with the plasma membrane (PM) and other organelles (Gallo et al., 2016; Scorrano et al., 2019). The list of such organelles includes mitochondria, peroxisomes, lysosomes, the Golgi complex, endosomes, and lipid droplets (LDs).
LDs are widely conserved organelles that can be induced and expanded in practically all cells (Walther and Farese, 2012; Henne et al., 2018; Olzmann, and Carvalho, 2019). LDs are responsible for cellular storage and regulated release of neutral lipid species, including triacylglycerol (TAG), cholesterol esters, retinyl esters, wax esters, and acyl-ceramide. The precise content of LDs often marks the cell type and its state and function. For example, LDs in adipocytes are highly enriched in TAG (Yen et al., 2008). The neutral lipid core of LDs is surrounded by a phospholipid monolayer (Tauchi-Sato et al., 2002). Such unique architecture of LDs has long prompted the hypothesis that LDs originate from the ER through budding of the ER outer leaflet (Murphy and Vance, 1999; Brown, 2001; Ohsaki et al., 2017), a process that can be recapitulated in vitro (Chorlay and Thiam, 2018; Chorlay et al., 2019).
In this review, we focus on the functional significance of ER-LD contacts in glycerolipid synthesis and turnover based on studies in yeast and animals. First, we provide a brief overview on the biochemical pathway for glycerolipid synthesis. We will then consider how different biochemical steps are compartmentalized, with an emphasis on ER-LD coupling during different phases of LD biogenesis and expansion. Other important aspects of ER-LD contacts in plants and during viral replication in animal cells have been considered in recent reviews (Herker and Ott, 2012; Pyc et al., 2017; Henne et al., 2018).
Activation of Fatty Acids
Fatty acids (FAs) are essential substrates for inter-/intracellular signaling, protein modification, energy supply, and membrane biogenesis. In most scenarios, in order to be reactive, a FA needs to be converted by an acyl-co enzyme A (CoA) synthetase (ACS) into an acyl-CoA in an ATP-dependent manner (Grevengoed et al., 2014). The acyl-CoA can then be used for TAG synthesis, phospholipid synthesis, protein modification, signaling, or energy production (Figure 1). Despite the necessity of FAs for protein and lipid homeostasis, excess accumulation of cellular free FAs (FFAs) is toxic. In skeletal muscle cells, the overload of FFAs contributes to insulin resistance. In hypertriglyceridemia, elevated level of serum FFAs induces apoptosis of cardiomyocytes and pancreatic β-cells, which might subsequently prompt heart failure and insulin-dependent diabetes, respectively (Schaffer, 2003).

Biosynthesis of triacylglycerol. The glycerol-3-phosphate and monoacylglycerol pathways are depicted. GPAT: glycerol-3-phosphate acyltransferase; AGPAT: 1-acylglycerol-3-phosphate acyltransferase; MGAT: monoacylglycerol acyltransferase; DGAT: diacylglycerol acyltransferase; BSCL2: Berardinelli–Seip congenital lipodystrophy 2; CoA: co enzyme A.
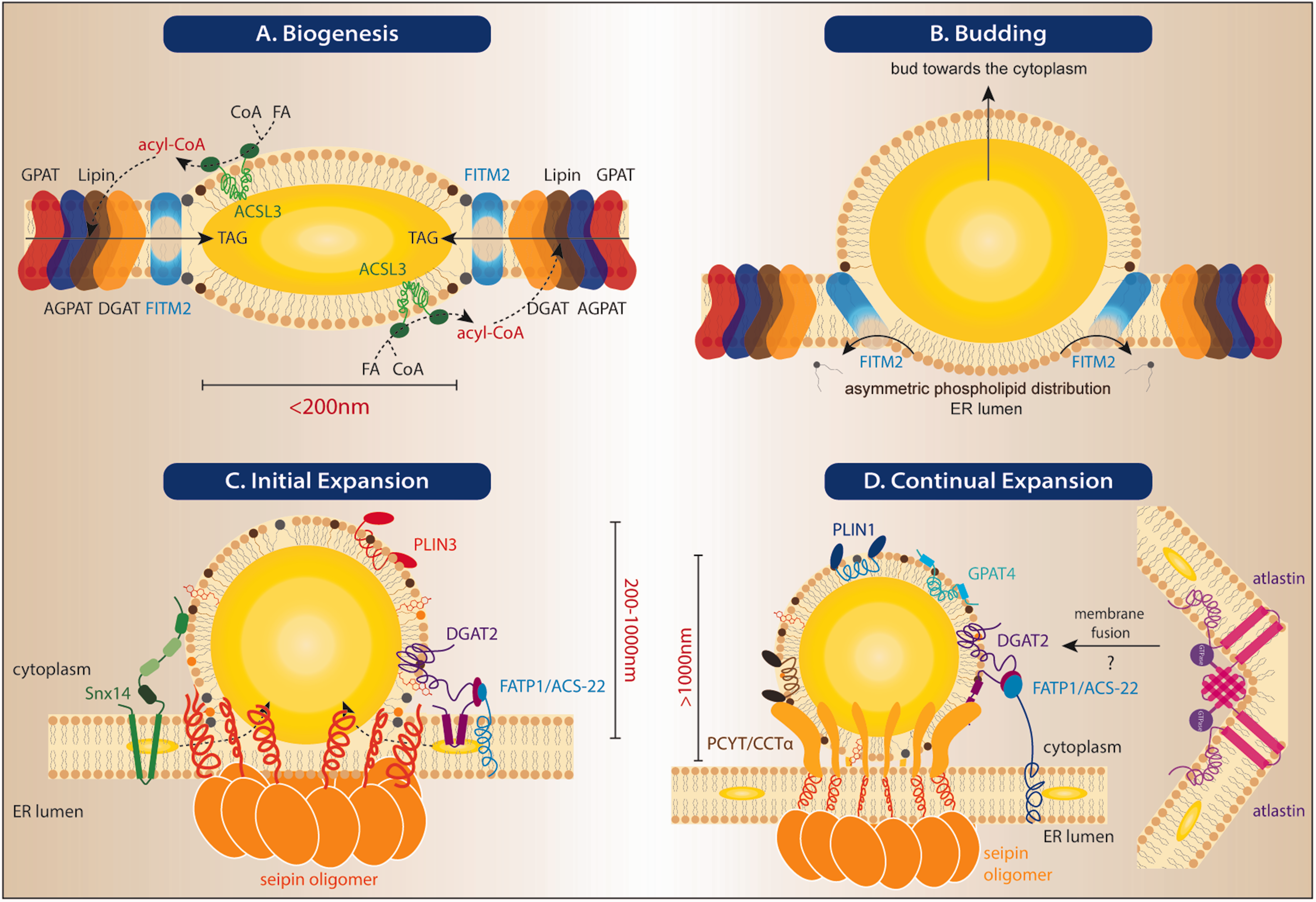
ER-LD contacts at different stages of LD biogenesis and expansion. (a) A nascent LD with a lens-like structure. TAG between the ER leaflets is synthesized by the ER-resident enzymes of the G3P pathway in this depiction. (b) The budding of an LD toward the cytoplasm. The directional budding is putatively organized by FIT orthologs. (c) An initial LD that remains attached to the ER membrane. The loading of lipid and protein cargoes is stabilized by oligomerized seipin orthologs, which may involve neutral lipids channeling from the ER to the preformed LD. Snx14 acts independently of seipin. PLIN3 is recruited to the LD surface