
Abstract
As a key sphingolipid metabolite, sphingosine-1-phosphate (S1P) plays crucial roles in vascular and immune systems. It regulates angiogenesis, vascular integrity and homeostasis, allergic responses, and lymphocyte trafficking. S1P is interconverted with sphingosine, which is also derived from the deacylation of ceramide. S1P levels and the ratio to ceramide in cells are tightly regulated by its metabolic pathways. Abnormal S1P production causes the occurrence and progression of numerous severe diseases, such as metabolic syndrome, cancers, autoimmune disorders such as multiple sclerosis, and kidney and cardiovascular diseases. In recent years, huge advances on the structure of S1P metabolic pathways have been accomplished. In this review, we have got a glimpse of S1P metabolism through structural and biochemical studies of: sphingosine kinases, S1P transporters and S1P receptors, and the development of therapeutics targeting S1P signaling. The progress we summarize here could provide fresh perspectives to further the exploration of S1P functions and facilitate the development of therapeutic molecules targeting S1P signaling with improved specificity and therapeutic effects.
Introduction – Sphingolipid Metabolism and S1P Signaling
Sphingolipids, named after the sphinx in Egypt to represent its mysterious role, were isolated from the hydrolysis products of phrenosin in the 1870s by Johann Thudichum (Thudichum, 1884; Spiegel and Milstien, 2003; Chun and Hartung, 2010). The chemical structures of sphingolipids attracted the attention of chemists in the early 20th century (Spiegel and Milstien, 2003). Then, its biological mysteries have been gradually unveiled to show irreplaceable roles in forming the cell membrane and functioning as signaling molecules in recent decades (Takabe et al., 2008). Complex sphingolipids, including glycosphingolipids and sphingomyelins, are structural components to form the mechanically stable and chemically resistant outer leaflet of eukaryotic cell membranes (Kumari et al., 2018). The general structure of sphingolipids is composed of a long-chain sphingoid base, O-linked to a polar head group on sn-1 site, and amide-linked to a fatty acid on sn-2 site of the backbone (Figure 1(a)). Variations of functional groups attached to the backbone determine the physical, chemical, and biological properties of sphingolipids. For instance, the phosphorylation of sphingosine, which is derived from the deacylation of ceramide, on the sn-1 hydroxyl group creates the lysophospholipid S1P (Figures 1(b) and 2).

General structures of sphingolipids. (a) Chemical structures of sphingolipids. The sphingoid base is shown in gray shadow. R1 and R2 representing functional groups are highlighted red and green, respectively. (b) The compositions of R1 and R2 in different types of sphingolipids are listed.

Metabolic pathways of S1P. S1P is generated by phosphorylation of sphingosine by sphingosine kinases (SPHK1 and -2) and converted to sphingosine by sphingosine phosphatases (SGPP1 and -2). Sphingosine is generated by deacylation from ceramide by ceramidase and converted back by ceramide synthase. Ceramide can also be produced through de novo synthesis or recycling from other sphingolipids. FTY720 (fingolimod) is a prodrug, which serves as a functional antagonist of S1P1 after phosphorylation to FTY720-phosphate by SPHK2.
The metabolism of sphingolipids in cells is complex and dynamic (Figure 2). There are three major pathways to generate ceramides. Firstly, sphingomyelinase breaks down sphingomyelins within the plasma membrane to release ceramides (Nganga et al., 2018). Secondly, the de novo pathway generates ceramide starting from less complicated substrates in the smooth endoplasmic reticulum (sER) (Lucaciu et al., 2020b). Condensation of serine and palmitoyl-CoA by serine palmitoyl-transferase, which is the limiting step of the pathway, generates 3-keto-dihydrosphingosine (Futerman et al., 1990; Satsu et al., 2013). In turn, 3-keto-dihydrosphingosine is reduced to dihydrosphingosines, which is then acylated by ceramide synthases to produce dihydro-ceramides (Linn et al., 2001; Galadari et al., 2006; Xu et al., 2006). The final step to produce ceramide is catalyzed by dihydro-ceramide desaturase (Hannun and Obeid, 2008). Thirdly, the salvage pathway in acidic subcellular compartments (Ditaranto-Desimone et al., 2003; Li et al., 2015 ), which facilitates the degradation of complex sphingolipids including sphingomyelin and glycosphingolipids, and contributes to the generation of ceramides (Takabe et al., 2008; Zhang et al., 2009; Tukijan et al., 2018). Ceramide is subsequently transported to the Golgi apparatus and further converted to other sphingolipids (Funato et al., 2002; Jain and Holthuis, 2017).
Ceramidases catalyze the reverse process to yield sphingosine by deacylation of ceramide (el Bawab et al., 2002). Additionally, sphingosine can be yielded from the diet (Ebenezer et al., 2017). Sphingosine can be further converted to S1P by sphingosine kinases (SPHK1 and -2) through phosphorylation (Olivera, 1993; Liu et al., 2000a; Gao and Smith, 2011). In contrast, dephosphorylation of S1P by S1P phosphatases (SGPP1 and -2) allows S1P to be transformed back to sphingosine (Le Stunff et al., 2002; Takabe et al., 2008; Tukijan et al., 2018). S1P lyase (SPL), S1P phosphatase, and three lysophospholipid hydrolases are responsible for S1P’s degradation (Tukijan et al., 2018). S1P lyase, which acts as a crucial regulator of S1P and other sphingolipid intermediates, is the last enzyme for S1P degradation at the ER membrane (Serra and Saba, 2010). To date, the extracellularly located broad substrate-specific lipid phosphate phosphohydrolases (LPPs) are the only known enzymes that can act as ecto-enzymes for S1P (Ksiazek et al., 2015).
