
Abstract
The endoplasmic reticulum (ER), the most pervasive organelle, exchanges information and material with many other organelles, but the extent of its interorganelle connections and the proteins that form bridges are not well known. The integral ER membrane protein vesicle-associated membrane protein-associated protein (VAP) is found in multiple bridges, interacting with many proteins that contain a short linear motif consisting of “two phenylalanines in an acidic tract” (FFAT). The VAP-FFAT interaction is the most common mechanism by which cytoplasmic proteins, particularly interorganelle bridges, target the ER. Therefore, predicting new FFAT motifs may both find new individual peripheral ER proteins and identify new routes of communication involving the ER. Here, we searched for FFAT motifs across whole proteomes. The excess of eukaryotic proteins with FFAT motifs over background was ≥0.8%, suggesting that this is the minimum number of peripheral ER proteins. In yeast, where VAP was previously known to bind 4 proteins with FFAT motifs, a detailed analysis of a subset of proteins predicted 20 FFAT motifs. Extrapolating these findings to the whole proteome estimated the number of FFAT motifs in yeast at approximately 50 to 55 (0.9% of proteome). Among these previously unstudied FFAT motifs, most have known functions outside the ER, so could be involved in interorganelle communication. Many of these can target well-characterized membrane contact sites; however, some are in nucleoli and eisosomes, organelles previously unknown to have molecular bridges to the ER. We speculate that the nucleolar and eisosomal proteins with predicted motifs may function while bridging to the ER, indicating novel ER–nucleolus and ER–eisosome routes of interorganelle communication.
Keywords
Introduction
The endoplasmic reticulum (ER) is the most widely distributed organelle in eukaryotic cells. Its functions are created in part by peripheral membrane proteins that reversibly bind its cytoplasmic surface. The sole widely documented mechanism by which cytoplasmic proteins target the ER is through a short linear motif (SLiM) called “two phenylalanines in an acidic tract” (FFAT; Loewen et al., 2003). Proteins with FFAT motifs communicate between the ER and many other compartments, including plasma membrane, Golgi apparatus, peroxisomes, and mitochondria (Hanada et al., 2003; Hynynen et al., 2005; Saita et al., 2009; De Vos et al., 2011; Murphy and Levine, 2016; Costello et al., 2017; Hua et al., 2017; Yadav et al., 2018). This fits the idea that SLiM interactions allow links to arise between seemingly distinct subcellular processes (Kim et al., 2014). Significantly, all proteins in a non-ER location with FFAT motifs have since been found to target both that location and the ER at the same time. They bridge between the two compartments at membrane contact sites (Murphy and Levine, 2016). Thus, finding a FFAT motif is significant because it suggests direct bridging to the ER, rather than the existence of a long-distance intracellular shuttle between two compartments (Kumagai and Hanada, 2019).
FFAT motifs bind to the major sperm protein (MSP, 120 residues) domain of vesicle-associated membrane protein-associated protein (VAP), a protein found in almost all eukaryotes. The MSP domain is linked by a flexible region to the cytoplasmic face of the ER (Skehel et al., 2000). The core of FFAT motifs is an extended peptide of seven residues, two of which bind into pockets in VAP (Figure 1(a); Kaiser et al., 2005). Prior to the engagement of the core, an acidic tract N-terminal to the core has a low-affinity charge-based interaction with the MSP domain (Furuita et al., 2010). All FFAT motifs that fitted the early, strict definition, have since been validated as interacting with VAP, except for one that is located in the lysosomal lumen. Subsequently, predicting new FFAT motifs has been made possible because the definition of FFAT motifs has been expanded through the discovery of new variants (Saita et al., 2009; Mikitova and Levine, 2012; Baron et al., 2014). Further sequence variation has been shown with substitution of anionic positions with serine or threonine (S/T in one letter notation), which allows reversible phospho-mimicking of motifs (Alpy et al., 2013; Kumagai et al., 2014; McCune et al., 2017; Johnson et al., 2018; Kirmiz et al., 2018).
Domain–SLiM interactions are hard to capture by standard protein–protein interaction techniques, and their discovery rate in databases is estimated at only ≤4% (Davey et al., 2017). Bioinformatics can predict new instances (Krystkowiak and Davey, 2017). Widening the definition of FFAT’s pattern allowed the prediction of many new human motifs, which were assessed against three criteria to reduce the number of false positives: (a) cytoplasmic location; (b) structure allowing formation of an extended loop, also described as an intrinsically disordered region (IDR); and (c) local conservation (Mikitova and Levine, 2012). Many of the FFAT motifs identified by bioinformatics have been verified in high-throughput protein interactomes (Hein et al., 2015; Huttlin et al., 2015), and some have been verified by specific mutagenesis (Costello et al., 2017; Kumar et al., 2018). To identify motifs that vary at multiple positions, a position weight matrix (PWM) can be used. The PWM for FFAT motifs produces a FFAT score for runs of 13 amino acids (7 core and 6 acidic tract preceding; Murphy and Levine, 2016). FFAT score varies from “perfect” motifs with FFAT score = 0, increasing with each suboptimal substitution (Figure 1(b); Murphy and Levine, 2016). Three sources of information contributed to the PWM: (a) residues in VAP interactors and their homologues, (b) the effect of substitutions on affinity for VAP, and (c) VAP-FFAT structures (Mikitova and Levine, 2012). A cutoff FFAT score ≤2.5 was chosen to reduce false positives, initially estimated at approximately 1% for this cutoff (Murphy and Levine, 2016). This PWM has been used to scan interactors of the two major human VAPs: VAPA and VAPB. These are 77% similar by sequence and share 80% of interactors (Huttlin et al., 2015), so they can be considered together. Forty percent interactors contained a motif with FFAT score ≤2.5, and 10% formed complexes with these, so 50% of VAP interactions are attributable to FFAT motifs (Murphy and Levine, 2016).
Here, we applied the PWM for FFAT motifs systematically across eukaryote proteomes using negative controls to estimate background. We predict that at least 0.8% of the proteome (50 proteins in budding yeast and 170 in humans) bind VAP. This estimate was confirmed by detailed consideration of a subset of yeast motifs. Some of the FFAT-positive proteins we predict act in compartments that have no known molecular link to the ER. As most known FFAT-positive proteins carry out their function while binding VAP, this indicates novel pathways of interorganelle communication.
Results
Proteins With Highly Optimal FFAT Motifs Are Already Known to Bind VAP
Every residue of every protein in six model eukaryotes (human, Drosophila melanogaster, Caenorhabditis elegans, budding yeast Saccharomyces cerevisiae, Arabidopsis thaliana, and Plasmodium falciparum) was scanned with the PWM we developed previously to identify FFAT scores for every sequence of 13 residues. The position with the lowest FFAT score in each protein was recorded as the FFAT score for that protein (Murphy and Levine, 2016). The overall distribution of FFAT scores was non-normal, and was similar across eukaryotes (Figure 2(a), and data not shown). Looking at proteins with extremely low FFAT scores, where VAP interactors are expected, the proteomes showed some variation, for example, Arabidopsis had fewer proteins with FFAT scores ≤1 (Figure 2(b)).