at this stage. Implication of a Rab18-SNARE-NAG-RINT1-ZW10 complex is also reported (not shown). (d) Continual LD expansion is facilitated by sustained engagement with the ER through oligomerized seipin orthologs and DGAT2-FATP1/ACS-22 interactions. Atlastins, which mediate membrane fusion of the ER, may be required to maintain ER tubules that are conducive to ER-LD interaction. TAG: triglycerides; GPAT: glycerol-3-phosphate acyltransferase; AGPAT: 1-acylglycerol-3-phosphate acyltransferase; DGAT: diacylglycerol acyltransferase; CoA: co enzyme A; ER: endoplasmic reticulum; FITM: fat storage-inducing transmembrane protein 2; ACS: acyl-co enzyme A synthetase; ACSL: ACS long-chain family member 3; FA: fatty acid; FATP1; fatty acid transporter protein 1; CCT: CTP:phosphocholine cytidylyltransferase; PCYT: phosphate cytidylyltransferase; PLIN3: Perilipin 3.
One effective way to alleviate lipotoxicity is to channel FFAs into TAG synthesis via their acyl-CoA intermediates (Listenberger et al., 2003). It should be noted that the accumulation of acyl-CoAs or TAG precursors are undesirable, as overexpression of long-chain ACSs in the heart induced lipotoxic cardiopathy (Chiu et al., 2001; Chiu et al., 2005). Furthermore, it was reported that the accumulation of di-saturated phosphatidic acid (PA) and diacylglycerol (DAG) following acute palmitic acid overload causes cellular lipotoxicity (Piccolis et al., 2019). It remained puzzling why disaturated glycerolipids could not be further converted to TAG for storage in LDs. Nevertheless, the importance of sequestering FFAs in the form of TAG, rather than its precursors, to defend against cellular lipotoxicity should be apparent.
Glycerolipid Synthesis
The synthesis of TAG relies on either the de novo glycerol-3-phosphate (G3P) or the monoacylglycerol (MAG) pathway (Figure 1; Kennedy, 1957; Lehner and Kuksis, 1996; Coleman and Lee, 2004). In the G3P pathway, G3P is sequentially modified by glycerol-3-phosphate acyltransferases (GPATs), 1-acylglycerol-3-phosphate acyltransferases (AGPATs), phosphatidic acid phosphatases (PAPs, also known as lipins), and diacylglycerol acyltransferases (DGATs). In the MAG pathway, MAG is modified by monoacylglycerol acyltransferases (MGATs) and DGATs. Common to both pathways, fatty acyl-CoA is used in each of the acylation steps. The notion that TAG synthesis occurs in the ER was largely derived from the initial discovery in the 1970s that specific acylation activities could be detected in microsomal membrane fractions (Bell and Coleman, 1980). The molecular characterization of individual enzymes and the increasingly sophisticated methods to ascertain their intracellular localization have enriched our understanding on how the synthesis of TAG in the ER can be coupled with its deposition into LDs.
Glycerol-3-Phosphate Acyltransferase
The conversion from G3P to lysophosphatidic acid by GPATs is often considered as the rate-limiting step for glycerolipid synthesis (Wendel et al., 2009). There are four mammalian GPATs documented to-date. In the 1970s, using density-gradient centrifugation and radioactive tracers, enzymatic activities of GPATs were mapped predominantly to mitochondria and microsomes (Yamashita and Numa, 1972; Monroy et al., 1973). However, it was not until 2006 and 2008 that the microsomal GPAT3 and GPAT4 were firstly identified, respectively (Cao et al., 2006; Chen et al., 2008; Nagle et al., 2008). Depletion of GPAT1, GPAT3, or GPAT4 individually was known to impair TAG synthesis, but none of these isoforms is essential for lipogenesis in mice models or human, likely a result of their functional redundancies (Wendel et al., 2009).