(Intra-)cellular S1P levels are a function of the dynamic relation to compartmental (including extracellular) S1P levels (Cartier and Hla, 2019). Different cellular stress conditions, the relative intracellular concentrations of S1P, sphingosine, and ceramide determine the cell fate through pathways described as the series of “drains” and “faucets” (Shaw et al., 2018). Ceramide is well characterized as a pro-apoptotic signal (Mizushima et al., 1996; Pettus et al., 2002; Lewis et al., 2018). Ceramidases hydrolyze ceramide to sphingosine to allow cellular escape from apoptosis (Nganga et al., 2018). S1P promotes cellular proliferation (Goetzl et al., 1999; Lewis et al., 2018). These discoveries suggest that S1P acts not only as a building block of cell membranes but also as a bioactive lipid mediator. From then on, S1P has been extensively studied and closely linked to a myriad of essential cellular processes including immune cell trafficking (Dorsam et al., 2003; Neubauer et al., 2016; Ebenezer et al., 2017), cell motility (Lee et al., 2001; Guerrero et al., 2011; Neubauer et al., 2016; Sanna et al., 2016; Lucaciu et al., 2020a), angiogenesis and vascular maturation (Liu et al., 2000b; Watson et al., 2010; Ohotski et al., 2014; Gaire et al., 2018; Lucaciu et al., 2020a), and neurogenesis (Mizugishi et al., 2005). Plasma S1P also helps to maintain vascular integrity and regulate vascular leaks (Baek et al., 2013a). Besides, S1P was identified as an early risk factor of lung cancer in an epidemiological study (Bagdanoff et al., 2009) and as a crucial mediator of cardio-protection (Billich et al., 2013).
The consequences of S1P signaling are not exclusively exerted within cells (Sanna et al., 2016; Xiong et al., 2019), but also by ligating to its five G protein-coupled receptors (GPCRs), designated as S1P1-5. Vascular endothelial cells comprise the predominant secretory source for S1P in the circulation (Chun et al., 2002; Pan et al., 2006; Sanna et al., 2016; Hur et al., 2017; Xiong et al., 2019). S1P receptors regulate cell proliferation, apoptosis, cell adhesion, cell motility, angiogenesis, and inflammation, by coupling different downstream heterotrimeric G proteins (Takabe et al., 2008; Lucaciu et al., 2020b). S1P1 prefers to bind to Gαi/o (Cyster and Schwab, 2012). S1P2, besides binding to Gαi/o, is also associated with Gαq, G12/13, and Gαs (Chun and Hartung, 2010; Cyster and Schwab, 2012). It couples most efficiently with G12/13, in the wake of activation of the small GTPase Rho (Gonda et al., 1999; Windh et al., 1999; Okamoto et al., 2000). S1P3 is reported to couple with Gαi/o, Gαq, and G12/13, and S1P4 and S1P5 couple to Gαs, Gαq, and G12/13 (Ancellin and Hla, 1999; Windh et al., 1999). This partially explains why S1P, such a simple molecule bears the potential to induce various systemic consequences (Figure 3).

Trafficking and signal transduction of S1P. S1P can act intracellularly to determine cell fate. S1P can also be exported via MFS transporters (Mfsd2b and Spns2) or ABC transporters and act on S1P receptors in autocrine or paracrine manners. In particular, Mfsd2b is expressed majorly in red blood cells and platelets, whereas Spns2 is highly expressed in lymphatic endothelial cells. The therapeutic molecule targeting S1P1, FTY720 phosphate, was developed for the treatment of multiple sclerosis. In the circulating system, S1P is transported by binding to ApoM. Five S1P receptors are belonging to the cell surface class A G protein-coupled receptor (GPCR) family and regulating different cellular responses, such as cell proliferation, apoptosis, cell adhesion, cell motility, angiogenesis, and inflammation.
Within the last two decades, atomic structures of many sphingolipid metabolizing enzymes were determined, including human sphingosine kinase 1 (Wang et al., 2013), three types of ceramidases (Inoue et al., 2009; Airola et al., 2015; Gebai et al., 2018; Vasiliauskaite-Brooks et al., 2018; Dementiev et al., 2019), human S1P receptor S1P1 (Hanson et al., 2012), bacterial homologs of serine-palmitoyltransferase (Raman et al., 2010), S1P lyase (Bourquin et al., 2010), some sphingolipids transfer proteins (Christoffersen et al., 2011; Blaho et al., 2015), and so on. However, there are still various other essential enzymes and receptors, such as ceramide synthases, sphingosine kinase 2, and S1P2-5, for which the structures and molecular mechanisms remain yet to be uncovered. Here, we catch a glimpse of the progress made in the metabolism, transportation, and sensing of S1P. From a structural viewpoint, this may pave the way for the development of therapeutic molecules targeting S1P signaling.
Molecular Mechanism of Sphingosine Kinases
To yield S1P in cells, SPHKs phosphorylate sphingosine on its primary hydroxyl group (Gustin et al., 2013). There are two SPHKs isoforms encoded in the human genome, designated as SPHK1 and -2 (Murakami et al., 2010; Gao and Smith, 2011). It appears that SPHK1 and -2 have important roles in cell fate determination (Maceyka et al., 2005) and angiogenesis (Mizugishi et al., 2005). The SPHK1/-2 double knockout mouse confers embryonic lethality due to detrimental effects on severe defects in angiogenesis and neurogenesis (Mizugishi et al., 2005). Apart from the large N terminal and internal insertions in the sequence of SPHK2, human SPHK1 and -2 share 48% primary sequence identity and 73% similarity, respectively. Although catalyzing the same reaction and having some functional redundancy (Allende et al., 2004), SPHK1 and -2 present different substrate specificities, tissue distributions, and subcellular localizations (Liu et al., 2003; Maceyka et al., 2005; Taha et al., 2006). For instance, SPHK1 is recruited to the plasma membrane in response to extracellular signals, such as TNFα, by generating S1P that binds to TNF receptor associated factor 2 and regulates cell survival (Alvarez et al., 2010; Jarman et al., 2010). While the S1P generated by SPHK2 stabilizes telomerase to enhance proliferation, it inhibits HDAC to maintain histone acetylation and to regulate PPARγ in nucleus and regulates the electron transport chain assembly in mitochondria (Hait et al., 2009; Panneer Selvam et al., 2015; Parham et al., 2015). Moreover, studies have shown abundant SPHK1 expression in several cancers (Van Brocklyn et al., 2005; Ruckhaberle et al., 2008) and inflammatory conditions (French et al., 2003). SPHK2 also shows the oncogenic function in various tumors, such as lymphoblastic leukemia (Weigert et al., 2009; Neubauer et al., 2016). The silencing of SPHK2 signaling prominently reduces tumor growth of human xenograft models in mice (Weigert et al., 2009). Enhancement of SPHK2 expression is related to the progression of non-small cell lung cancer and multiple myeloma (Gao and Smith, 2011; Ebenezer et al., 2016), and the blockage of SPHK2 expression induces cell death and increases sensibility to various cancer cell types (Sankala et al., 2007). Thus, SPHK inhibitors bear the potential to alter mitochondria function, regulate S1P signaling, and prevent cellular immortality in cancer (Adams et al., 2020).