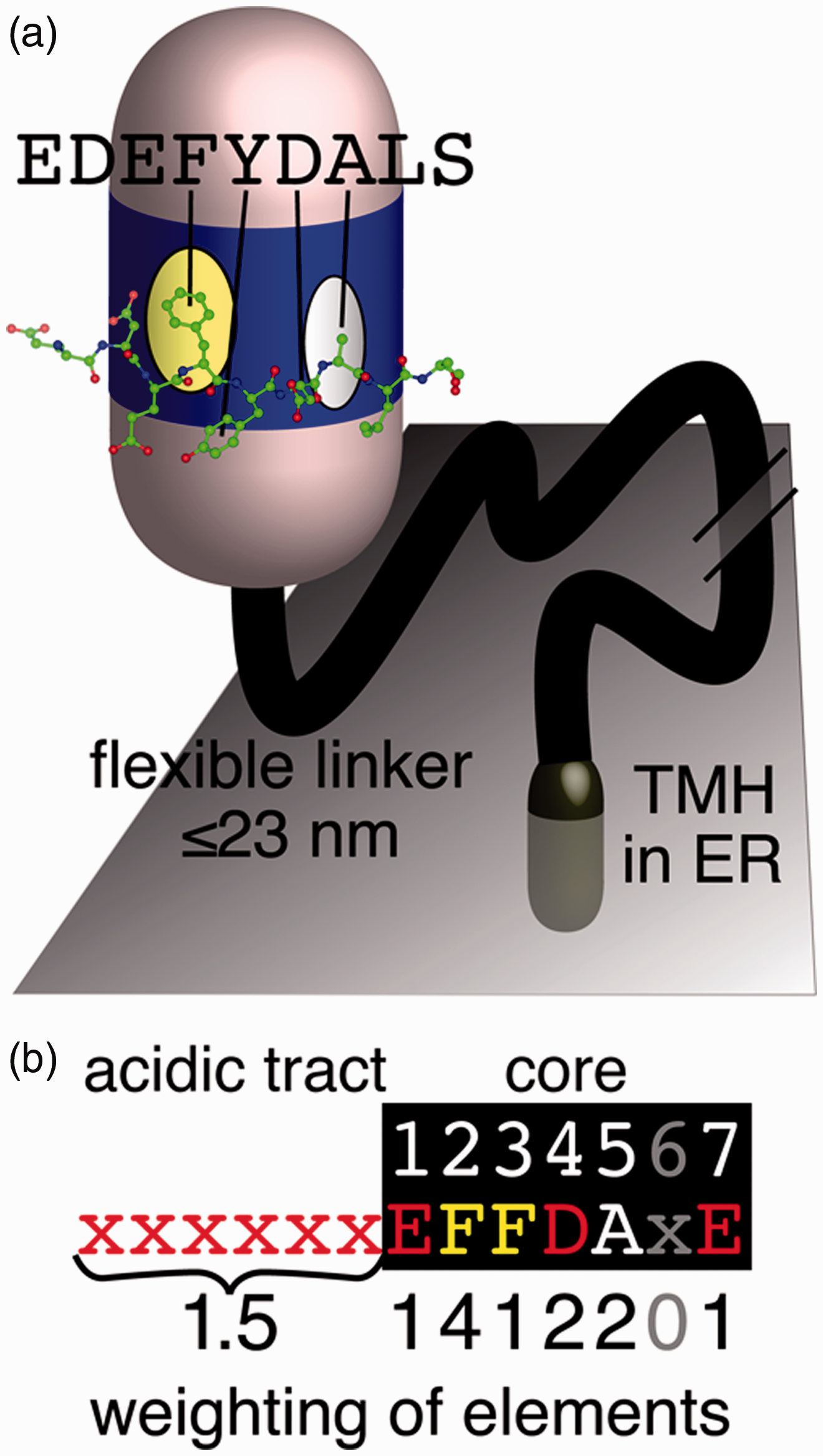
The conserved VAP-FFAT interaction. (a) A FFAT motif forms an extended loop that binds to a conserved basic face of the MSP domain of VAP. VAP also has a linker of approximately 100 residues that includes a coiled-coil section in some species and can maximally extend to ≥20 nm (not shown), and a tail-anchoring TMH in the ER membrane (Murphy and Levine, 2016). The basic face of VAP’s MSP domain contains two pockets: one of which binds an aromatic residue (F/Y) and the other binds a small noncharged residue (A, C, G, S, and T). These are the second and fifth residues, respectively, of the seven-residue core of the FFAT motif. Peptide from ORP1L FFAT peptide crystallized in complex with VAP in PDB:1Z9O (Kaiser et al., 2005). (b) The weights of the seven elements scored by the FFAT motif. An ideal motif (six upstream = acids, then seven core residues = EFFDAXE) has FFAT score = 0. Suboptimal substitutions at each position in the motif are assessed by the PWM: six elements are all the positions in the core except one, the seventh element is the overall acidity of the upstream residues (Murphy and Levine, 2016). TMH = transmembrane helix; ER = endoplasmic reticulum.

Three percent of eukaryotic proteins have FFAT scores ≤2.5. (a) Distributions of FFAT scores in human and yeast (Saccharomyces cerevisiae). FFAT scores are assigned as described in “Methods” section. The means and standard deviations are yeast: 4.6 ± 1.1, human 4.5 ± 1.1; the distributions are statistically similar (χ2 0.998). (b) Proportion of six eukaryotic proteomes at different levels of low FFAT score, from highly optimal (top, range 0.0–1.0) to less optimal (bottom, =2.5), including the average, unweighted for proportion of proteins in each of the 6 proteomes, and standard deviation. (c) Proportions of yeast and human proteins with motifs with highly optimal FFAT scores (range 0.0–1.0) showing if they are known interactors of VAP. (d) Proportion of proteins identified as binding VAP in yeast and human according to their FFAT scores (range 0.0–6.0). Inset shows detail in the range FFAT scores 1.5 to 3.0. Dotted line indicates the average across the two species. Identification of proteins as VAP interactors declines rapidly as FFAT score increases: 59% for FFAT score ≤1.0 (human 11/17, yeast 2/5), 8% for 1.0 < FFAT score ≤ 2.0 (human 17/180, yeast 3/62), and 3% for 2.0 < FFAT score ≤ 3.0 (human 11/440, yeast 5/146). FFAT = two phenylalanines in an acidic tract; VAP = vesicle-associated membrane protein-associated protein.
We next determined the correlation between low FFAT score and documented physical interaction with VAP, combining data from the two species with the best documented protein–protein interaction networks: human and yeast. For the human VAP interactome, we included VAPA, VAPB, and also MOSPD2. The latter is a newly described variant of VAP known to bind only a minority of the interactors of VAPA/B, possibly because it has not been studied in as much detail, or because the region of its MSP domain that binds FFATs is divergent from VAPA/B (Di Mattia et al., 2018). The three VAPs have 341 documented physical interactors (1.7% of proteome; Chatr-Aryamontri et al., 2017). Yeast has two VAPs, the major form Scs2p and a minor homologue Scs22p, with 74 physical interactors between them (1.2% of proteome). All of the proteins with FFAT score = 0.0 (two in yeast, six in human) are already known to bind VAP. This is to be expected because the sequences of proteins with the lowest FFAT scores were used to create our PWM, so by definition their FFAT scores are low. The two yeast proteins in this group are the transcriptional regulator Opi1p and the oxysterol binding protein-related protein (ORP) homologue Osh1p (Table 1(a) and (b)). For FFAT scores marginally higher (range 0.5–1.0), 2 of the 6 yeast proteins (Table 1(a) and (b)) and 13 of the 17 human proteins are documented to bind VAP (65% overall, Figure 2(c)). The motifs in the four missing human proteins (FRPD1, S22AF, TSYL2, and ORC2) were all identified previously by SCAN-PROSITE (Mikitova and Levine, 2012). The four missing yeast proteins (Bbc1p, Kri1p, Seg2p, and Ypr097wp; Table 1) are considered later.
Yeast Proteins With Motifs With FFAT Scores ≤1.5.

Note. FFAT = two phenylalanines in an acidic tract; ER = endoplasmic reticulum; VAP = vesicle-associated membrane protein-associated protein; ORP = oxysterol binding protein-related protein; PUF = protein of unknown function; VID = vacuolar import and degradation .
aFour motifs already known.
bFour proteins already shown to bind VAP.
cHHsearch was used to predict domains for all yeast proteins, using settings described previously (Fidler et al., 2016).