1-Acylglycerol-3-Phosphate Acyltransferase
In contrast to GPATs, specific roles of AGPAT1 and AGPAT2 have been shown in mice and human. Knockout of Agpat1 led to premature death of most mice with reduced body fat (Agarwal et al., 2017). However, it is yet to be documented whether mutations in AGPAT1 associate with human diseases. Mutations in Agpat2 were mapped in human patients with congenital generalized lipodystrophy (CGL; Agarwal et al., 2002), which is an autosomal recessive disorder characterized by the marked reduction of adipose tissues since birth (Patni and Garg, 2015). Similar to obese individuals, CGL patients develop metabolic disorders such as severe insulin resistance, hepatic steatosis, and hypertriglyceridemia, likely a result of adipocyte dysfunction. Curiously, the amount of adipose tissue in new-born Agpat2–/– mice was normal but diminished postnatally as a result of cell death (Cautivo et al., 2016). This observation suggests that one or more of AGPAT2 products are vital for sustaining the structure or function of adipocytes.
Lipin
In mammals, there are three lipin family members, which serve as PA phosphatases (PAPs). Lipin-1, but not lipin-2/3, is essential for lipogenesis in mice (Péterfy et al., 2001; Zhang et al., 2019). The dephosphorylation from PA to DAG was originally identified in 1957 using chicken liver extracts (Smith et al., 1957). Enzymatic activities of PAPs were then mapped to microsomes, mitochondria, cytosol, and lysosomes in cell extracts (Wilgram and Kennedy, 1963; Bell and Coleman, 1980). Compared with others in the G3P pathway, PAP in purified membranes was reported as the least active enzyme (Fallon et al., 1977). It was therefore inferred that PAPs catalyze the rate-limiting step of glycerolipids synthesis (Bell and Coleman, 1980). Nonetheless, instability of PAPs during fractionations might also account for their low activities in vitro (Takeuchi and Reue, 2009).
In yeast, the first phosphatase specific to PA was purified to near homogeneity in 1989 (Lin and Carman, 1989; Carman, 2019). This phosphatase (annotated as Pah1 in yeast) was subsequently cloned and characterized (Han et al., 2006). Loss of Pah1 disrupts lipogenesis but accumulates PA, causing abnormal phospholipid synthesis and membrane expansion (Santos-Rosa et al., 2005; Pascual et al., 2013). The first lipin, lipin-1, was identified in 2001 in fatty liver dystrophic (fld) mice (Péterfy et al., 2001), but the molecular function of lipin-1 was elusive. By comparing the primary sequence and enzymatic activities, the homology between Pah1 and lipin-1 was confirmed (Han et al., 2006). Deficiency of lipin-1 in mice also leads to lipodystrophic disorders, similar to symptoms noted earlier in CGL patients (Péterfy et al., 2001). However, humans with lipin-1 deficiency are not lipodystrophic (Zeharia et al., 2008; Takeuchi and Reue, 2009).
The regulation of Pah1/lipin-1 activities is intriguing. In yeast, Pah1 is primarily inactivated by phosphorylation and activated by dephosphorylation (Carman, 2019). The selective phosphorylation excludes Pah1 from the membrane, impairs the catalytic activities of Pah1, or promotes Pah1 degradation (Carman, 2019). The dephosphorylation of Pah1 by the Nem1-Spo7 membrane complex recruits Pah1 to membranes (Santos-Rosa et al., 2005; Karanasios et al., 2010; Choi et al., 2011). Metazoan orthologs of Nem1 (C-terminal domain nuclear envelope phosphatase 1; Kim et al., 2007) and Spo7 (nuclear envelope phosphatase 1-regulatory subunit 1 or TMEM188; Han et al., 2012) were also identified, but their roles in lipogenesis remain to be fully characterized.
Monoacylglycerol Acyltransferase
MGATs were originally identified based on their sequence homology with DGAT2 (Yen et al., 2002; Cheng et al., 2003; Yen and Farese, 2003). They are primary enzymes for the esterification of MAG into DAG, most notably for the absorption of dietary MAG in intestinal epithelial cells. The DAG product of MGATs can then be fed to DGATs for further esterification into TAG, thus completing the monoacylglycerol pathway. Heterologous expression of MGATs in cultured cells indicated that they are primarily localized to the ER (Yen et al., 2002; Cheng et al., 2003; Yen and Farese, 2003), which enables their functional coupling with DGATs (Jin et al., 2014). In addition, MGAT1 has been reported to localize to LDs (Lee and Kim, 2017). MGAT1 and MGAT2 showed tissue restricted expression in humans and mice (Yen et al., 2002; Yen and Farese, 2003). Interestingly, a third MGAT, MGAT3, is expressed in the intestine of humans but not mice (Cheng et al., 2003; Yue et al., 2011). A link between MGAT1 and hepatic steatosis has been suggested in some but not all mouse models (Soufi et al., 2014; Agarwal et al., 2016; Yu et al., 2016). However, the precise role of MGAT1 under normal and challenged conditions awaits further studies. In contrast, a physiological role of MGAT2 in promoting TAG synthesis in the mouse intestine has been convincingly demonstrated in knockout models (Nelson et al., 2014).
Diacylglycerol Acyltransferase
As the intermediates (PA and DAG) in the G3P pathway are also shared by phospholipid synthesis (the Kennedy pathway or phosphatidylinositide cycle), DGATs thus become key determinants in directing lipid flux toward TAG synthesis (Yen et al., 2008). In addition, the G3P and MAG pathways converge at DGATs. Therefore, the subcellular localization of DGATs dictates the site of TAG accumulation. There are two DGATs in mammals, DGAT1 and DGAT2. The microsomal activity of DGATs in livers was firstly noted in the 1960s (Wilgram and Kennedy, 1963), but it was not until 1998 that the first DGAT (DGAT1) was identified (Cases et al., 1998). Dgat1–/– mice are resistant to diet-induced obesity but viable (Smith et al., 2000). DGAT2 was subsequently cloned in 2001 to account for the residual DGAT activity in Dgat1–/– mice (Cases et al., 2001). Dgat-–/– mutants die soon after birth and are more lipopenic than Dgat1–/– mice in brown adipose tissue and liver (Stone et al., 2004). Human patients with DGAT1 deficiency develop congenital diarrheal disorders but not lipodystrophy (Haas et al., 2012). A missense mutation in DGAT2 was identified in patients with inherited peripheral neuropathy (Hong et al., 2016), but there is currently no clinical evidence that humans devoid of DGAT2 are viable.