Apo and inhibitor bounded structures of human SPHK1 are available in Protein Data Banks (Wang et al., 2013; 2014). Based on the sequence and structural alignments, human SPHK1 belongs to the phosphofructokinase (PFK)-like superfamily, sharing the same protein fold with NAD kinases, diacylglycerol kinases (DGKs), and ceramide kinases, but not other lipid kinases, such as phosphatidylinositol-3 kinase (PI3K) (Wang et al., 2013). Five motifs (C1-C5) are highly conserved between SPHK1, -2 and ceramide kinase (CERK) (Wang et al., 2013). Among these motifs, the C4 domain varies the most, suggesting a possible molecular basis of substrate specificity (Yokota et al., 2004; Wang et al., 2013). SPHK1 exhibits the overall fold of two lobes, the nucleotide-binding site in the N-lobe and a hydrophobic lipid-binding pocket in the C-lobe, whereas the catalytic motif (S/G)GDG is positioned in between (Figure 4(a)). The lipid-binding pocket, a J-shape hydrophobic tunnel that can accommodate substrates with 14 to 18 carbon alkyl chains, is largely buried inside the kinase (Figure 4(b)). Although it is hard to distinguish the suitable length of the acyl chain from the omitted map observed in the structure (PDB code: 3VZB), the head group of sphingosine fits the density well and coordinates with the surrounding residues through three pairs of hydrogen bonds (Figure 4(c)). The nucleotide-binding site is also varified by obtaining the SPHK1-ADP-inhibitor complex structure (PDB code: 3VZD) (Wang et al., 2013). The SPHK1 structural model, bound with ATP and sphingosine, was generated based on the structures described above. The γ-phosphate of the ATP, as a nucleophile, attacks the primary hydroxyl group of sphingosine, resulting in the phosphoryl transfer. The conserved Asp81 in SPHK1 and -2, as a general base, deprotonates and increases the nucleophilicity of the primary hydroxyl group of sphingosine (Wang et al., 2013). Some molecular modeling works raised other possibilities on the catalyzing mechanism of SPHK1, suggesting D178 as the catalytic residue, since inhibitors may interact with D81 (Baek et al., 2013a). Thus, an atomic resolution complex structure of SPHK1 with both sphingosine and ATP analog is needed to confirm the catalytic residues and reveal the precise catalyzing mechanism of S1P phosphorylation.

Structures of human SPHK1. (a) Human SPHK1 exhibits the overall fold of two lobes, designated N-/C-lobe, with a hydrophobic lipid-binding pocket highlighted in gray shadow, and a nucleotide-binding site highlighted in orange shadow. (b) Surface representation of sphingosine binding pocket. Sphingosine is shown in yellow sticks. (c) Detailed interactions between sphingosine and SPHK1. Hydrogen bonds are highlighted in brown dashes. (d) Superposition of sphingosine and three competitive inhibitors in SPHK1 structures. (e) Chemical structures of SPHK1 inhibitors.
SPHK1 inhibitors have been investigated with scrutiny since SPHK1 was identified as a potential therapeutic target in many diseases (Lynch et al., 2016; Cao et al., 2018). There are several types of SPHK inhibitors, including lipidic, non-lipidic, and natural products, which are in use or under development for different diseases (Kono et al., 2000a; 2000b; French et al., 2003; Salma et al., 2009; Pitman et al., 2015). Most of the early developed inhibitors, such as Safingol (Buehrer and Bell, 1992), dimethyl-sphingosine (DMS) (Yatomi et al., 1996), and trimethyl-sphingosine (TMS) (Endo et al., 1991), are sphingosine analogs with poor potency and selectivity. Although Safingol showed the therapeutic potential in certain solid tumors and was applied to phase I clinical trials (NCT00084812) (Dickson et al., 2011), it had severe off-targets on protein kinase C (PKC) and ceramide synthase (CerS) (Schwartz et al., 1995). DMS and TMS also showed cross activities with SPHK2 (Liu et al., 2000a) and CERK (Sugiura et al., 2002), making them difficult to decipher the role of SPHK1. Soon afterward, four different types of non-lipidic small molecules, named SKI-I, -II, -III, and -IV, were developed with sub-micromolar to micromolar inhibition of SPHK1 specifically (French et al., 2003). These compounds, showing anti-tumor activities in vivo without obvious toxicities, shed light on SPHK inhibitors to be developed as anticancer drugs. Some other non-lipidic SPHK1 inhibitors, such as PF-543 (Schnute et al., 2012), compound 23 (Baek et al., 2013b), and RB-005 (Baek et al., 2013b), have been identified through different approaches as well. To date, three non-lipidic inhibitors, all occupying the sphingosine binding pocket competitively, have been co-crystallized with SPHK1. The superposition of SPHK1 structures bound with different inhibitors suggests the convergent inhibitory mechanism with unique interaction features (Figure 4(d) and (e)). Although these inhibitors showed significant inhibitions of SPHK1 activity in vitro and favorable PK/PD properties, the therapeutic effects in patients remain to be further explored. The first selective SPHK2 inhibitor, ABC294640, reducing the total amount of S1P in the nucleus (Ebenezer et al., 2017), also completed phase I clinical trials for the treatment of advanced solid tumors (NCT01488513) (French et al., 2010; Gestaut et al., 2014).
S1P Degradation by S1P Lyase
The degradation of S1P, mainly through interconversion with sphingosine or irreversible degradation to phosphoethanolamine (PE) and trans-2-hexadecenal, synergistically matches S1P synthesis according to the metabolic demand (Kumar and Saba, 2009; Lucaciu et al., 2020b). Dephosphorylation of S1P to form sphingosine is mainly employed by two S1P phosphatases (SGPP1/2). S1P lyase (SPL) and lipid phosphate phosphohydrolases (LPPs) act as the crucial enzymes for S1P degradation in the intracellular and extracellular spaces, respectively. Here we only focus on SPLs as their structures have been well characterized. Further structural studies on SGPP1/2 and LPPs are needed.
