
Abstract
A growing body of evidence supports the idea that organelles talk to each other. This communication is characterized by either physical interactions, functional associations mediated by signaling molecules, or both. This flow of information allows the orchestration of proper cellular metabolic responses to the ever-changing extracellular environment. Mitochondria, the cell’s principle metabolic factories, have emerged as a major player that not only influence the functions of other organelles, such as the endoplasmic reticulum, nuclei, and lysosomes, but as recently shown by our group, mitochondria also receive functionally critical information from lysosomes. This process was found to be mediated by the lysosome-associated mechanistic target of rapamycin complex 1, another major regulator of cellular metabolism. As discussed here, disruption of this lysosome-to-mitochondria signaling pathway may underlie the early pathogenesis of Alzheimer’s disease.
Interorganelle communication is an emerging topic in cell biology, but little is known about the underlying mechanisms, its functional consequences, or its potential role in disease. Mitochondria play a critical role in cellular metabolism by regulating cellular bioenergetics, calcium, and reactive oxygen species homeostasis, cell death, and others (Nunnari and Suomalainen, 2012). These phenomena require not only coordinating cellular energy demands to nutrient availability, mitochondrial biogenesis, and degradation but also involve the orchestrated expression of >1,000 nuclear-encoded genes and those encoded by mitochondrial DNA (mtDNA) (Gustafsson et al., 2016). The coordination of this daunting task has been fundamental for the successful evolution and survival of the eukaryotic cell. Unsurprisingly, mitochondrial dysfunction plays a major role in several diseases including Alzheimer’s disease (AD), type-2 diabetes, and cancer, yet the precise molecular mechanisms involved remain poorly defined.
Interorganelle Communication Comes in Different Flavors
In yeast, mitochondria are continuously interacting with other organelles, such as vacuoles, nuclei, peroxisomes, and the endoplasmic reticulum (ER) (Murley and Nunnari, 2016). This process involves complex molecular machineries located in the membranes of those organelles. ER–mitochondria encounter structure, or ERMES (Murley and Nunnari, 2016), is the best characterized example. ERMES regulates phospholipid transport between mitochondria and the ER and elegantly emphasizes functional roles for interorganelle contacts (Murley and Nunnari, 2016). Although ERMES is fundamentally important for yeast, it is not conserved in metazoans, suggesting that mammalian cells use alternative mechanisms to regulate functional connections between mitochondria and ER.
Several forms of interorganelle communication have been detected in mammalian cells. Contact sites between ER and mitochondria, and between lysosomes and mitochondria have been found, and transfer of signaling molecules between nuclei and mitochondria, and from lysosomes to mitochondria support information flow and functional regulation of organelles. There are excellent recent reviews discussing these topic (Murley and Nunnari, 2016; Wong et al., 2019). Here, we will describe a novel functional interaction between lysosomes and mitochondria and discuss how its disruption might be an early signature of AD.
Nutrient-Induced Mitochondrial Activation: A Novel Form of Communication From Lysosomes to Mitochondria
Lysosomes and mitochondria play essential roles in cellular metabolism. Increasing evidence demonstrates how both organelles regulate each other’s functions. This cross-regulation is further evidenced by the fact that alterations in one can lead to reciprocal dysfunction (Wong et al., 2019).
The serine-threonine kinase, mechanistic target of rapamycin, is a constituent of two multiprotein complexes, mechanistic target of rapamycin complex 1 (mTORC1) and mTORC2, which respond to cell surface sensors that detect insulin, growth factors and nutrients, like amino acids. Both mTOR complexes regulate fundamental cellular responses, such as protein synthesis, cell cycle progression, and autophagy (Saxton and Sabatini, 2017). The mTOR complexes and corresponding regulators are found in various subcellular compartments, including nuclei, Golgi, peroxisomes, lysosomes, and mitochondria. The mechanisms that regulate this diverse distribution and their functional significance are not fully understood. One of the best characterized mechanisms regulating mTORC1 involves its activation at the lysosomal surface by insulin or amino acids. Activation of mTORC1 at that location upregulates translation; synthesis of proteins, lipids, and nucleotides; and downregulation of autophagy (Saxton and Sabatini, 2017). Lysosomal mTORC1 activity also regulates lysosome biogenesis and function by a mechanism involving nuclear translocation of the transcription factor, TFEB. There are also studies showing functional connections between mTOR and mitochondrial activity. mTORC1 coordinates mitochondrial metabolic stress responses in muscle cells (Khan et al., 2017), directly regulates mitochondrial metabolism in jurkat cells, and indirectly regulates mitochondrial biogenesis and function through the 4EBP-eIF4E signaling pathway (Morita et al., 2017). In Caenorhabditis elegans, lysosome-derived lipid messengers were found to regulate mitochondrial function. This process was shown to require the coordinated action of lysosomes, nuclei, and mitochondria and was also found to play a role in aging (Ramachandran et al., 2019). As mTOR plays key roles in aging and aging-related diseases, these observations open the possibility that mTOR orchestrates a multiorganelle response in aging and consequently, its disruption may underlie disease pathogenesis.