There had been attempts to demonstrate the benefit DGAT1/2 inhibition as a strategy to treat hepatic steatosis and dyslipidemia. It is reported that depletion (knockdown or knockout) of DGAT2, but not DGAT1, alleviates diet-induced hepatic steatosis in mice (Choi et al., 2007; Gluchowski et al., 2019). In the obese state, selective inhibition of DGAT2 by small molecules profoundly perturbs the deposition and secretion of hepatic TAG in mice, but the effect is moderate in monkeys (McLaren et al., 2018). Nevertheless, simultaneous inhibition of both DGAT1 and DGAT2 impairs TAG synthesis in both species (McLaren et al., 2018).
Apart from major enzymes in the TAG synthesis pathways, two proteins, with functions less characterized, are also essential for cellular fat storage in mammals. One is fat storage-inducing transmembrane protein 2 (FITM2) and the other one is Berardinelli–Seip congenital lipodystrophy 2 (BSCL2)/seipin.
Regulators of Adipogenesis
Fat Storage-Inducing Transmembrane protein
FITM2 was initially identified as a target gene of peroxisome proliferator-activated receptor-α (PPARα) and PPARγ (a master regulator of adipogenesis; Kadereit et al., 2008). FITM2 is an ER membrane-resident protein, depletion of which impairs the fat accumulation in cultured adipocytes, zebrafish, and mice (Kadereit et al., 2008; Miranda et al., 2014). Loss of FITM2 orthologs in yeast appears to disrupt LD biogenesis (Choudhary et al., 2015). Purified FITM2 in detergent micelles showed high binding affinity to TAG but not to DAG (Gross et al., 2011). Recent reports suggested that FITM2 is a lipid phosphatase (Hayes et al., 2017; Becuwe M, et al., 2018. FIT2 is a lipid phosphate phosphatase crucial for endoplasmic reticulum homeostasis. bioRxiv, 291765). It has been proposed that FITM2 synthesizes lipid mediators that stabilizes the tubular network of the ER, as the ER membrane is prone to aggregation in SUM159 (a breast cancer cell line) cells devoid of FITM2 (Becuwe M, et al., 2018). FIT2 is a lipid phosphate phosphatase crucial for endoplasmic reticulum homeostasis. bioRxiv, 291765). A missense mutation in FITM2 was mapped in patients with deafness-dystonia syndrome (Zazo Seco et al., 2017); however, the connection between FITM2 mutations and lipogenesis dysfunctions in humans awaits further confirmation.
BSCL2/seipin
BSCL2/seipin was firstly identified in human patients with CGL2, which is the most severe form of lipodystrophy (Magré et al., 2001). Seipin plays a highly conserved role in lipid homeostasis in eukaryotes (Cartwright and Goodman, 2012). Similar to CGL2 patients, Bscl2–/– mice also develops lipodystrophic abnormalities (Chen et al., 2009). The defective adipocyte differentiation could be rescued by PPARγ agonist in vitro (Chen et al., 2009, 2012), suggesting seipin may act upstream of PPARγ (Fei et al., 2011a). Knockout of seipin diminishes neutral fat level in fat bodies (the major fat storage tissue in flies) of Drosophila as well, accompanied with the ectopic accumulation of LDs in salivary glands and midgut (Tian et al., 2011). In yeast, a seipin complex (Fld1-Ldb16) is essential for regulating LD abundance at log phase and maintaining the regular LD morphology at stationary phase (Szymanski et al., 2007; Fei et al., 2008; Wang et al., 2014; Grippa et al., 2015). Moreover, it is recently reported that Caenorhabditis elegans seipin mutants accumulate abundant tiny LDs in the intestine (the major fat storage tissue in worms; Cao et al., 2019). Despite the recognition of seipin as a master regulator, the mechanisms by which seipin regulates lipogenesis and alleviates lipotoxicity cell-autonomously or cell-nonautonomously are not fully understood. In addition to its cytoplasmic function, seipin has been reported to operate in the nucleus at the inner nuclear membrane-nuclear LD bridge (Romanauska and Köhler, 2018). An intimate correlation between PA abundance and seipin was documented in yeast, flies, and mice (Fei et al., 2011b; Liu et al., 2014; Han et al., 2015; Wolinski et al., 2015), as seipin may physically interact with lipin-1 and modify its activity (Sim et al., 2012). Alternatively, it is also proposed that seipin physically interacts with GPATs and antagonizes their activity to adjust the adipocyte level of PA (Pagac et al., 2016). Accordingly, partial rescue of adipogenesis in Bscl2–/– mice by GPAT3 deficiency has been reported (Gao et al., 2020).