Eukaryotic SPLs associate with the ER through the N-terminal transmembrane helix. The catalytic domains of eukaryotic SPLs are evolutionary conserved and operate on the cytosolic leaflet of the ER membrane (Ikeda et al., 2004). Numerous bacterial and mammalian SPL structures have been determined. The bacterial and yeast homolog structures of StSPL (Symbiobacterium thermophilum SPL) (Figure 5(a)) and Dpl1p (yeast SPL) were solved first to characterize the substrate and co-factor binding sites to elucidate the mechanism of S1P degradation (Bourquin et al., 2010). Unsurprisingly, the human SPL exhibits the ideal dimerization state and, for each protomer, an overall r.m.s.d (root-mean-square deviation) of 0.8 Å over 400 Cα atoms in an overlay of the human SPL and Dpl1p (Weiler et al., 2014). SPLs are members of the pyridoxal phosphate (PLP)-dependent superfamily. Here we use StSPL structures to present the interaction of PLP and the catalytic reaction of SPLs. PLP covalently links to the residue K311 in substrate-free structures (Figure 5(b)). The phosphate group of PLP is well coordinated by residues G168, T169, H310 in one protomer, and S353 from the adjacent protomer through hydrogen bonds (Figure 5(c)). The pyridinium ring forms a hydrogen bond with D274 and faces H201 to form cation-pi interaction (Figure 5(b)). The consistent phosphate ions, which are close to PLP and interact with residues Y105 in one protomer and N126, H129, and S353 in the adjacent protomer, are observed in all structures and may mimic the head group recognition of S1P (Bourquin et al., 2010) (Figure 5(c)). The PE bound structure of StSPL also demonstrates substrate-binding residues such as A103, Y105, H129, and K317 (Figure 5(d)). Besides, the Schiff base structure of PE-PLP unveils the mechanistic convergence of S1P degradation with the classic PLP-dependent decarboxylation reactions (Figure 5(d)). The substrate S1P could replace K311 of StSPL, which initially forms an internal aldimine with PLP, to be a Schiff base partner of PLP. Then the retro-aldol cleavage occurs by nucleophilic attack on S1P, releasing hexadecenal. The following re-protonation of the quinonoid intermediate will lead to the release of PE and allows the active site to revert to the original state.

Structures of bacterial S1P lyase StSPL. (a) Dimer structure of StSPL (PDB code: 3MAD) is shown, in which one protomer is in cylindrical mode (white) and the other protomer is in surface mode (lightblue). (b) Detailed interactions between K311-PLP and the surrounding residues. Hydrogen bonds are highlighted in brown dashes. (c) Detailed interactions between PLP, phosphate ion, and the surrounding residues in one protomer of PE bound S1P lyase structure (PDB code: 2MAU). (d) Detailed interactions between PE-PLP and the surrounding residues in the other protomer of PE bound S1P lyase structure (PDB code: 2MAU). (e) Compound 31 acts as an inhibitor to block the narrow substrate entrance of StSPL (PDB code: 4Q6R). Two perpendicular views are shown, in which the protein is in surface mode and compound 31 (yellow) and PLP (green) are in stick mode.
Since inhibition of SPL causes T cell sequestration and immunosuppression (Schwab et al., 2005), SPL is considered as an important therapeutic target in the treatment of auto-immune diseases such as multiple sclerosis and rheumatoid arthritis (Bagdanoff et al., 2009; Fleischmann et al., 2012; Billich et al., 2013). Several SPL specific inhibitors have been developed and their complex structures with SPL were determined in the past few years, including compound 31 with human SPL, and compound 1 and -2 with StSPL surrogate. SPL dimer only leaves a narrow hydrophobic tunnel linking to its active site, so the inhibitors bind at the entrance of this tunnel through hydrophobic interactions (Figure 5(e)). Because a structure of SPL in complex with S1P is lacking, it is not clear if these inhibitors occupy the S1P binding site competitively or block the entry of S1P. The precise S1P binding mode may help in designing small molecule inhibitors with improved specificity and efficacy. Nevertheless, these structural observations shed light on possibilities to study the development of SPL inhibitors for therapeutic purposes.
Molecular Mechanism of Ceramidases
Ceramidases hydrolyze membrane ceramide into sphingosine, which in turn is phosphorylated to form S1P, to regulate S1P/ceramide ratio and multiple cellular processes. Based on the primary sequences, subcellular localizations, functions, and pH preferences, five ceramidases are classified into three categories (acid, neutral, and alkaline). Neutral ceramidase (ASAH2) is crucial for the digestion of dietary sphingolipids (Kono et al., 2006), regulation of the level of sphingolipid metabolites in the intestinal tract (Symolon et al., 2004), and protection against inflammatory cytokines (Kono et al., 2006). Alkaline ceramidases, including ACER1-3, mediate cell differentiation by controlling sphingosine and S1P (Sun et al., 2008), DNA damage-induced cell death (Xu et al., 2016), and cell proliferation (Hu et al., 2010). Acid ceramidase (ASAH1), which hydrolyzes lysosomal ceramide into sphingosine, is the best-characterized member of the family due to the association with Farber disease, the extremely rare autosomal recessive lysosomal storage disease (Schuchman, 2016). The structural characteristics of ceramidases were extensively explored in the last decade. All three categories of ceramidases exhibit different structural folds and perform ceramide hydrolysis activities through distinct mechanisms. ASAH1 belongs to the N-terminal nucleophile (Ntn) superfamily of hydrolases (Pei and Grishin, 2003; Schulze et al., 2007), which are synthesized as a proenzyme and activated through auto-cleavage (Brannigan et al., 1995). Three structures of mammalian ASAH1 (Gebai et al., 2018) indicate that both proenzyme and active states of ASAH1 are similar regarding overall structure and subunit organization. However, auto-cleavage results in conformational changes that uncover a hydrophobic groove (13 Å) leading to the active site that probably accommodates ceramide (Figure 6(a)). The deprotonated catalytic residue Cys143 of human ASAH1 is employed for both auto-cleavage and ceramide hydrolysis, although the precise mechanisms are different. The active site exposure mode was also observed in the human ASAH2 structure previously (Airola et al., 2015). The major difference is that the larger tunnel (20 Å) uncovered in ASAH2 could accommodate ceramide with longer acyl chains (Figure 6(b)). Besides, ASAH2 is a single-pass transmembrane protein on epithelial cell membranes and functions at neutral pH (Kono et al., 2006). It belongs to a unique structural family, which displays little sequence homology to other proteins, and catalyzes ceramide hydrolysis mediated by Zn2+ ion. ACERs, the integral membrane proteins comprised of seven transmembrane helices, are less well understood in part because of the hydrophobicity nature. By analyzing the structure of human ACER3, the large hook-shaped cavity is entirely embedded in the membrane (Figure 6(c)). Computational docking and molecular dynamic simulation results suggest that the carbonyl group and primary alcohol of ceramide could directly interact with Zn2+ ion. Considering the function of crystallographic water in the active site as a nucleophile attacking the carbonyl of ceramide, a general acid-base catalytic mechanism was proposed (Vasiliauskaite-Brooks et al., 2018).