Few of the Many Proteins With Suboptimal FFAT Motifs Are Known to Bind VAP
The number of proteins containing FFAT motifs that score 1.5, 2.0, or 2.5 rose steeply, so that across the six model eukaryotes 3.5% of proteins have suboptimal FFAT motifs with such scores (Figure 2(b) and Supplementary Tables 1A, 2, and 3 ). With higher FFAT scores, the proportion already identified as interacting with VAP declined: 11%, 6.7%, and 2.7% for FFAT score = 1.5, 2.0, and 2.5, respectively (Figure 2(d)). This could reflect a genuine decrease in the proportion of proteins with these motifs that ever binds VAP. Alternately, the copurification and coprecipitation methods of major high-throughput studies (Hein et al., 2015; Huttlin et al., 2015) could be too harsh to identify interactions between VAP and suboptimal motifs (Davey et al., 2017). Interactions between domains and SLiMs tend to have lower affinity than the interactions between domain pairs (Kim et al., 2014).
Estimating the Proportion of Proteins With Suboptimal FFAT Motifs That Interact With VAP
Among the eukaryotic proteins that have suboptimal FFAT motifs, we used two approaches to estimate the proportion that are irrelevant, equivalent to random background. In the first approach, we analyzed species without VAP, which have no selective pressure for FFAT motifs, so they may act as negative controls. Organisms from the two noneukaryotic kingdoms (bacteria and archaea) have been used before for this purpose (Meszaros et al., 2012). In addition, we identified a small group of variant unicellular eukaryotes that lack VAP: Euglena gracilis, Spironucleus salmonicida, and Paratrypansoma confusum. The levels of suboptimal FFAT motifs were lowest in bacteria and similar in archaea and VAP-negative eukaryotes. For FFAT scores ≤1.0, the controls had approximately 10% observed motifs as VAP-positive eukaryotes. This proportion rose to 40% to 50% for motifs with FFAT score = 2.5 (Figure 3(a)).
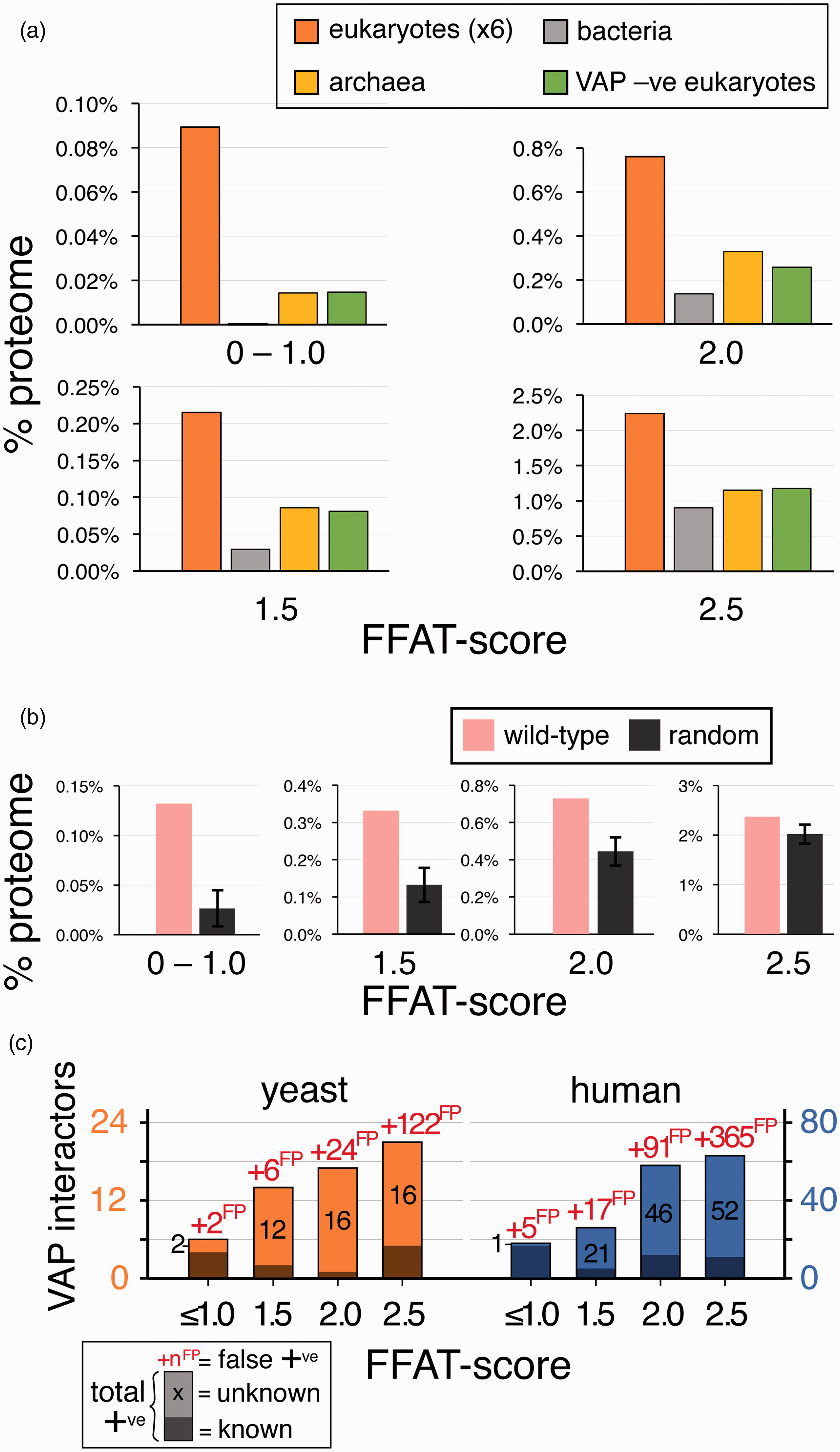
Comparison of the observed level of motifs with low FFAT scores with expected levels. (a) Levels of motifs with low FFAT scores in groups of bacteria and archaea, and VAP-negative eukaryotes across a range of low FFAT scores, from highly optimal (0.0–1.0, top left) to less optimal (=2.5, bottom right). In each case, average levels in eukaryotes are included for comparison. For species names, see “Methods” section. All data points are the average across all proteins in that class. (b) Levels of motifs with low FFAT scores in randomized yeast proteins (10 randomizations, showing means and standard deviations) compared with observed levels in yeast, from highly optimal (0.0–1.0, left) to less optimal (=2.5, right). (c) Predicted total number of FFAT motifs in yeast and human proteomes, based on the background of false positives (numbers in red as “xyzFP”) calculated from randomization data. Motifs are subdivided into those already known (dark shading) and those predicted (light shading, black numbers). FFAT = two phenylalanines in an acidic tract; VAP = vesicle-associated membrane protein-associated protein.
The second estimation of the background level of low FFAT scores was achieved by randomizing VAP-negative proteomes. To check this randomization acted as expected, we compared the numbers of motifs before and after randomization in VAP-negative proteomes, with the expectation that there would be no difference. Four of the 13 proteomes had more motifs after randomization. Among the nine others, we tested for an excess of motifs over background using the “N−1” χ2 test (Campbell, 2007) to compare total number of motifs at different levels of FFAT score: There was no significant excess of observed motifs over expected (lowest p value = 0.08, Supplementary Table 1B ). By comparison, all VAP-positive genomes had excess motifs with p values varying from 10−4 to 10−20 ( Supplementary Table 1B , bottom). This showed that the randomization is a useful way to estimate background.
The background from randomization of VAP-positive proteomes was 76% for all FFAT score ≤2.5, mainly because it was 85% for FFAT score = 2.5 (Figure 3(b)). This background is higher than the background estimated from VAP-negative organisms, and even this more conservative prediction of FFAT motifs still produces a minimum estimate that approximately 0.8% of proteins are positively selected above background to have motifs with FFAT score ≤2.5 (50 in yeast and 170 in human). The two background estimations agreed that the proportion of VAP interactors is higher where the FFAT score is lower, with rates of false positives starting low but rising steeply as FFAT score increases from 1.5 to 2.5 (40%–85%, respectively, Figure 3(c)).