LDs and the ER: A Long-Lasting Marriage
According to a prevailing theory, newly synthesized TAGs and other neutral lipids accumulate initially between the two leaflets of the ER membrane. Asymmetric budding of the outer ER leaflet toward the cytoplasm then marks a definitive step of LD biogenesis. The subsequent apposition of the ER membrane to LD surface throughout the development of LDs is commonly recognized as the ER-LD contact (Schuldiner and Bohnert, 2017; Wu et al., 2018). The ultrastructural feature of the ER-LD tethering was firstly examined in white adipocytes isolated from rat epididymal fat pad, where a close apposition of an enlarged LD to the ER membrane was noticed (Cushman, 1970). This was confirmed by additional studies by electron microscopy (Novikoff et al., 1980; Blanchette-Mackie et al., 1995; Robenek et al., 2009). More recently, these pioneering studies with fixed samples were elaborated by the use of new fluorescence microscopy techniques on live samples (Valm et al., 2017), thus rapidly enhancing our knowledge on ER-LD contacts.
Based on published work, a four-step model for LD development can be summarized as follows: (a) biogenesis, (b) budding, (c) initial expansion, and (d) continual expansion (Figure 2). Each of these steps requires the translocation and enrichment of specific proteins or complexes at ER-LD contacts. However, additional, perhaps mechanistically distinct, steps for LD development (i.e., LD budding by coat protein complex I (COPI)/Arf1 and LD-LD fusion by Fsp27/Cidec; Gong et al., 2011; Jambunathan et al., 2011; Wilfling et al., 2014), for which ER-LD contacts were dispensable, will not be discussed in this review.
ER-LD Crosstalk During LD Biogenesis
As noted earlier, the biogenesis of LDs initiates at the ER membrane. LDs at this stage are commonly described as nascent, with diameters less than 200 nm (Wang et al., 2016). Indeed, formation of nascent LDs can only be halted by completely blocking neutral lipid synthesis. To study the ER-LD crosstalk at this stage, biogenesis of LDs was stimulated in synchronized (or delipidated) cells lacking any detectable LDs. In yeast, genetic mutants devoid of all neutral lipid synthesis enzymes (i.e., Dga1 and Lro1 synthesize TAGs, Are1 and Are2 synthesize sterol esters) were employed (Sandager et al., 2002; Hayes et al., 2017). The spatial and temporal organization of the ER on LD production could then be examined in this mutant by galactose-inducible expression of Lro1, which was sufficient to induce LD biogenesis (Jacquier et al., 2011). In cultured cells, delipidation was performed by pre-incubation in serum-free medium (Kassan et al., 2013), lipoprotein-free medium (Salo et al., 2016), or lipoprotein-free medium in combination with inhibitors against DGAT1/2 (Salo et al., 2019). The kinetics of nascent LDs was then readily monitored by the addition of oleate. In delipidated mammalian cells, ACS long-chain family member 3 (ACSL3), which is originally distributed in the entire ER, becomes rapidly enriched at nascent LDs upon oleate loading (Kassan et al., 2013). This process was plausibly regulated by seipin and syntaxin 17, as studies noted that loss of either seipin (Salo et al., 2016) or syntaxin 17 (Kimura et al., 2018) perturbed the proper targeting of ACSL3. Until now, ACSL3 appears to be the only ACSL that is enriched at ER-LD contacts (Grevengoed et al., 2014).
ER-LD Crosstalk During LD Budding
LD budding toward the cytoplasm follows the initial accumulation of TAG between the membrane leaflets of the ER. This asymmetric process is putatively regulated by FITM2, as erroneous budding of neutral lipid structures toward the luminal side of the ER was seen in yeast, 3T3-L1 cell lines and C. elegans lacking the FITM2 orthologs (Choudhary et al., 2015). Follow-up studies suggest DAGs, marked by peptide sensors, are dispersed in wild-type yeast but partially enriched at ER-LD contacts in yeast lacking FITM2 orthologs (Choudhary et al., 2018). It was therefore inferred that DAG consumption by FITM2 at ER-nascent LD contacts potentiated LD budding toward a legitimate direction (Choudhary et al., 2018). Attempts have been made to link the recently proposed phosphatase activity of FITM2 with LD budding (Hayes et al., 2017; Becuwe M, Bond LM, Mejhert N, Boland S, Elliott SD, Cicconet M, Liu XN, Graham MM, Walther TC, Farese RV (2018). FIT2 is a lipid phosphate phosphatase crucial for endoplasmic reticulum homeostasis. bioRxiv, 291765). A recent in vitro analysis suggests that the asymmetry in membrane surface tension contributed to the directional budding of LDs from the phospholipid monolayer with less tension (Chorlay and Thiam, 2018). The reduction of surface tension could be achieved by the insertion of proteins or lipids (Chorlay et al., 2019). It was speculated that rather than consuming DAG, FITM2 dephosphorylates phospholipids into DAG specifically at the luminal side of the ER. The reduction of phospholipids at the luminal leaflet reduces the relative tension of the cytoplasmic leaflet, thereby promoting LD budding toward the cytoplasm (Chorlay et al., 2019).