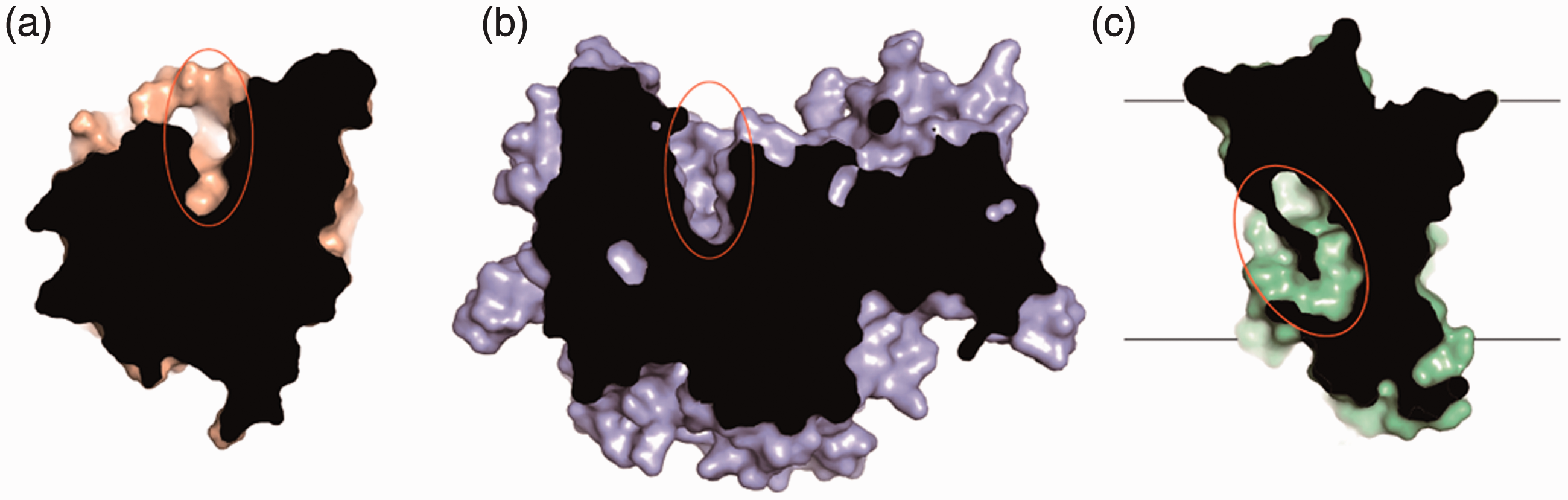
Ceramide binding pockets of ceramidases. (a) Human active ASAH1 structure (PDB code: 5U7Z) is shown on wheat surface. The uncovered ceramide binding pocket is circled. The depth of the pocket is about 13 Å. (b) Human ASAH2 structure (PDB code: 4WGK) is shown on purple surface. The uncovered ceramide binding pocket is circled. The depth of the pocket is about 20 Å. (c) The human ACER3 structure (PDB code: 6G7O) is shown on palegreen surface. The hook-like ceramide binding pocket buried in the membrane is circled.
A number of ceramide-mimicking inhibitors of ceramidase (first-generation inhibitors) have been developed in the past two decades. N-Oleoylethanolamide (NOE) was first used to increase the cellular ceramide level and induce apoptosis in various cell lines (Sugita et al., 1975). However, the low potency and poor selectivity limit its therapeutic use (Grijalvo et al., 2006; Houben et al., 2007). Then, a series of NOE analogs were developed as potent and selective lysosomal acid ceramidase inhibitors (Grijalvo et al., 2006; Bedia et al., 2008). Besides, some lipophilic aromatic ceramide analogs, such as D-e-MAPP, B13, and LCL compounds, were developed to induce apoptosis via inhibition of ceramidases as anticancer agents (Bielawska et al., 1992; Samsel et al., 2004; Szulc et al., 2008). The rational design of first-generation inhibitors is limited by lacking enough heteroatoms on ceramide, making it difficult to increase much higher potency than their natural progenitor. The only exception is SABRAC, which is the irreversible inhibitor likely forming a covalent bond with the enzyme (Camacho et al., 2013). The first-generation inhibitors help to highlight the therapeutic potentials of inhibiting ceramidases in cancer treatments. The non-ceramide-like inhibitors (second-generation inhibitors), usually obtained by high throughput screening, are more likely potent and drug-like than ceramide-mimicking compounds. Series of second-generation inhibitors of ceramidases have been developed. Cerenib-2, a representative quinolinone-based compound showed dose-dependent inhibition to ceramidase, led to ceramide accumulation, and sphingosine and S1P reduction (Draper et al., 2011). Carmofur and its derivatives, the novel acidic ceramidase inhibitors, act synergistically with standard anti-cancer therapeutics to inhibit cancer cell proliferation (Realini et al., 2013). However, the short half-life times of these compounds in vivo hinder their therapeutic use despite the strong inhibition (Ouairy et al., 2015). Then, the benzoxazolone carboxamide compounds are screened as the irreversible inhibitors of intracellular acidic ceramidase. The representative compound 17a efficiently inhibits acidic ceramidase activity and is metabolically stable in vivo (Pizzirani et al., 2015).
Mechanism of S1P Transport
S1P prompts its physiological roles through two mechanisms, binding to its intracellular targets or by extracellular secretion. In this review, the focus is drawn on the secretion of S1P and its subsequent transport in the circulation. Hematopoietic cells and endothelial cells comprise the major source of extracellular S1P (Fukuhara et al., 2012; Vu et al., 2017). Since it contains a negatively charged phosphate group, S1P cannot diffuse freely but has to be transported across the cell membrane through active transport. Several types of transporters have been identified in the past two decades.