Using Families of Fungal Orthologues to Determine Whether the Motif in an Individual Protein Is Subject to Preferential Conservation
To study VAP interactors in one organism, we chose yeast because its proteome is small and better documented than other eukaryotes. Inclusion criteria for determining if motifs are functionally relevant have been developed previously: (a) location in the cytoplasm, not inside an organelle or secreted; (b) predicted to form an extended loop, not in a helix or sheet, particularly if the secondary structural element is conserved and in a globular domain and if it has been confirmed experimentally (crystal structure, etc.); and (c) preferential conservation of motifs that are not excluded by the other two criteria among closely related homologues, ideally orthologues (Mikitova and Levine, 2012; Murphy and Levine, 2016).
This third criterion was previously carried out by manual examination of homologues, particularly of the key core residues of the motif (#2/4/5). Here, we developed the criterion to measure conservation statistically. Families of orthologues of each yeast protein have been created by tools such as OrthoDB (family size n = 18–700, median 150, Table 2(a); Kriventseva et al., 2015). We tested if the rate of occurrence of motifs among orthologues was significantly above background, which was calculated from randomized sequences, as done for proteomes above. Based on the statistical robustness of randomizing VAP-negative control proteomes ( Supplementary Table 1B), our cutoff for statistical significance of excess of motifs with FFAT at different scores (≤2.5/2.0/1.5/1.0) was p ≤ .01, with borderline excess where p values lie between .001 and .01, and confirmed excess for p ≤ .001.
We first tested this approach on the four yeast proteins with known FFAT motifs: Osh1/2/3p and Opi1p. As these sequences were used to create our PWM, by definition, their FFAT scores are low, nevertheless they illustrate the use of families of orthologues. For Osh1p (FFAT score = 0.0), which includes Osh2p (FFAT score = 1.0) as a paralogue in the same family, 99% of orthologues had motifs with FFAT score ≤ 2.5, indicating uniform conservation of the VAP-Osh1/2 interaction. By comparison, only 74% of Osh3p orthologues (FFAT score = 1.0) had motifs with FFAT score ≤2.5. Another 15% were short sequences representing fragments of longer Osh3p sequences that were readily identifiable in sequence databases. All of those contained motifs with low FFAT scores (data not shown). The remaining 10% Osh3 orthologues all had degenerate FFAT-like motifs at the same position as the motif in Osh3p. For example, Meyerozyma guilliermondii has GFVDAQD without acids upstream. Motifs with tryptophan substituted at Position 2 (which cannot arise from a point mutation of F or Y) were found both in Taphrinomycotina, a branch of the Ascomycota (Pneumocystis, Schizosaccharomyces pombe, etc.) and in Agaricomycetes, a branch of the Basidiomycota (Fomitopsis, Coprinopsis, etc.), but not in the closely related Ustilaginomycotina. As these Position 2 = tryptophan FFAT-like motifs are evolutionarily separated (Stajich et al., 2009), this substitution appears to have evolved in Osh3 orthologues on multiple occasions.
In the family of orthologues of Opi1p (FFAT score = 0.0), only 13% of proteins had a motif with FFAT score ≤ 2.5. This created a statistical excess (p = 10−4 for motifs with scores ≤2.0) of far less statistical significance than for Osh3 (p = 10−26) or Osh1 (p = 10−44). Many of the Opi1 orthologues have an alternate name: “related to clock-controlled protein 8 (ccg-8).” ccg-8 is a Neurospora crassa transcription factor that regulates an aspect of lipid metabolism different from that regulated by Opi1p in S. cerevisiae (Xue et al., 2019). FFAT motifs are absent from all proteins named ccg-8. This indicates that the fungal orthologue families have subfamilies with diverged function. To address this, we first looked at a narrower orthology family from the Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology database (Kanehisa et al., 2004), excluding all ccg-8s. This had a higher incidence of FFAT motifs (28%), but the smaller size of the family meant that there was no greater statistical significance ( Supplementary Table 4 ). We then created a custom-made family using BLAST to identify proteins closer to the budding yeast Opi1p, but with only 15% to 20% overall sequence identity, and no selection for the FFAT motif in the BLAST search. These proteins had a 48% incidence of motifs with FFAT scores ≤2.5 (most FFAT scores = 0.0) and a much higher statistical significance (p = 10−10, Supplementary Table 4 ).
Overall, this indicates that low discovery rates of a motif among Opi1p orthologues did not result from a lack of sensitivity (i.e., not false negatives). Instead, there appears to have been evolutionary pressure to interact with VAP only in some ecological niches, reducing the statistical significance of excess motifs in a family that covers a wide range of organisms.
Ten New Predicted VAP Interactors in 23 Yeast Proteins Identified With FFAT Scores 1.0 or 1.5
To predict new FFAT motifs, first we considered proteins with FFAT score = 1.0 (n = 4) and = 1.5 (n = 20; Table 1(a) and (b)). Among these 24 proteins, Erb1p had previously been identified as an interactor of yeast VAP (Ho et al., 2002). This protein contained a motif that met all our criteria (p = 10−7), strongly suggesting a VAP-FFAT interaction in this family. Among the newly identified 23 proteins, our criteria excluded 13 of the candidate proteins with motifs with FFAT scores = 1.5 (Table 2(a)). Seven proteins were excluded by location or structure of the region containing the motif. Five of these had weak (or no) conservation (Asg7p, Exo1p, Ost1p, Pet123p, and Ste13p; Table 2(a)). In contrast, two highly homologous proteins (Tub1p and Tub3p) were excluded because the motifs are in a helix even though the motifs were universal among their orthologues. All α-tubulins, including in humans, apparently have the same inaccessible FFAT-like sequence (Murphy and Levine, 2016).
Application of Exclusion Criteria to Potential FFAT Motifs in Yeast.

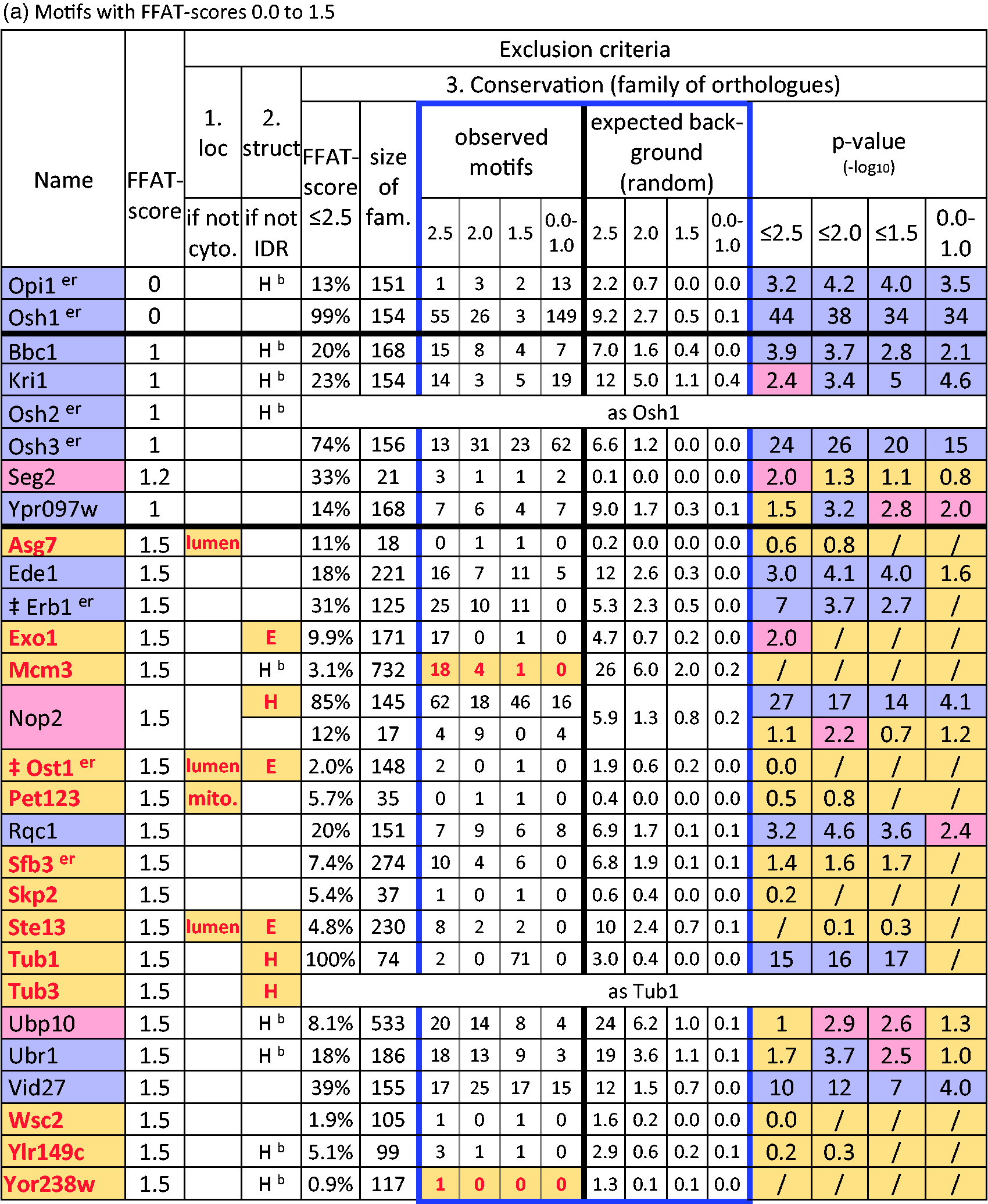