ER-LD Crosstalk During the Initial Expansion of LDs
Once the nascent morphology is determined, LDs are committed to the initial expansion while maintaining membrane continuity with the ER. This step is supported by the establishment of additional ER-LD contacts. Seipin is considered critical in maintaining such contacts during LD expansion (Salo et al., 2016), possibly by stabilizing the peculiar membrane curvature at the neck region of the ER-LD junction (Grippa et al., 2015). By cryo-electron microscopy, structures of Drosophila and human seipin were determined, and both studies suggested the luminal loop region of seipin forms oligomers with ∼60Å central tunnels (Sui et al., 2018; Yan et al., 2018). The homo-oligomers were hypothesized to stabilize ER-LD contacts and assist the channeling of TAG or PA. A predicted hydrophobic helix in the luminal loop of seipin exhibited higher affinity to monolayered membranes than bilayered ones (Sui et al., 2018). In Drosophila and mammalian-cultured cells, seipin promotes the initial maturation of LDs (300–500 nm diameter; Wang et al., 2016), which is dependent on its dynamic association with TMEM159/Promethin/LD assembly factor 1 (LDAF1; Castro et al., 2019; Chung et al., 2019). LDAF1 is a putative ortholog of yeast Ldo proteins that act with seipin (Eisenberg-Bord et al., 2018; Teixeira et al., 2018). At basal level, seipin appeared to bind LDAF1 via its hydrophobic helix. Upon FAs loading, TAG accumulating between the two leaflets of the ER membrane preferentially binds seipin and displaces LDAF1 to the LD surface (Chung et al., 2019). It is unclear how LDAF1 assists the contact between seipin and LDs. Curiously, the loss of LDAF1 increases the expression level of seipin but impaired the contacts between seipin and LDs, resulting in excess seipin foci in the bulk ER (Chung et al., 2019). Further data suggest that seipin and LDAF1 participate at additional distinct steps for LD homeostasis. For example, depletion of LDAF1 delayed the formation of LDs and mildly diminished their abundance (Chung et al., 2019). However, LDAF1 deficiency did not alter LD morphology, unlike seipin deficiency (Sui et al., 2018). This might be due to the heterogeneous composition of seipin puncta as not all of them were marked by LDAF1 (Chung et al., 2019).
It is noteworthy that seipin is not essential for the expansion of all LDs as supersized LDs are present in yeast, cultured mammalian, and Drosophila cells and primary human fibroblasts that are devoid of seipin orthologs (Fei et al., 2008; Salo et al., 2016; Wang et al., 2016). Depending on the model system, aberrant expansion of LDs in response to the loss of seipin may be dependent on GPAT3 (Pagac et al., 2016) or GPAT4/AGPAT3/DGAT2 (Wang et al., 2016). In line with this, recent studies in C. elegans indicated that seipin only associates with a subset of LDs and facilitates their expansion (Cao et al., 2019). The loss of the C. elegans seipin ortholog causes an accumulation of nascent LDs but does not necessarily affect the proper expansion of all LDs (Cao et al., 2019).
In hepatoma cells, ACSL3 promotes initial LD expansion (Fujimoto et al., 2007), in addition to its role in LD biogenesis. In 3T3-L1 preadipocytes, initial expansions of LDs also require the ER-LD tethering established by LD-resident Rab18 and the ER-resident SNARE/NAG-RINT1-ZW10 complex (Xu et al., 2018), a process shown to be regulated by DFCP1 (also known as ZFYVE1; Li et al., 2019). In SUM159 cells, depletion of Rab18 did not evidently affect the morphology of stimulated LDs (Jayson et al., 2018). Nevertheless, recent evidence suggested that endogenous Rab18 preferentially engages ER-LD contacts and supports the initial LD expansion, in a compensatory response to seipin deficiency (Salo et al., 2019). It would be of interests to investigate whether the disparate observations on Rab18 reflect cell-type specific functions.
Mammalian Snx14 is a sorting nexin orthologous to yeast Mdm1 (regulator of LD biogenesis at junctions between the nuclear ER and vacuole under nutritional stress; Hariri et al., 2017). It has recently been reported that loss of Snx14 in U2OS osteosarcoma cells attenuates the expansion of most, if not all, nascent LDs (Datta et al., 2019). In seipin knockout cells, Snx14 can still be recruited to ER-LD contacts. Furthermore, overexpression of Snx14 fails to correct the aberrant LD morphology that stems from seipin deficiency, suggesting Snx14 and seipin act independently (Datta et al., 2019). It will be interesting to establish whether Snx14 and seipin regulate distinct populations of LDs. If so, depletion of both Snx14 and seipin may confer synthetic phenotypes.
A number of proteins, such as atlastins, reticulons, and spastin, serve as key regulators of ER morphology (Hu et al., 2011). Given the importance of ER-LD contacts in promoting the initial expansion of LDs, depletion of these proteins unsurprisingly perturbs LD number or morphology (Park et al., 2010; Klemm et al., 2013; Falk et al., 2014; Papadopoulos et al., 2015). Atlastins (named as ATLNs in C. elegans) are required for maintaining the proper tubular ER network, primarily by mediating either fusion or tethering of the ER membrane (Hu et al., 2009; Niu et al., 2019). Atlastins are dispersed in the general ER when expressed at physiological levels (Klemm et al., 2013). In C. elegans, ATLN-1 is critical for the initial expansion of LDs but not essential for their biogenesis (Klemm et al., 2013). Intriguingly, a mutant form of ATLN-1 capable of sustaining proper ER morphology but defective in promoting LD expansion was noted (Klemm et al., 2013). Therefore, it is plausible that the regulatory function of ATLN-1 on the ER and LDs can be uncoupled.
ER-LD Crosstalk During the Continual Expansion of LDs
Following their initial expansion, LDs continue to grow in close proximity to the ER. However, their membranes are not necessarily contiguous. The surface identity of mature LDs is mostly distinctive from the ER, as demonstrated by the preferential enrichment of perilipins (Wolins et al., 2005). In Drosophila S2 cells, GPAT4, AGPAT3, and DGAT2 appear to move from the bulk ER to ER-LD contacts to support continual LD expansion upon lipid loading (Wilfling et al., 2013). Notably, GPAT4 and DGAT2 translocate through ER-LD contacts and eventually support LD expansion locally on the LD surface. This step is likely regulated by Arf1/COPI (Wilfling et al., 2014), but it remains unclear why the COPI machinery (primarily formed at the cis-Golgi membrane and ER-Golgi intermediate compartment) is involved in the context of ER-LD contacts. One possibility is that ER-Golgi intermediate compartment-LD contacts serve as sites for continual LD expansion.