We recently described a previously unknown communication pathway between lysosomes and mitochondria (Norambuena et al., 2018) (Figure 1). Using two-photon fluorescence lifetime microscopy for label-free imaging of mitochondrial activity in live cultured neurons and measuring oxygen consumption in live mouse brain with multiparametric photoacoustic microscopy (Norambuena et al., 2018), we showed that stimulation of lysosome-associated mTORC1 with amino acids or insulin (Saxton and Sabatini, 2017) directly regulates mitochondrial oxidative metabolism by increasing oxygen consumption, reactive oxygen species, and ATP production (Norambuena et al., 2018). This nutrient-induced mitochondrial activation (NiMA) was completely reversed by genetically or pharmacologically interfering with mTORC1. Furthermore, NiMA was completely insensitive to the expression of two main mTORC1 downstream regulators of protein synthesis, S6K and 4EBP-eIF4E pathway. In addition, NiMA was insensitive to fatty acid transport into mitochondria, suggesting that autophagy-regulated mechanisms are not involved. Importantly, NiMA was insensitive to reduced expression of the essential mTORC2 subunit, rictor, indicating that mTORC2 is not required for NiMA (Norambuena et al., 2018). These observations are intriguing as mTORC1 and mTORC2 codistribute on lysosomes, mitochondria and other subcellular compartments, and regulate each other through negative and positive feedback loops (Saxton and Sabatini, 2017).
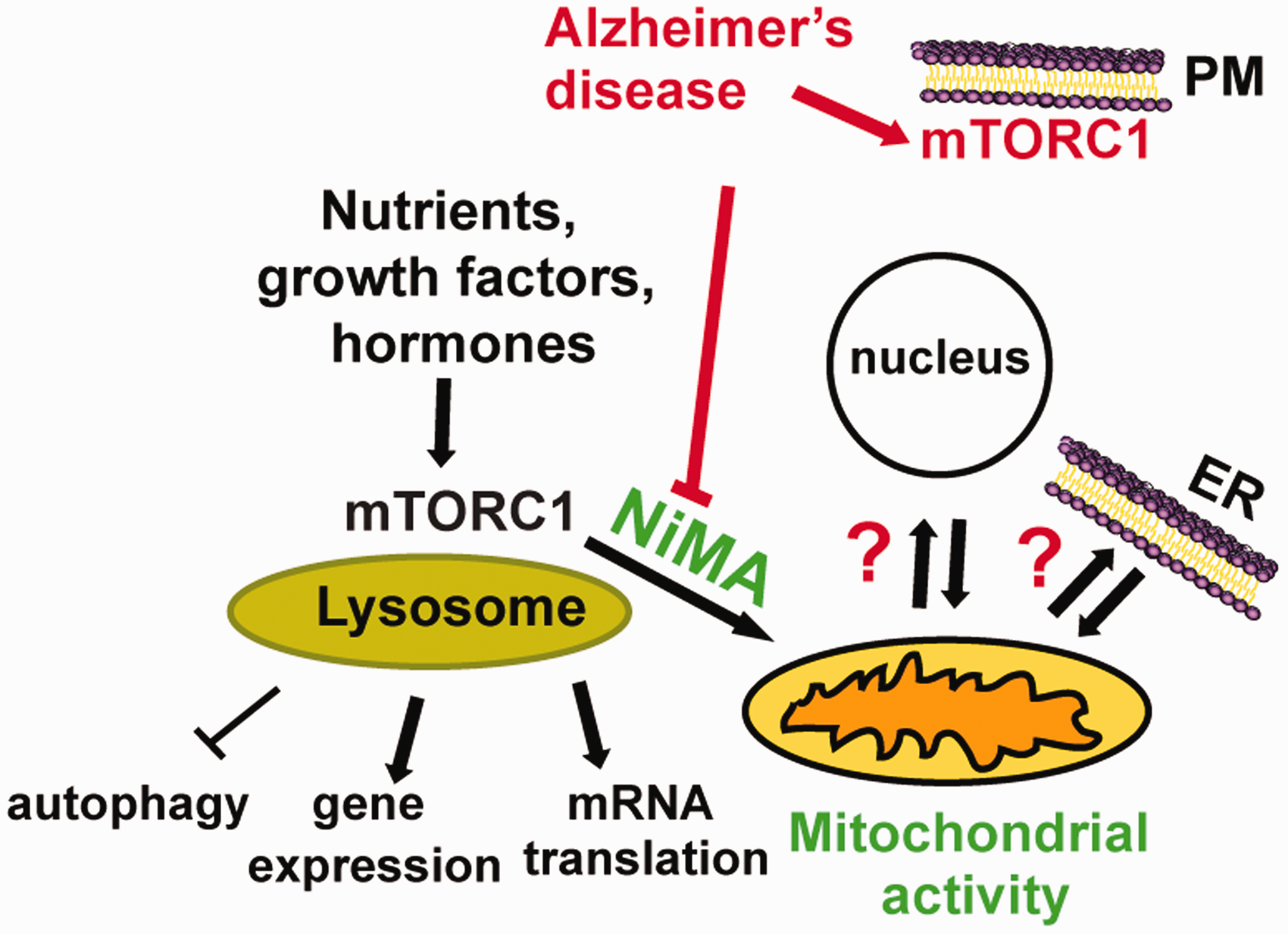
Nutrient-induced mitochondrial activity (NiMA), a novel lysosome-to-mitochondria signaling pathway. Nutrient stimulation of the lysosome-associated mTORC1 triggers mitochondrial oxidative metabolism. This novel form of interorganelle communication from lysosome to mitochondria is blocked by amyloid-β oligomers, which stimulate mTORC1 kinase activity at the plasma membrane. Dysregulation of the NiMA pathway could affect other aspects of interorganelle communication (question marks) and may underlie the pathogenesis of diseases, such as AD. NiMA = nutrient-induced mitochondrial activation; ER = endoplasmic reticulum; mTORC1 = mechanistic target of rapamycin complex 1.