Note. Sequences in order of increasing FFAT score. Red text, yellow fill indicates motif excluded and exclusion criterion: Location: lumen = translocated; mito. = mitochondrion; empty: cytoplasm. Structure: H helix, E sheet (red confirmed, black predicted, Hb indicates helix between domains); empty: loop. Conservation: exclusion either if observed motifs ≤ expected (numbers of motifs in red) or if all p values for excess motifs with FFAT score ≤2.5/2.0/1.5/1.0 ≥ 0.01 (i.e., −1*log10 p value < 2.0). Display of p value: nonsignificant (p > 10−2): yellow; borderline (10−3 ≤ p < 10−2): pink; confirmed excess (p ≤ 10−3): blue. The most significant χ2 is used to color the protein name. χ2 is not valid if all expected values = 0, and not given if only one motif was observed (“/”). NOP2 has motifs of 2 main types: one in a confirmed helix and others in a loop. Kxd1 and Tor1 have two motifs with FFAT score = 2.5. Superscripter = known on ER; FFAT = two phenylalanines in an acidic tract; IDR = intrinsically disordered region.
aErb1 and Ost1 have been previously identified as binding Scs2, yeast vesicle-associated membrane protein-associated protein (VAP) (Chatr-Aryamontri et al., 2017).
bEpo1, Num1, and Pbi1 have been reported to bind Scs2p, but potential FFAT motifs have not been tested (Chao et al., 2014; Riekhof et al., 2014).
Six proteins were excluded because they failed the conservation criterion. Two of these had fewer motifs with low FFAT scores in the orthologue family than expected: Mcm3p and Yor238wp (Table 2(a)). Three had low numbers of motifs similar to the expected numbers (Skp2p, Wsc2p, and Ylr149cp). The final protein, Sfb3p, had a nonsignificant excess of motifs with FFAT score 1.5 to 2.5 within a large family of orthologues (7% of 274, p = 0.02 = 10−1.7, Table 2(a)). Limiting Sfb3 orthologues to the narrower group of Ascomycota increased the rate of occurrence of motifs with low FFAT scores but reduced the significance (16% of 43, p = 0.06 = 10−1.2, Supplementary Table 4 ), so there is a statistical trend for an excess of motifs in Sfb3 orthologues, but they fail to meet the statistical criterion.
After these exclusions, 10 new proteins with FFAT score = 1.0 or 1.5 met all the criteria for new predictions of FFAT motifs: Bbc1p, Ede1p, Kri1p, Nop2p, Rqc1p, Seg2p, Ubp10p, Ubr1p, Vid17p, and Ypr097wp (Table 2(a) and described later). The positive prediction rates for motifs with FFAT scores = 1.0 and 1.5 were 100% and 32%, respectively. The latter is lower than the frequency of motifs we had predicted (60%), but with the low numbers involved the difference is not statistically significant.
Three of the predictions only reached borderline statistical significance (p value between .001 and .01): Seg2p, Ubp10p, and Nop2p. For Seg2p, the low significance was most likely because the protein family is small (n = 21). Ubp10p orthologues showed a small excess of motifs over background with FFAT scores 1.5 to 2.0 in a large orthologue family. Like Opi1p, the motif showed more significant conservation in a subfamily of Ubp10 orthologues from Ascomycota only ( Supplementary Table 4 ). Nop2 was unique among the proteins we studied in having two types of motifs with low FFAT scores in different orthologues: The vast majority (97%), including in S. cerevisiae, had a motif in a helix near the start of the RNA methyltransferase domain (Yakubovskaya et al., 2012). A minority (12%) had a second motif with FFAT score as low as 1.0 in a highly anionic IDR, and one orthologue had a motif after the methyltransferase domain. After excluding motifs with helical structure, the minority in IDRs were still significantly in excess over background (Table 2(a)). This predicts that Nop2 orthologues functionally interact with VAP. This implies that the potential motif in Nop2p is conserved to interact with VAP (see “Discussion” section).
These 10 proteins with putative FFAT motifs, together with Erb1p, are implicated in diverse cellular activities (Figure 4). Two (Rqc1p and Ubr1p) have diverse cytoplasmic functions that are already linked to the ER. Four are involved in processes that have previously been linked to VAP-FFAT interactions: (a) Bbc1p and Ede1p act in clathrin-coated endocytosis, which is also linked to VAP and ORPs via Myo2p (Encinar Del Dedo et al., 2017); (b) Ypr097wp localizes to the vacuole (the yeast equivalent of the lysosome) and resembles a sorting nexin, so it may parallel the binding of human SNX2 to VAP (Dong et al., 2016); and (c) Vid27p localizes to puncta resembling late (or multivesicular) endosomes and it has an ill-defined function in endocytosis, similar to human proteins at ER–endosome contacts (Rocha et al., 2009; Saita et al., 2009). The five other hits are involved with cellular components not previously known to be linked to VAP: Erb1p, Kri1p, Nop2p, and Ubp10p assemble ribosomes in nucleoli; Seg2p stabilizes eisosomes.

Cellular distribution of proteins with FFAT motifs. Likely location of 24 yeast proteins with FFAT motifs, including 4 known, all 11 with FFAT scores ≤1.5, and a sample of 9 others with FFAT scores 2.0 or 2.5. Each protein is colored by the statistical significance of the χ2 test of the number of motifs in its orthologue family over background (Table 2 and see left hand Key): pink = borderline significance, increasingly dark gray = increasing significance. To reflect known localization to multiple contact sites, Ent3, Epo1, Osh1, Sec2, and Vps13 appear more than once. Contacts between the ER and other cellular components are colored according to the level of prior knowledge (see right hand Key): light yellow (VAP-FFAT bridge known in yeast), dark yellow (VAP-FFAT bridge known, but not in yeast), orange (contact known, but no VAP-FFAT bridge in any cell type), and red (contact unknown). Note that no yeast FFAT proteins have been predicted yet that target lipid droplets or peroxisomes. MVE = multivesicular endosome; FFAT = two phenylalanines in an acidic tract; VAP = vesicle-associated membrane protein-associated protein; ER = endoplasmic reticulum.
Seven New Predicted VAP Interactors in a Sample of 28 Yeast Proteins Identified With FFAT Scores 2.0 and 2.5
Even though yeast has a relatively small proteome for a eukaryote, there are too many proteins with FFAT scores 2.0 and 2.5 (n = 44 and 143, respectively, Supplementary Table 1A ) to consider them all here in detail. Therefore, we sampled these groups ( Supplementary Table 1C ). The basis for this sampling was firstly to include all proteins previously identified as interactors of yeast VAP and secondly to randomly sample the remaining proteins.