Mfsd2b, which belongs to the major facilitator superfamily (MFS), is an orphan transporter that is expressed in erythrocytes and platelets (Vu et al., 2017). Mutations of the conserved D97 and T159 in human Mfsd2b resulted in a loss of S1P export activity (Vu et al., 2017). About ∼42-54% of the S1P reduction in the plasma was also observed in Mfsd2b-/- mice (Vu et al., 2017). Through a comprehensive lipidomics analysis, S1P was specifically accumulated in Mfsd2b knockout red blood cells and platelets (Vu et al., 2017). As a blood-borne lipid mediator, S1P is more abundant in circulatory fluids than tissue fluids (referring to the S1P gradient) to facilitate lymphocyte egress both from lymph nodes into the lymph, but also from the lymphatic system into the blood system, respectively (Fukuhara et al., 2012; Lucaciu et al., 2020a). However, the abolishment of S1P export through Mfsd2b did not affect lymphocyte egress and trafficking (Allende et al., 2004), indicating a functional overlap with other means of S1P export. In that regard, spinster homolog 2 (Spns2), originally identified in zebrafish as an S1P transporter (Kawahara et al., 2009), contributes approximately 25–50% to lymphatic S1P through lymphatic endothelial cells in humans while it is dispensable for contributing to plasma S1P (Fukuhara et al., 2012; Nagahashi et al., 2013; Mendoza et al., 2017). It plays a crucial role in S1P export that regulates lymphocyte egress and trafficking (Mendoza et al., 2012) by creating an S1P gradient (Schwab et al., 2005). Spns2 knockout mice featured an increase in mature thymic T cells, whilst decreased numbers of T cells in the hematopoietic system and secondary lymphoid organs were observed (Fukuhara et al., 2012). A possible reason is that the export of S1P by SPNS2 is also essential for the survival of circulating naive T cells (Mendoza et al., 2017).
Moreover, some ATP-binding cassette (ABC) family transporters have been reported to export S1P, including ABCA1 in astrocytes (Sato et al., 2007), ABCC1 in mast cells (Mitra et al., 2006), ABCG2 in breast cancer cells (Takabe et al., 2010), and others in platelets and erythrocytes (Kobayashi et al., 2006; Kobayashi et al., 2009). However, the downregulation of these ABC transporters did not decrease the S1P levels in plasma, rendering them debatable in terms of physiological contribution to S1P transport. Another ABC transporter, the cystic fibrosis transmembrane regulator (CFTR), was also reported to be involved in the uptake of S1P, dihydrosphingosine 1‐phosphate (dhS1P), and lysophosphatidic acid (Brown et al., 2014).
The structural and biochemical studies of transmembrane proteins have been drawn attention to but difficult to study because of the technical challenges in the past decades. Thus, the molecular basis of S1P export crossing the membrane remains unclear. Recently, a crystal structure of HnSPNS, the bacterial homolog from Hyphomonas neptunium shared 18% of sequence identity and 29% similarity with human Spns2, was reported at 3.1 Å in an inward-facing conformation (PDB Code 6E9C) (Zhou et al., 2019) (Figure 7(a)). Similar to other MFS transporters, HnSPNS consists of 12 transmembrane α-helices, assembling two structural repeats with pseudo-symmetry: the N- and C-terminal domains by TM1-6 and TM7-12, respectively (Figure 7(a)). Among species from bacteria to mammals, the most conserved residues gather in the N-terminal domain of Spinster proteins (Zhou et al., 2019). Notably, the contacts between TM2 and -4 in the inner cavity, where the best-characterized residues E129 and R122 are located, are significantly conserved. Besides, 13 of 25 residues composing this cavity are highly hydrophobic. There is an obvious and continuous omitted density in the center of the cavity surrounded by those conserved residues, such as R42, R122, F71, Y277, and Y371. Taken together, an acidic hydrophobic small molecule may be accommodated into the evolutionarily conserved cavity (Figure 7(b)), suggesting S1P may serve as a potential substrate for eukaryotic Spinster proteins.

Structure of bacterial spinster homolog HnSPNS. (a) The overall architecture of HnSPNS (PDB code: 6E8J) is shown in two perpendicular views, in which the side view is in cylindrical mode and the periplasmic view is in electrostatics mode. The N-/C-domains are colored wheat and lightblue, respectively. The inner cavity is circled in orange. (b) Conserved residues in the inner cavity of spinster homologs’ are shown in the yellow stick mode. The putative S1P binding pocket is shadowed in orange.
Once exported from cells, S1P must bind to apolipoproteins and albumin in the bloodstream, due to the hydrophobic characteristics, to be effectively transported. More than half of the S1P molecules are occupied by the apolipoprotein M (ApoM), associated with high-density lipoprotein (HDL) in the plasma (Christoffersen et al., 2011; Blaho et al., 2015). From the crystal structure of ApoM in complex with S1P, the hydrophilic groups of S1P are mainly recognized by R98, W100, and R116 via direct hydrogen bonds, whereas other water-mediated interactions also contribute to the interactions (Christoffersen et al., 2011). As a similar strategy of inhibiting S1P transporters, targeting S1P, ApoM, or S1P-ApoM complexes in the plasma are possible ways for anti-angiogenic therapeutics. Some antibodies targeting plasma S1P have been developed. However, it is controversial to deplete plasma S1P due to its possible athero-protective effects (Poti et al., 2014), antihypertensive functions via S1P1/3 signaling (Cantalupo et al., 2017), endothelial-sealing effect through signaling to endothelial junctions (Xiong and Hla, 2014), and cardio-protective functions in several cardiovascular diseases such as hypertrophic heart disease, myocardial infarction and chronic heart failure (Cartier and Hla, 2019; Jozefczuk et al., 2020). Therefore, the balance of S1P in both cancer and cardiovascular disease models still needs to be better characterized.
S1P Signaling via S1P Receptors
There are five S1P receptors, designated as S1P1–5, encoded in the human genome and activated by the endogenous ligand S1P. All five S1P receptors belong to the cell surface class A G protein-coupled receptor (GPCR) family, but regulate different cellular responses, such as cell proliferation, apoptosis, cell adhesion, cell motility, angiogenesis, and inflammation, by coupling different downstream heterotrimeric G proteins (Takabe et al., 2008). Among S1P1-5, S1P1 stands out due to its non-redundant functions. In particular, S1P1 mediates the egress of T and B cells from the thymus and secondary lymphoid tissues (Ebenezer et al., 2017; Lucaciu et al., 2020a), making it a potential therapeutic target similar to SPL. Thus, the structure and biochemical properties of S1P1 have been studied extensively.