NiMA Inhibition: A Seminal Step in AD Pathogenesis
Mammalian mtDNA encodes for essential components of the oxidative phosphorylation system. The study of the molecular mechanisms that regulate replication, transcription, and expression of mtDNA is a growing field (Gustafsson et al., 2016). Interestingly, a recent work has provided evidence that mtDNA replication in human cell lines can be regulated by ER–mitochondrial contact sites (Lewis et al., 2016). However, the question of how extracellular cues and energy demands affect mtDNA maintenance is still unknown. In the course of our studies on the functional consequences of the NiMA pathway, we also found that nutrient-mediated activation of lysosomal mTORC1 inhibit mtDNA synthesis (evaluated as EdU uptake into mtDNA nucleoids). To the best of our knowledge, this is the first example for regulation of mtDNA synthesis by extracellular cues. These results are also puzzling as the mTORC1/4EBP/eIF4E pathway has been shown to increase translation of mitochondrial fission process 1 mRNA and mitochondrial fission (Morita et al., 2017). As mtDNA replication needs to be completed before mitochondrial fission occurs, it is attempting to speculate that activation of mTORC1 inhibits mtDNA synthesis by a rapid translation-independent mechanism (Norambuena et al., 2018), thus “labeling” mitochondria that will proceed through division following an mTORC1-mediated translational process (Morita et al., 2017). Thus, lysosomal mTORC1 couples nutrient availability to mtDNA replication and activity, functionally connecting these two organelles.
Soluble amyloid-β oligomers (AβOs) are one of the main causative agents in AD. AβOs alter microtubule-mediated mitochondrial transport and may directly affect mitochondrial dynamics at synapses by interacting there with mitochondria (Manczak et al., 2011). AβO-induced mitochondrial dysfunction thereby drives progressive loss of synaptic activity and cell death, two major cellular features of AD. We recently found that AβOs activate mTORC1 at the plasma membrane (PM) in neurons (Norambuena et al., 2017), which leads to neuronal cell cycle reentry, a pathological pathway linked to neuronal death in AD (Norambuena et al., 2017) (Figure 1). In addition, we found that AβO-mediated activation of mTORC1 at the PM not only blocks NiMA but also dysregulates mtDNA replication (Norambuena et al., 2018) (Figure 1). As alterations in neuronal mtDNA maintenance account for brain energy metabolism deficiencies and inflammasome activation, alterations of the NiMA pathway may represent a mechanistic link connecting metabolic alterations, such as insulin resistance and inflammation, to impaired mitochondrial DNA maintenance in diseases such as AD. Thus, these novel AβO-mediated effects on NiMA pose exciting questions about molecular mechanisms that integrate nutrient signaling, mitochondrial dysfunction, and AD.
A detailed molecular mechanism regulating NiMA is not yet fully understood and several outstanding questions remain open. For example, do nutrients directly regulate mitochondrial function through an unknown mTORC1 substrate or it is supported by a third organelle? Does NiMA involve lysosome–mitochondria (Wong et al., 2019) or ER–mitochondria contact sites (Lewis et al., 2016)? (Figure 1). Thus, identifying molecular machineries regulating interorganelle function is not only an exciting topic in cell biology but also represents an important opportunity to develop new biomarkers and therapies for early detection and treatment of human diseases.
Footnotes
Acknowledgments
We apologize to all authors whose outstanding contribution is not cited in this article due to space limitations.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors are grateful for financial support from the Alzheimer’s and Related Diseases Research Award Fund (grant 17-5 to AN), the Owens Family Foundation (GSB), NIH/NIA (grant RF1 AG051085 to GSB), the Cure Alzheimer’s Fund (GSB), the Alzheimer’s Association (grant ZEN-16-363266 to GSB), Webb and Tate Wilson (GSB), The Virginia Chapter of the Lady’s Auxiliary of the Fraternal Order of Eagles (GSB), and the University of Virginia President’s Fund for Excellence (GSB).