In C. elegans, blocking the peroxisomal β-oxidation pathway induces giant LDs in intestinal cells (Zhang et al., 2010). The aberrant expansion of LDs is likely an adaptive mechanism for sequestering toxic long-chain FAs, which cannot be efficiently catabolized when peroxisomal function is compromised. This process requires ER-LD contacts bridged by the C. elegans orthologs of DGAT2 (annotated as DGAT-2) and FA transporter protein 1 (which belongs to the ACS family, and annotated as ACS-22 in C. elegans; Xu et al., 2012). In the differentiated intestinal cells of wild-type C. elegans, red fluorescent mRuby::DGAT-2 fusion proteins expressed from single-copy transgenes are found predominantly on the LD surface (Xu et al., 2012). Enrichment of DGAT-2 on LDs in C. elegans also holds true when a green fluorescent GFP::DGAT-2 fusion protein is expressed from the endogenous dgat-2 locus (Mak lab, unpublished data). In contrast, ACS-22/FA transporter protein 1 is primarily dispersed in the general ER but profoundly enriched with DGAT-2 at ER-LD contacts upon the need of continual LD expansion (Xu et al., 2012).
Seipin promotes the continual expansion of LDs as well. Although seipin-positive puncta have been widely observed in mitotic yeast and tissue-cultured cells, GFP fusion proteins of both the worm seipin ortholog (named SEIP-1) and human seipin localize to yarn ball-like peri-LD cages surrounding only a subset of LDs in differentiated C. elegans intestinal cells (Cao et al., 2019). The enrichment of seipin to peri-LD cages, which are ER subdomains that tightly associate with LDs, promotes continual LD expansion. Interestingly, the enrichment of seipin is dependent on polyunsaturated gamma-linoleic acid (C18:3n6) and microbial cyclopropane FAs (cyclo-FAs, including C17:0Δ and C19:0Δ; Cao et al., 2019). How gamma-linoleic acids and cyclo-FAs recruit seipin to peri-LD cages remains unclear. However, this set of results hint at a possible mechanism that coordinates organelle remodeling with lipid metabolism in response to elevated FFAs. In models other than C. elegans, seipin oligomers mostly appear as puncta at ER-LD contacts (Szymanski et al., 2007; Salo et al., 2016; Wang et al., 2016). It is likely that peri-LD cages are mature forms of seipin-containing ER-LD contacts. The discrepancy may be explained by the different FA composition between C. elegans and other cellular models. For example, C. elegans is capable of synthesizing polyunsaturated FAs from scratch and ingesting cyclo-FAs from its Escherichia coli diet (Figure 3; Watts and Browse, 2002; Watts, 2009). Polyunsaturated FAs and cyclo-FAs are therefore the two most abundant FA species present in TAGs and phospholipids of C. elegans (Watts and Browse, 2002; Cao et al., 2019). Polyunsaturated and cyclopropanated acyl chains in combination with varied phospholipid headgroups likely enhance the diversity of membrane curvatures, thereby raising the possibility of engaging a wider spectrum of integral membrane proteins.

The source and composition of cellular FAs in Caenorhabditis elegans. Cyclopropane FAs (cyclo-FAs) are exclusively converted in Escherichia coli from monounsaturated FAs by the CFA. C. elegans absorbs cyclo-FAs in the intestine after ingesting E. coli. PUFAs are synthesized endogenously in C. elegans from saturated FA substrates. Cyclo-FAs and PUFAs are the most abundant FA species in C. elegans raised in the laboratory. CFA: cyclopropane-fatty-acyl-phospholipid synthase. FA: fatty acid; PUFA: polyunsaturated FA; FAT: fatty acid transporter.
ER-LD Contacts Get Company
ER-LD contacts can also be observed at the periphery of a cell. A Snx14 ortholog in Drosophila, Snazarus, is recently reported to bridge ER-LD contacts with the PM in fat bodies (Ugrankar, 2019). During the maturation of small peripheral LDs (pLDs, as opposed to large cytosolic medial LDs, mLDs) in fat bodies, Snazarus, like Snx14, is anchored to the ER and interacts with LDs with its C-terminal nexin domain (Datta et al., 2019; Ugrankar, 2019). Curiously, a middle phosphoinositide-binding domain specifically tethers Snazarus to the PM as well, resulting in tricontacts between LDs, the ER, and PM (Ugrankar, 2019). The significance of these Snazarus-regulated peripheral LDs remains unclear. Nevertheless, it is plausible that a subpopulation of LDs participates in the rapid accommodation of newly absorbed FAs. Alternatively, the pLDs in fly fat bodies may be primed for FA mobilization in support of adipocyte signaling.
Similar tricontacts was also seen in lactating mammary gland epithelial cells that actively export cytosolic LDs during milk production (Masedunskas et al., 2017). In this case, the establishment of LD-PM contacts requires the interaction between butyrophilin, which is anchored to the PM, and adipophilin/PLIN2, which coats the LD surface (McManaman et al., 2007). This interaction is further stabilized by the recruitment of cytosolic xanthine oxidoreductase (McManaman et al., 2007). More recently, CideA and seipin were also reported to control lactation, possibly by regulating the maturation and expansion of LDs in the mammary gland (Wang et al., 2012; Monks et al., 2016; El Zowalaty et al., 2018). The precise molecular steps at which CideA and seipin support LD export at multiorganelle junctions will benefit from dynamic superresolution microscopy.
Electron microscopy studies have long revealed contacts between the ER, LDs, and mitochondria in adipocytes (Novikoff et al., 1980; Blanchette-Mackie et al., 1995). The molecular basis for the maintenance of such contacts appears to be distinct in white and brown adipocytes. Normal function of brown adipocytes requires mitochondria–LD contacts, maintained by Perilipin 5 and mitofusin 2 (Boutant et al., 2017; Benador et al., 2018). In contrast, the outer mitochondrial membrane protein mitoguardin 2 has been reported to use separate domains to facilitate LD–mitochondria and ER–mitochondria contacts in white adipocytes (Freyre et al., 2019). Loss of mitoguardin 2 attenuates TAG synthesis and adipocyte differentiation, suggesting the functional importance of such tri-organelle contacts (Freyre et al., 2019).