Among 13 proteins we examined with FFAT score = 2.0, 1 had previously been shown to bind yeast VAP: Utp9p. This protein contained a motif that just failed the statistical significance criterion possibly because the family of orthologues is small (n = 13, p = 0.04 = 10−1.5) suggesting that the VAP-FFAT interaction is conserved at a rate below the sensitivity of our approach. Among the other 12 proteins in this group, 3 were excluded by structure or location and 4 by lack of conservation. Five met all criteria for predicting an active motif (p < 0.01), two with borderline statistical significance (Efr3p and Sxm1p), and three with the stronger significance (Ecm13p, Ent3p and Msh6p, Table 2(b), top). Changing the orthologue families for Efr3p and Sxm1p to Ascomycota increased the proportion of proteins positive for motifs but did not increase their statistical significance ( Supplementary Table 4 ). For the sample of proteins with motifs with FFAT score = 2.0, the positive prediction rate 5/12 (42%) was close to the predicted rate (40%, Figure 3(b) and (c)). This implies a further 13 motifs would be found among the 31 proteins with FFAT score = 2.0 we did not study.
Among 19 proteins with FFAT score = 2.5 we examined, 3 (Epo1p, Num1p, and Pbi1p) have been shown to bind yeast VAP previously, though their predicted FFAT motifs have not been tested (Chao et al., 2014; Riekhof et al., 2014). Our analysis predicted that both Epo1p and Num1p have motifs that meet all the criteria for being an active FFAT motif, albeit with borderline significance in terms of conservation (Table 2(b), bottom). However, the motif in Pbi1p is in a known helix and did not meet the conservation criterion (Table 2(b), bottom). Among the other 16 proteins, 6 proteins were excluded by structure or location criteria and 8 by lack of sequence conservation. Four of these just missed our significance criterion (p ≤ 0.05 but p > 0.01, like Utp9p see earlier), including integral ER proteins (Mga2p and Ubp1p), a trafficking protein (Kxd1p) and a polarity protein (Aim44p; Table 2(b)). A subfamily of sequences more closely related to Mga2p showed no greater statistical significance ( Supplementary Table 4 ). This left two proteins that met all the criteria for predicting a FFAT motif: Sec2p and Vps13p (Table 2(b), bottom). The positive prediction rate among motifs with FFAT score = 2.5 (2/16, 13%) is similar to our proteome-wide estimate (15%, Figure 3(b)). Applying this rate to all other 124 yeast proteins with FFAT scores = 2.5 would produce a further 16 motifs.
The nine predicted motifs with FFAT scores 2.0 or 2.5 are in proteins localized to multiple cellular components that communicate with the ER, functioning in membrane traffic (Ent3p, Sec2p, and Vps13p), cytoskeleton and polarity (Epo1p and Num1p), PI4P synthesis at the plasma membrane (Efr3p), nuclear components (Msh6p and Sxm1p), and a cytoplasmic protein of unknown function (Ecm13p; Figure 4).
These findings create an overall picture of the number of FFAT motifs in yeast: 4 previously known, 3 in 5 partially characterized VAP interactors, and 17 we predict by detailed study. Extrapolating from the sample of proteins with FFAT scores 2.0 to 2.5 indicates the remainder of the proteome contains a further 29 proteins with motifs that meet all our criteria. However, with 155 proteins yet to be studied in these groups, this number has considerable margin for error. Thus, the bioinformatics approach predicts that the number of yeast proteins with FFAT motifs is approximately 50 to 55 (0.9% of the whole proteome), >10-fold higher than currently tested and verified.
Comparison of These Predictions With the MEME Suite Automated Pipeline
We next asked if an automated pipeline for SLiM discovery would perform just as well as the custom-made PWM. For this, we used the MEME Suite, which creates ungapped SLiMs from sequences and then searches for other occurrences in unrelated sequences (Bailey et al., 2009), and has been used to identify motifs in contact site biology (Bean et al., 2018). Seeded with a family of FFAT-positive proteins (here we used ORPs), MEME identified their shared FFAT motifs as the seven-residue core, together with acidic flanking residues both upstream and downstream ( Supplementary Table 5A ). We then used the FIMO (find motifs) module in MEME to identify motifs that fit this statistical model. To understand the significance of hits at the whole proteome level, we searched both the actual yeast proteome and randomizations of the yeast proteome, as we had done for the PWM. Motifs that match the MEME model most closely (with p values as low as 2 × 10−12) were only found in the actual proteome. With increasing p values, the background hit rate increased, eventually approaching that of the observed proteome ( Supplementary Table 5B ), which is the same as we had observed for our PWM. The excess number of strong hits to the matrix in the actual proteome compared with random background was 37 ± 16 (number of randomizations = 4), which is approximately 70% of the level estimated by the PWM.
At the level of individual proteins, there was variation after the sixth hit ( Supplementary Table 5C ), and the rank order of yeast hits correlated only partially with that produced by FFAT scores (r = 0.3). Approximately 20% of the top 3% of MEME hits contained residues that would prevent productive interaction with VAP (e.g., L2, E5). Thus, while the automated tool MEME generated a list of hits with much less work than our approach, in detail its predictions are less informative.
Widespread Motifs Found in Nonfungal Homologues in 30% of Yeast Proteins With a Motif
Finally, we looked for conservation of yeast FFAT motifs in other species for the 20 proteins with newly predicted FFAT motifs; 14 have nonfungal families, of which 6 had a clear excess of FFAT motifs (Table 3). Excess FFAT motifs occur in all eukaryotic kingdoms for Vps13, Erb1 (alternate name BOP1 in other species), and Ubr1 (orthologue called UBR4 in mammals; data not shown). For Nop2, motifs are found in animal homologues but not significantly in plants. For Kri1, motifs are found in plants not animals. Vid27 homologues are only found in plants and alveolates, both of which have more motifs than fungal homologues. Twelve of the 20 new yeast hits have human homologues, three of which have motifs with low FFAT scores: VPS13 = 0.0 and NOP2 and UBR4 = 2.5.
Using Fungal Hits to Search for FFAT Motifs in Nonfungal Homologues.


Note. Alternate names for human homologues and range of species searched are in column 3. For some families, different species groups were searched separately (indicated in column 4). Coloring, p values, and exclusions as in Table 2. Where different groups of species within an overall family have different significance levels, only the most significant group is colored (column 4). FFAT = two phenylalanines in an acidic tract. “/” indicates that χ2 is not valid or only motif was observed.
Discussion
We studied one SLiM in one organism in detail. The potential FFAT motifs we predicted need verification of ER targeting dependent on VAP. One question we cannot exclude is whether these sequences might be selected for another reason. Many eukaryotic SLiMs combine aromatic and charged residues. One overlaps with the FFAT motif: “FxDxF,” which sorts endocytic cargo (Brett et al., 2002; Dinkel et al., 2016). It is predicted that many more SLiMs will be discovered in the future (Tompa et al., 2014), and some may overlap FFAT more closely.
Maximizing Accurate Detection With a PWM
There are some issues with our PWM. First, it overvalues the known VAP interactors used to create it. Position 3 of FFAT motifs exemplifies this: while the original motifs varied little here, crystal structures showed little interaction with VAP (Kaiser et al., 2005), and divergent residues were later found (Saita et al., 2009; Baron et al., 2014). This raises a second problem: The PWM weights need verification by systematic measurement of how different substitutions affect affinity, especially combinations of multiple substitutions, which have only rarely been studied (Furuita et al., 2010; Mikitova and Levine, 2012). Phage display of peptides, particularly variants of FFAT, might address this in the future. For example, the finding of repeated substitution of F2 with tryptophan in Osh3 suggests the possibility that tryptophan at this position is not completely inhibitory to VAP interaction. This could be tested experimentally.
Searching for Motifs in Orthologues
Application of a PWM to large families of orthologues provided evidence that powerfully influenced the assessment of motifs. In some cases, the OrthoDB families were too inclusive: subfamilies of proteins more closely related to the Opi1p and Ubp10p produced more excess motifs that were more statistically significant. Therefore, improved accuracy in curating orthologue families would increase sensitivity of this approach.