The crystal structure of S1P1 was yielded using the classic lipidic cubic phase crystallization method with the addition of a T4-lysozyme fusion, which stabilizes the conformation of the intracellular side of the receptor (Thorsen et al., 2014) and increases the possibility of crystal contacts (Chun et al., 2012; Hanson et al., 2012). The receptor was co-crystallized in complex with the sphingolipid mimic antagonist ML056, which is selected to stabilize the receptor by constraining the extracellular side of the receptor. Multiple microcrystal datasets are merged to reach the acceptable completeness to determine the S1P1 structure at 2.8 Å resolution. S1P1 adopts the identical fold of typical inactive GPCRs (Figure 8(a)), which are composed of seven transmembrane helices (TMs) (Figure 8(b)). However, the structural analysis reflects some unique features for lipid binding. Firstly, the extracellular loops ECL1-3 and the N-terminal region are coordinated to exclude the antagonist ML056 from the extracellular solvent. Meanwhile, there is a cleft between TM1 and TM7 facing the membrane bilayer. Similar structural features are also observed in other lipid receptors, such as cannabinoid receptors and CRTH2 (Pei et al., 2008; Hurst et al., 2010; Wang et al., 2018a) (Figure 8(a)). These observations suggest a common mechanism that these lipidic molecules, such as S1P, cannabinoid, and prostaglandins, are first integrated into the lipid bilayer before binding to the receptor occurs (Hua et al., 2016; Shao et al., 2016; Wang et al., 2018a). ML056 mimics the zwitterionic nature of S1P and forms equivalent contacts in the orthosteric binding pocket of S1P1 (Figure 8(b)). The benzene ring and acyl chain of ML056 are surrounded by numbers of hydrophobic residues, such as F125, F210, W269, and F273 on TM3 and TM5-7, respectively (Figure 8(b)). Residues N101 and E201 interact with the amine group of ML056, while Y29, K34, and R120 coordinate the phosphate group (Figure 8(b)). Together, these interactions provide a high-affinity binding site for ML056 or S1P (Hanson et al., 2012). Considering the similar structures of S1P and ML056 but distinct functions as agonist or antagonist, the activation of S1P1 may depend on the conformations of the acyl chain as well as the lengths of ligands. Besides, S1P1 is reported to be activated by CD44 and aPC (activated Protein C), whilst inhibited by CD69, S1P2, and LPA1 (Lucaciu et al., 2020b). However, determination of the structure only in an inactive conformation, is insufficient to elucidate the activation mechanism of S1P1. Taking advantage of the recent development of cryo-electron microscopy, several GPCR structures in complex with G proteins and β-arrestin have been determined (Scheerer and Sommer, 2017; Hilger et al., 2018; Huang et al., 2020). The S1P1 structure in complex with agonist and G proteins is worth studying to better understand the mechanism of activation.

Structure of human S1P1 (a) The S1P1 structure is shown in surface mode. The palegreen color represents the N-terminal helix as a “lid” to make the orthosteric binding site occluded. The cleft toward the cell membrane is shadowed in orange. Two perpendicular views are shown. (b) The detailed interaction of ML056 with the surrounding residues. ML056 is colored yellow. The aromatic residues that interact with the hydrophobic tail of S1P are highlighted in purple. The polar residues that interact with the hydrophilic head of S1P are highlighted in magenta. The hydrophobic and hydrophilic regions of the orthosteric binding site are shadowed in purple and wheat, respectively.
Currently, the therapeutic molecules targeting S1P1 can be divided into two classes: the lipid-like S1P mimic such as FTY720-P or the non-lipid-like molecules such as BAF-312 (Siponimod) and RPC-1063 (Ozanimod) (Zemann et al., 2006; Pan et al., 2013; Scott et al., 2016). S1P1-5 show complete conservation of residues (N101 and E201 in S1P1) for S1P head group recognition, which is reflected in the lack of selectivity of S1P mimicking drugs. For instance, FTY720 is an agonist for receptors S1P3-5 as well, causing severe off-target side effects like bronchoconstriction and cardiovascular dysfunction (Guerrero et al., 2010). In contrast, the residues in interacting with the acyl chain of S1P among five S1P receptors are significantly distinct, making a promising strategy to screen the selective molecule targeting the specific S1P receptor(s). Notably, the binding pockets of S1P1 and S1P5 are nearly identical, resulting in the lack of ligand specificity between the two receptors. For instance, Siponimod and Ozanimod target both S1P1 and S1P5. By improving the specificity, allosteric modulators rather than orthosteric agonists should be better choices. Recently, an increasing number of S1P1 agonists have been developed, including ponesimod (ACT-128800, Actelion), cenerimod (ACT-334441, Idorsia), mocravimod (KRP-203, Kyorin Pharmaceutical and Novartis), CS-0777 (Daiichi Sankyo), AUY954, CS-2100, CYM5442, GSK1842799, RP001, SEW2871, Syl948, Amgen 8 (TC-G 1006), and Amgen 14 (TC-SP 14) (Pan et al., 2006; Zhang et al., 2009; Gaire et al., 2018). Notably, Novartis developed NIBR-0213 as a potent and selective S1P1 antagonist with an IC50 value of 2.5 nM to human S1P1 (Quancard et al., 2012).
Moreover, S1P1 is essential for endothelial cell functions (Xiong and Hla, 2014), indicating its potential as an agent for endothelial protection. However, the activation of S1P1 by commercialized drugs causes receptor over-desensitized and a long-lasting internalization. This results in the damage of endothelial function and limits the therapeutic potential of these molecules on the endothelial axis. A recently identified biased S1P1 agonist SAR247799 may act as a possible S1P1-targeted endothelial-protective agent. SAR247799 can activate S1P1 in a G protein-biased manner instead of recruiting β-arrestin, which will minimize the receptor internalization. The amelioration of coronary endothelial dysfunction in a pig model and the protection of renal function in a rat model were observed (Poirier et al., 2020). Although more characterizations are needed, the discovery of a biased S1P1 agonist shows great potential to expand indications via distinct S1P signaling transduction pathways.
Summary and Discussion
The sphingolipid metabolism has attracted the attention of structural biologists for the past three decades. On the one hand, the available structural information unveils the recognition specificities of S1P to different targets demonstrating possible reactions, transportation, and mechanisms of sensing. This explains the biochemical and clinical observations at the atomic level and sheds light on the prospect of developing therapeutic molecules. On the other hand, we have only obtained limited structural snapshots of executors in the sphingolipid metabolism. Many questions concerning the regulation of the metabolism of sphingolipids and the impact of this information in the development of therapeutics remain unanswered. It is needed to build the whole picture of sphingolipid metabolism from a structural viewpoint.