The establishment of ER-LD contacts may also be linked to peroxisomes. Recent studies suggest that the biogenesis of LDs and pre-peroxisomal vesicles may converge at specific ER subdomains (Schrul and Kopito, 2016; Joshi et al., 2018; Wang et al., 2018). Specifically, loss of both seipin and Pex30 (a membrane-shaping protein enriched at pre-peroxisomal vesicles) in yeast synthetically restricted the morphogenesis of LDs and caused growth defects (Wang et al., 2018). Such tricontacts between the ER, LDs, and preperoxisomes hint at a mechanism for the detoxification of lipid intermediates that arise from the biogenesis of LDs and peroxisomes.
ER-LD Contacts During Lipid Mobilization
The depletion of cellular energy or a need for membrane phospholipid synthesis triggers the utilization of neutral lipids that have been stored in LDs. Such lipid mobilization, or lipolysis, necessitates the remodeling of ER-LD contacts. As discussed before, the ER and LD surface maintain relatively distinct proteomes, plausibly because of a gating mechanism that prevents the ready exchange of ER and LD resident proteins. However, the restriction on protein movement is lifted during lipolysis. In yeast, using one of the TAG synthesis enzymes Dga1p as a reporter, it was shown to move readily from LDs to the ER when lipogenesis is inhibited and lipolysis triggered (Jacquier et al., 2011). In another context, when high demand for membrane phospholipids during the exponential phase prompts lipid mobilization from LDs, the retrograde movement of Dga1p was shown to require the ER membrane protein Ice2p (Markgraf et al., 2014). Taken together, a two-step model for lipolysis can be formulated that involves LDs capture by ER resident proteins, which is followed by the restoration of membrane continuity between LDs and the ER. It should be noted that, in addition to promoting ER-LD contacts, Ice2p is also required for ER-PM contacts (Quon et al., 2018).
In mammalian hepatocytes, TAG stored in cytoplasmic LDs is mobilized to yield substrates for TAG re-synthesis and packaging into very-low-density lipoprotein particles in the ER lumen for export (Gibbons et al., 2000; Quiroga and Lehner, 2018). The secretion of very-low-density lipoprotein particles in the fed state is triggered by insulin. Recent evidence suggests that insulin signaling in hepatocytes promotes the recruitment of ADP-ribosylation factor 1 (ARF1) and phospholipase D1 to the LD surface (Rai et al., 2017). Phospholipase D1 promotes the conversion of LD surface phosphatidylcholine into phosphatidic acid (PA). It has been proposed that the elevation of PA level serves two purposes: (a) recruitment of the kinesin 1 motor that transports LDs to the cell periphery for their engagement with the ER prior to lipolysis and (b) stabilization of ER-LD membrane junctions of high curvature as the two organelles re-engage (Kumar et al., 2019). The latter postulate is similar to the one used to account for PA over-representation in seipin-deficient cells in which nascent LDs that are prevented from full maturation remain attached to the ER via a membrane neck (Han et al., 2015; Wolinski et al., 2015). In both cases, the ability of PA to accommodate high negative curvature without packing defects was invoked.
Future Perspectives
The recognition that LDs maintain close contact with the ER has been built on the foundation of biochemical and cell biological studies, accumulated over many decades. Key questions remain in our quest to understand ER-LD contacts. For example, are there cell type-specific ER-LD contacts? Do subtypes of ER-LD contacts exist in the same cell? What are the gatekeepers flanking the contacts and how are they specifically enriched there? Structurally, the interaction between phospholipid bilayers (the ER) and monolayers (LDs) is the most peculiar type of membrane contacts. Although a continuity of the two membranes is commonly appreciated (often linked by the ER-LD neck (Salo et al., 2019) or the ER-LD bridge (Wilfling et al., 2013), depending on the stage of LD maturation), the surface protein and lipid signatures remain invariably distinct, suggesting the presence of gatekeepers at ER-LD contacts. To identify novel components that reside at ER-LD contact sites, the use of APEX2 fusion proteins seems prudent. APEX2 is an engineered peroxidase that facilitates biotinylation of proteins that are in its close proximity (Lam et al., 2015). By fusing a catalytic dead adipose triglyceride lipase (ATGL) mutant or PLIN2 to APEX2, the LD proteomes of U2OS and Huh7 cells have been comprehensively analyzed and new connections between LDs and the ER were revealed (Bersuker et al., 2018). APEX2 also allows the visualization of its fusion partner by electron microscopy (Lam et al., 2015). As such, APEX2 has been used to determine the localization of ER-LD contact proteins, Snx14 (Datta et al., 2019), seipin (Cao et al., 2019) and ORP5 (Du et al., 2020), in mammalian cells. The split APEX2 tag is an exciting new development (Han et al., 2019). In theory, it should allow the demonstration of molecular interaction of partner proteins at organelle contact sites. Nevertheless, the reconstitution of functional APEX2 will depend on the relative configuration and mobility of partner proteins in their native membrane environment. We expect the study of ER-LD contacts to continue to benefit from additional technical advances in genome editing (for the facile insertion of coding sequence of functional tags), mass spectrometry, and superresolution microscopy (Dickinson and Goldstein, 2016; Baddeley and Bewersdorf, 2018; Bersuker and Olzmann, 2018; Krahmer and Mann, 2019; Manna et al., 2019; Peng et al., 2019).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Hong Kong Research Grants Council.