Nop2p is unique because the yeast protein, like almost all fungal NOPs, has a motif that fails to meet our criteria because it occurs in a helix in the methyltransferase domain. However, other motifs in a minority of Nop2 orthologues cast doubt on the exclusion. These additional motifs in an anionic IDR (Figure 5(a)) were significantly in excess over the background of the whole family (p = .006, Table 2(a)). This has implications for the motifs in the methyltransferase domain both in fungi (consensus EFFEANE) and mammals (hNOP2 called NSun6, consensus EFLEANE), but not in plants (consensus ELIEAFE) and bacteria (consensus ALLAALN). One explanation for the motif in yeast and human is that its function is unrelated to VAP in the methyltransferase domain. An alternative explanation is that the first helix of the domain identified in crystal structures is dynamic (Figure 5(b)) and can switch to an unstructured loop. This scenario speculates that sequences that match FFAT motifs have been positively selected in opisthokonts in a region with variable structure that is permissive for FFAT formation. This would explain the definite but low rate of motifs with low FFAT scores in bacterial NOP2 homologues (Table 3).

The FFAT motif in Nop2p is located in a helix. (a) FFAT motifs occur at three sites in the fungal family of Nop2 orthologues. Top: overall domain map, showing the positon of motifs relative to the central methyltransferase domain and to the intrinsically disordered N-terminus, with its conserved basic (blue) and acidic (red) IDRs. The three locations for the 13 residues of motifs with FFAT scores ≤2.5 are indicated with yellow bars, showing the proportion of orthologues they are found there, including one motif found to the C-terminus of the folded domain. Bottom: the methyltransferase domain is divided into three parts: N-terminal extension (“N”, gray, dashed border) ending in the FFAT motif core (7 residues, dark gray), RNA Recognition Motif (RRM, blue), and catalytic core (rainbow green to red); the domain associates with a PseudoUridine synthase and Archaeosine transglycosylase (PUA) domain (rainbow blue to green, dotted border). In yeast, the PUA domain is in a separate protein, encoded by Nip7p, while animal homologues contain a PUA domain in cis. (b) Side and top views of methyltransferase domain of NSun6, a human homologue of Nop2, shown with colors and borders as in (a), from crystal structure 5WWQ (Liu et al., 2017). Four residues of the FFAT motif are folded into a single turn of a helix and then three in a linker to the RRM motif. FFAT = two phenylalanines in an acidic tract.
Estimates of the Background Rate of FFAT Motifs
Background levels of FFAT motifs were estimated from VAP-negative proteomes and sequence randomization. The main issue with VAP-negative proteomes is their variability. For example, 4.5% of proteins in the halophile archaeon Haloterrigena turkmenica have FFAT scores ≤2.5. The level of low FFAT scores (more than eukaryotes) was not changed by randomizing the proteome (data not shown). This effect is associated with halophiles having high proportions of acidic residues and few bases to survive in their highly saline (2–4M salt) habitat (Lanyi, 1974). VAP-negative eukaryotes may represent better controls than prokaryotes, but they are separated from the bulk of eukaryotes by long phylogenetic branches, so their proteomes may vary for other unknown reasons.
For our randomizations, all residues in each protein were treated the same. In the future, we might attempt to improve this, randomizing residues in folded domains separately from those in IDRs, which are enriched for charged residues (Davey et al., 2012). A different way to avoid mixing IDRs with folded domains is to randomize within tiled windows longer than the motif. However, sequences rich in D/E with a minority of F/Y may produce a low FFAT score randomly. For example, the FFAT score of . . . DDDDDDD[F/Y]DD@DD . . . (where @ is A/C/S/T) is 1.0 to 1.5. Therefore, sliding windows would overestimate background levels of FFAT motifs because of their inherent repetitive nature.
Evolution of SLiMs
The ability of FFAT motifs to arise simply by the appearance in an acidic tract of F/Y and a small residue at +3 shows how FFAT motifs might evolve, especially as Y and A can both result from point mutations of D codons. We assume that motifs that occur in isolation have evolved relatively recently. Like any mutation, they may have no productive function initially and could be counterselected if deleterious. This may have occurred in the highly anionic motor protein Dyn1, orthologues of which have many fewer motifs than the same sequences randomized.
Among the eukaryotes we studied, A. thaliana lacks proteins with the lowest FFAT scores (Figure 2(b)). Could the plant VAP-FFAT interaction be marginally different from yeast/human (opisthokonts)? Comparison of motifs in A. thaliana with those in yeast and human showed that the cores were similar, but upstream residues were less acidic in plants (data not shown). This suggests that plant VAP might have a compensatory difference from yeast and human VAP.
Can VAP Bind Approximately 0.9% of the Proteome?
There are pleiotropic phenotypes in cells lacking VAP which are consistent with dysfunction of many pathways in combination (Loewen and Levine, 2005; Di Mattia et al., 2018). However, the finding that cells still grow without VAP implies that loss of VAP-FFAT interactions mainly leads to reduced efficiency, which can be compensated by over-expression (Hanada et al., 2003). There are far less copies of VAP than the sum of its interactors, so only a fraction of FFAT motifs can be active at any one time. Part of the explanation might be that FFAT motifs interact only weakly (micromolar Kd), so they only bind VAP when multimerized. In addition, they may be masked by intramolecular interactions regulated by post-translational modifications (Alli-Balogun and Levine, 2019), as indicated by considerable diffuse background of green fluorescent protein-tagged FFAT-positive proteins (Breker et al., 2014). If the active FFAT motifs in total saturate VAP, then competitive binding will depend on both affinity (related to FFAT score) and avidity determined by local concentration. Thus, interaction may favor proteins concentrated near the ER, such as on an adjacent organelle.
FFAT Motifs at Contact Sites in Yeast
Most of the proteins with FFAT motifs newly predicted here are located on organelles that are already known to use VAP-FFAT interactions to communicate with the ER either in yeast or other cells (Figure 4) (Phillips and Voeltz, 2016). Some are involved in functions apparently outside the ER, but their location allows them to access it, for example, the intranuclear proteins Sxm1 and Msh6, which can access VAP that diffuses to the inner nuclear envelope (Beilharz et al., 2003; Brickner and Walter, 2004; James et al., 2019).
There are two yeast organelles that contain proteins with putative FFAT motifs where ER contact is not well-characterized: nucleolus and eisosome. For the nucleolus, we had four hits. Among these, Erb1p and Nop2p localize partly to the nuclear envelope, so may be constitutively associated with VAP, while Kri1p and Ubp10p are diffuse in the nucleolus (Breker et al., 2014). For the eisosome, Seg2 has a motif with a low FFAT score as does its paralogue Seg1p. Eisosomes are grooves 50 nm deep and 1 µm long in the yeast plasma membrane that sense tension (Berchtold et al., 2012). The majority of eisosomes are well separated from the nearest element of the cortical ER network (150–250 nm). This has been the basis for considering eisosomes to be unlinked to the ER (Stradalova et al., 2012). However, approximately 20% eisosomes are ≤50 nm distant from cortical ER (Stradalova et al., 2012), making it possible for Seg2p or Seg1p to bridge from eisosomes to ER. Mutating putative FFAT motifs in both Seg1/2p could probe this potential ER–eisosome bridging.