To begin, a mass of data shows that SPHK isoforms differ not only in cellular locations and in regulations, but also exhibit distinct substrate specificities. What is the structural basis for SPHK substrate selectivity? What is the stereochemical nature of inhibitor head groups in SPHK inhibition? Although biochemical data suggest that SPHK2 may be more tolerant of changes in hydrophilic groups than SPHK1 (Adams et al., 2020), the detailed conformations of SPHKs and organizations of surrounding residues are not revealed. From a wider perspective, the determination of substrate and inhibitor selectivity of SPHKs confers specific targeting in various cancers with elevated expression levels of both SPHK1 and SPHK2. It also facilitates the investigation of the physiological roles in the homeostatic regulation of S1P-dependent signaling.
After that, as we reviewed above, ceramidases and ceramide synthases are critical regulators that maintain intracellular ceramide, sphingosine, and S1P homeostasis. Although crystal structures and catalytic residues of all acid, neutral and alkaline ceramidases have been characterized, the structural based therapeutic inhibitor development is lagging behind. The development of novel inhibitors of ceramidases is boosted by the availability of high-throughput ceramidase assays. However, too little is known about the exact functions of ceramide in triggering downstream cellular signaling. The novel inhibitors may promote the solid mechanistic study of ceramide biology, which is the prerequisite for the development of therapeutics.
Then, none of the ceramide synthases’ structures have been determined yet. In mammals, ceramides are synthesized by a family of six ceramide synthases (CerSs), each of which uses acyl-CoAs of distinct chain lengths for N-acylation(Raichur, 2020). From a metabolic perspective, these enzymes occupy a unique niche in that they catalyze the synthesis of ceramide via both de novo and salvage pathways. Although numbers of mutagenesis study and in silico predictions demonstrated the importance of the Lag1p motif in CerS activity (Turpin-Nolan and Bruning, 2020), detailed structures and topology of CerSs are needed to verify many unclear questions, including the precise functional domains accommodating acyl-CoAs and sphingosines, the substrate specificity of CerS isoforms, and regulation and inhibition mechanisms of CerSs. In addition, several studies have suggested that CerS activity can be modulated by homo- and heterodimer formation (Laviad et al., 2012), hence the interface of CerS dimers also awaits to be characterized.
Furthermore, there are still plenty of chances for S1P transportation studies from a structural viewpoint. Firstly, the structural information of Spinster proteins is still limited. The alternating access model of MFS transporters, the well-accepted hypothesis in explaining the transportation crossing the membranes, suggests at least three conformations including outward open, occluded, and inward open as intermediate states. Without obtaining complete structural information in the transporting cycle, it is impossible to unveil the transportation mechanism at the molecular level. Thus, more structures of Spinster proteins, especially in humans, are still worth getting due to the low sequence similarity among HnSPNS and other species. Secondly, the incomplete omitted electron density in HnSPNS is most likely the contaminant from the cell-culturing medium. Lacking the detailed recognition of S1P with its transporters, the rational design of specific inhibitors targeting S1P transportation crossing the membrane is contrived. Moreover, the structural information and biochemical characterizations of Mfsd2b and those ABC transporters with S1P are even less. Pharmacologically targeting S1P transporters would be an alternative way of inhibiting the activation of S1P to its receptors or intracellular responses if uptake was inhibited. These inhibitors may act as anti-angiogenic and anti-lymphangiogenic drugs in cancers or inhibit the “inside-out signaling” of the SPHK1/S1P axis (French et al., 2003; Anelli et al., 2010). It is of great importance to elucidate these structures to guide rational drug design as well as provide insight into the underlying mechanisms.
Besides, compared to S1P1, functions of S1P2-5 are less characterized. For instance, it is reported that conjugated bile acids (CBAs) and FAM19A5 were other activators of S1P2 (Studer et al., 2012; Wang et al., 2018b). For better elucidating the functions of these receptors, numerous selective agonists and antagonists targeting S1P2-5 were developed. JTE-013, a selective antagonist targeting S1P2 with an IC50 of 17 nM, was widely used to study possible functions of S1P2 (Ikeda et al., 2003). AB1, the analog of JTE-013 with improved potency, inhibited the growth of neuroblastoma xenografts more effectively (Li et al., 2015). There is a lack of in vivo studies of S1P2 selective agonists, such as CYM5520, CYM5478, and XAX-126 (Satsu et al., 2013). The S1P3 antagonists, such as CAY10444 (Koide et al., 2002), TY-52156 (Murakami et al., 2010), SPM-354 (Sanna et al., 2016) were also developed. However, the function of S1P3 in inflammation remains controversial, as both pro- and anti-inflammatory effects were observed (Hirata et al., 2014; Zhao et al., 2016). The physiological function of S1P4 is also poorly understood. So the selective agonists ML178, ML248 (Guerrero et al., 2010), CYM50260 (Onuma et al., 2017), and benzo-thiophene analogs (Hur et al., 2017) or S1P4 antagonists CYM50358 and compounds ground on a 5-aryl furan-2-arylcarboxamide scaffold (Guerrero et al., 2011; Hur et al., 2017) are expected to provide insights into the functional studies of S1P4. Besides, A-971432 is a selective S1P5 agonist (Hobson et al., 2015). Since the structure-based optimizations of orthosteric and allosteric modulators have been applied to many GPCRs (Congreve et al., 2017; Christopher et al., 2019; Xu et al., 2020), it is vital to obtain more structures of S1P receptors in different conformations bound with diverse ligands.
At last, the importance and complexity of sphingolipids are also exemplified by recent studies, such as the divergent impact of sphingolipid species and pathological backgrounds (Saville and Fuller, 2020) and the linkage of SPHKs and S1P receptors to the level of various blood cell traits (Astle et al., 2016). With the discoveries of novel physiological and pathological roles of sphingolipids, the importance of the molecular mechanistic understandings will get increased over time.
Data
All structural coordinates are downloaded from Protein Data Bank (www.rcsb.org). The figures representing structures are generated by PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.)
See Table 1 for abbreviations and Table 2 for definitions.
Abbreviations.

Definitions.

Footnotes
Acknowledgments
R. R. is supported in part by Kobilka Institute of Innovative Drug Discovery and Presidential Fellowship at the Chinese University of Hong Kong, Shenzhen.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest for the research, authorship, and/or publication of this review article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the National Natural Science Foundation of China (Project No. 31971218), and Shenzhen Science and Technology Innovation Committee (Projects No. JCYJ-20180307-151618765 and JCYJ-20180508-163206306)