Contact Sites in Humans and Beyond
Conserved ER–nucleolar relationships are already known for the integral inner nuclear envelope protein RRS1 both in yeast (Horigome et al., 2011) and human cells, where it mediates targeting of ER proteins to nucleoli (Sutherland et al., 2004; Carnemolla et al., 2009). We found an excess sequences with low FFAT scores in nucleolar proteins in different kingdoms: ERB1 in all eukaryotes, KRI1 in fungi and plants, and NOP2 (with the caveats above) in fungi and animals (Table 3). These should be the focus of future study into the relationship between the ER and nucleolus. Given that proteins with FFAT motifs typically engage VAP when they function to form bridges between the ER and other compartments, we speculate that there is ER–nucleolar bridging. ER–nucleolus bridging in yeast is easy to conceptualize because the nucleolus forms a crescent adherent to approximately 50% of the inner nuclear envelope (Thiry and Lafontaine, 2005). By contrast, nucleoli in plants and animals are spherical structures deep in the nucleoplasm. Despite this, ER–nucleolar contact could be achieved by the tubular nucleoplasmic reticulum, which extends from the inner nuclear envelope deep into the nucleus of animal and plant cells (Malhas et al., 2011; Ohsaki et al., 2016). Our data suggest that nucleoplasmic reticulum may directly contact nucleoli. Alternatively, there may be no bridging, and the nucleolar proteins with FFAT motifs have inactive pools targeted to the nuclear periphery. Therefore, motifs with low FFAT scores in proteins with unexpected locations (nucleoli and likewise eisosomes) prompt further study of the relationship between the ER and other organelles.
Methods
Producing FFAT Scores
The scoring system is identical to that used previously (Murphy and Levine, 2016). In brief, to produce FFAT scores, suboptimal substitutions in any core of seven residues were allocated a score in a PWM (Table 4(a)). Some sequences in databases contain undetermined residues (identified as “X”). These were scored as inhibiting VAP binding. The six residues upstream were assessed as a single unit for charge (Table 4(b)), and the estimate of overall negative charge was scaled (Table 4(c)) and combined with the core to create the FFAT score. This represents the sum total of suboptimal scores across 13 continuous residues. FFAT scores were binned into 0.5 unit bins: “0” = 0 to 0.25, “0.5” = 0.251 to 0.75, “1.0” = 0.751 to 1.25, and so on. For proteomes and families of sequences, the scoring system was enacted in Python. The code for the scripts will be made available on GitHub or on request from the authors.
Suboptimal Scores for Substitutions in Core of FFAT Motif.

Note. (a) Position weight matrix applied to core to convert sequence into scores and (b) and (c) estimation of charge in six residues in possible acidic tract upstream of core and conversion into scores.
Sequences
Proteome-wide sequences were obtained from Uniprot. Control proteomes were as follows: bacteria—Escherichia coli, Mycobacterium tuberculosis, Staphylococcus aureus, and Mycoplasma hominis; archaea—Pyrococcus horikoshii, Methanosarcina mazei, Thermoplasma acidophilum, Archaeoglobus fulgidus, Sulfolobus solfataricus, and Methanocaldococcus jannaschii; and VAP-negative eukaryotes—Euglena gracilis, the diplomonad Spironucleus salmonicida, and the kinetoplastid Paratrypansoma confusum. Randomization was carried out within each protein, repeated 10-fold to obtain a mean and standard deviation for FFAT scores. Families of proteins orthologous to yeast hits were obtained from Uniprot using the OrthoDB identifier of the yeast protein ( Supplementary Table 1D ). All lists were filtered to remove entries flagged as fragments and also with “identity: 0.9” filter, or “identity: 0.5” if there were more than 1,000 sequences. Where no OrthoDB family was available (Ypt7 and Seg2), we searched for homologues using BLAST filtered to nr90 (Ypt7p), or for iterative searches we used HHblits three iterations (Seg2p) at the Tuebingen Toolkit (Zimmermann et al., 2018). Similarly, subfamilies made by KEGG and Orthologous MAtrix (OMA) (Kanehisa et al., 2004; Schneider et al., 2007) used the KEGG/OMA identifiers. For oxysterol binding protein homologues to submit to MEME, nonredundant proteins containing the Pfam domain PF01237 (Oxysterol_BP) were downloaded from Uniprot and submitted (at meme-suite.org/tools/meme), with the resulting matrix forwarded to FIMO (http://meme-suite.org/tools/fimo; Bailey et al., 2009).
Analysis of Potential Motifs in Yeast
Criteria applied to each potential FFAT motif included two used previously: location and secondary structure (Murphy and Levine, 2016). Location was determined by combining two sources of data: (a) topology prediction from signal sequences (Signal4.0) and transmembrane domains (TMHMM2.0 and TOPCONS; Kahsay et al., 2005; Petersen et al., 2011; Tsirigos et al., 2015) and (b) reconsideration of topology predicted in (a) in the light of functional requirements implied by domain analysis in HHsearch (Soding et al., 2005), which is available online at the Tuebingen Toolkit as HHpred (Zimmermann et al., 2018). Predicted secondary structure was in three states (helix, sheet, and unstructured loop), which was produced by PSI-PRED 3.0 as part of HHsearch (Buchan et al., 2013).
For the third criterion, conservation of the motif in closely related species, excess motifs with low FFAT scores in families of orthologue were estimated by comparing numbers of observed motifs with FFAT scores ≤2.5 (and ≤2.0, ≤1.5, and ≤1.0 in parallel) with the average numbers of motifs in randomized proteins by the “N−1” χ2 test that compares the (ad−bc)2(N−1)/mnrs with the χ2 distribution with one degree of freedom, as described (Campbell, 2007). Multiple incidences of motifs with FFAT scores ≤2.5 per protein were included, that is, statistics refer to every residue, with no reduction to the most optimal FFAT score per protein. The most significant of the four tests (excess ≤2.5, ≤2.0, ≤1.5, or ≤1.0) was assumed to apply (Tables 2 and 3). A cutoff p ≤ 10−2 was used (Table 2, final four columns: cutoff ≥2 when expressed as –log10). The values of p between 10−2 and 10−3 were considered borderline. Any protein family with only one motif or fewer motifs than the background at a particular FFAT score was excluded as having too low numbers to assess, and with only two motifs it was considered of borderline significance. The level of motifs with low FFAT scores in all randomized families together was similar to that in the randomized yeast proteome (data not shown).
For data mining, documented physical interactions of VAPA/VAPB/MOSPD2 and Scs2/Scs22 were obtained from BIOGRID. Where published literature is not informative on intracellular targeting, we used high-throughput localization studies at the LOQATE database (Breker et al., 2014), which includes genome-wide libraries of all yeast proteins tagged with fluorescent proteins at both N- and C-termini (Weill et al., 2018).
Analysis of Potential Motifs in Nonfungal Relatives of Yeast Hits
Twenty lists of nonfungal homologues of 14 yeast hits were constructed from UniProt Reference Clusters (UniRef, nonredundant as above) based either on OrthoDB families or on combinations of domains ( Supplementary Table 1E ). Fragments were removed, and sequences were filtered for nonredundancy as above. Observed motifs with low FFAT scores were compared with the number of motifs after sequence randomization (n = 10), including multiple incidences per protein as above.
Supplemental Material
CTC883136 Supplemental Material1 - Supplemental material for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum
Supplemental material, CTC883136 Supplemental Material1 for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum by John A. Slee and Timothy P. Levine in Contact
Supplemental Material
CTC883136 Supplemental Material2 - Supplemental material for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum
Supplemental material, CTC883136 Supplemental Material2 for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum by John A. Slee and Timothy P. Levine in Contact
Supplemental Material
CTC883136 Supplemental Material3 - Supplemental material for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum
Supplemental material, CTC883136 Supplemental Material3 for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum by John A. Slee and Timothy P. Levine in Contact
Supplemental Material
CTC883136 Supplemental Material4 - Supplemental material for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum
Supplemental material, CTC883136 Supplemental Material4 for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum by John A. Slee and Timothy P. Levine in Contact
Supplemental Material
CTC883136 Supplemental Material5 - Supplemental material for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum
Supplemental material, CTC883136 Supplemental Material5 for Systematic Prediction of FFAT Motifs Across Eukaryote Proteomes Identifies Nucleolar and Eisosome Proteins With the Predicted Capacity to Form Bridges to the Endoplasmic Reticulum by John A. Slee and Timothy P. Levine in Contact
Footnotes
Acknowledgments
The authors thank Martin Frisher (Keele University) and Matt Hayes (UCL Institute of Ophthalmology) for helpful discussions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the Bioinformatics and Biological Resources Fund of the Biotechnology and Biological Sciences Research Council UK (BB/M011801/1).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.