
Abstract
Intracellular cholesterol transport is a complex process involving specific carrier proteins. Cholesterol-binding proteins, such as the lipid transfer protein steroidogenic acute regulatory-related lipid transfer domain-3 (STARD3), are implicated in cholesterol movements between organelles. Indeed, STARD3 modulates intracellular cholesterol allocation by reducing it from the plasma membrane and favoring its passage from the endoplasmic reticulum (ER) to endosomes, where the protein is localized. STARD3 interacts with ER-anchored partners, notably vesicle-associated membrane protein-associated proteins (VAP-A and VAP-B) and motile sperm domain-containing 2 (MOSPD2), to create ER–endosome membrane contacts. Mechanistic studies showed that at ER–endosome contacts, STARD3 and VAP proteins build a molecular machine able to rapidly transfer cholesterol. This review presents the current knowledge on the molecular and cellular function of STARD3 in intracellular cholesterol traffic.
Keywords
Introduction
Steroidogenic acute regulatory (StAR)-related lipid transfer domain-3 (STARD3) was identified using a bedside to bench approach. It was isolated from human breast biopsies in a screen designed to identify new genes implicated in breast cancer. In early publications, STARD3 was named Metastatic Lymph Node clone 64 (MLN64), as it was the 64th hit isolated in the screen (Tomasetto et al., 1995). Its current name STARD3 was given with respect to its homology with the StAR protein, also referred to as STARD1 (Alpy & Tomasetto, 2005; Stocco & Clark, 1996). On the human genome, the STARD3 gene lies next to the oncogene human epidermal growth factor receptor 2 (HER2) on chromosome 17q11-12 (Tomasetto et al., 1995). Amplification of this chromosomal subregion explains the co-expression between STARD3 and HER2 in breast cancer cells (Alpy et al., 2003; Bièche et al., 1996; Tomasetto et al., 1995). Remarkably, several studies showed that STARD3 contributes to the growth of HER2-positive cancer cells, but given that both proteins do not directly interact the molecular mechanism remains unclear (Alpy & Tomasetto, 2014b). Over the last years, the basic function of STARD3 has been clarified. Notably, it was showed to be a cholesterol transporter working at contact sites between endosomes and the endoplasmic reticulum (ER) (Alpy et al., 2013; Wilhelm et al., 2017). In this article, we review the literature on STARD3 structure and function on intracellular sterol transfer.
STARD3 Is a Lipid Transfer Protein From the START Protein Superfamily
STARD3 contains an MLN64 N-terminal (MENTAL) domain, anchoring the protein to the endosome membrane, a central two phenylalanines in an acidic tract-like motif (FFAT-like) suggesting the binding to resident proteins from the ER, and a C-terminal StAR-related lipid transfer (START) domain able to transfer cholesterol (Figure 1(a)).

STARD3 protein organization. (a) Schematic representation of STARD3 protein containing a conserved MENTAL domain made of four transmembrane helices (blue), an FFAT-like motif (red), and a START domain (magenta). (b) STARD3 is positioned in endosomes and the START domain is in the cytosol. (c) Models of cholesterol transfer by the START domain. (d) The MENTAL domain mediates the formation of homo- and heterodimers or oligomers with STARD3NL. These interactions may create microdomains enriched in cholesterol at the surface of endosomes. These microdomains may serve to concentrate cholesterol next to the START domain or as sensing domains. MENTAL: MLN64 N-terminal domain; FFAT: two phenylalanines in an acidic tract motif; START: steroidogenic acute regulatory-related lipid transfer domain; STARD3: steroidogenic acute regulatory-related lipid transfer domain-3; STARD3NL: STARD3 N-terminal like.
The START Lipid Transfer Domain
The presence of the START domain at the C-terminus qualifies STARD3 as a lipid transfer protein (LTP) from the START superfamily (Figure 1; Alpy & Tomasetto, 2014a; Ponting & Aravind, 1999; Soccio & Breslow, 2003). The START domain of STARD3 adopts an alpha-helix/beta-strand grip fold structure with an internal cavity matching the size and hydrophobicity of a cholesterol molecule (Figure 2(a); Letourneau, Lefebvre, Lavigne, & LeHoux, 2015; Tsujishita & Hurley, 2000). The affinity of the START domain for cholesterol is around 1.5 µM, a low range consistent with a transport activity (Tsujishita & Hurley, 2000). Surprisingly, the domain structure was similar to that of the birch pollen allergen Bet v1 (Horvath et al., 2016; Tsujishita & Hurley, 2000). Since then, the Bet v1 structure revealed the existence of a large superfamily of structurally related proteins. Eleven Pfam families were determined to be members of the Bet v1-like superfamily also known as the SRPBCC (START/RHO_alpha_C/PITP/Bet_v1/CoxG/CalC) ligand-binding domain superfamily (Iyer, Koonin, & Aravind, 2001; Radauer, Lackner, & Breiteneder, 2008). Among them, two families are distributed in all three superkingdoms: the polyketide cyclase and the activator of heat shock protein 90 (Hsp90) ATPase homologue 1 families. The ring hydroxylases and the CoxG families are only present in bacteria and archaea. Two families are specific to eukaryotes: Phosphatidylinositol transfer protein family is present in most eukaryotes, while the Bet v1 family is only present in plants. Of interest, while START proteins were thought to be absent from yeast, in silico analyses, based on both sequence comparison and secondary structure prediction, showed recently that a large family of eukaryotic proteins contains a domain resembling the START domain (Gatta et al., 2015; Khafif, Cottret, Balague, & Raffaele, 2014). This family comprises three human proteins (Aster-A, B, and C; encoded by the GRAMD1A, B, and C genes, respectively) and six proteins from yeast (Lam1p/Ysp1p, Lam2p/Ysp2p, Lam3p/Sip3p, Lam4p, Lam5p, and Lam6p/Lct1; Gatta et al., 2015). Moreover, these novel START-like containing proteins bind and transport sterols (Gatta et al., 2015; Horemcamp, Valverde, Nunnari, & Reinisch, 2018; Murley et al., 2015; Murley et al., 2017; Sandhu et al., 2018). It was tempting to speculate that the structure of the START-like domain first defined by Gatta et al. (2015) shares the Bet v1 fold. This assumption was confirmed recently by the crystal structure of the START-like domains of Lam2, Lam4, and Aster-A, which have a similar overall structure to the one of STARD3 and STARD4 (Horenkamp, 2018; Jentsch et al., 2018; Sandhu et al., 2018; Tong, Manik, & Im, 2018; Figure 2(b)). Interestingly, the isolated sterol-binding domains of Lam2p, Lam4, and Aster-A proteins were co-crystallized with sterols, which was present in the ligand-binding pocket created by the concave beta-sheet, and the long C-terminal helix (Horenkamp et al., 2018; Jentsch et al., 2018; Sandhu et al., 2018; Tong et al., 2018). These data strengthen previous predictions regarding the orientation of the sterol molecule in the START domain, and thus the orientation of the START domain in relation to the membrane for sterol loading and unloading (Figure 2; Murcia, Faraldo-Gomez, Maxfield, & Roux, 2006; Tsujishita & Hurley, 2000). However, we can speculate that the mode or the efficiency of transfer differs between the STARD and the Lam/Aster/Gram proteins. Indeed, in the Lam/Aster/Gram subfamily, the START-like domain is embedded in the protein and the C-terminal helix is not free while all the STARD proteins have a free C-terminal helix.
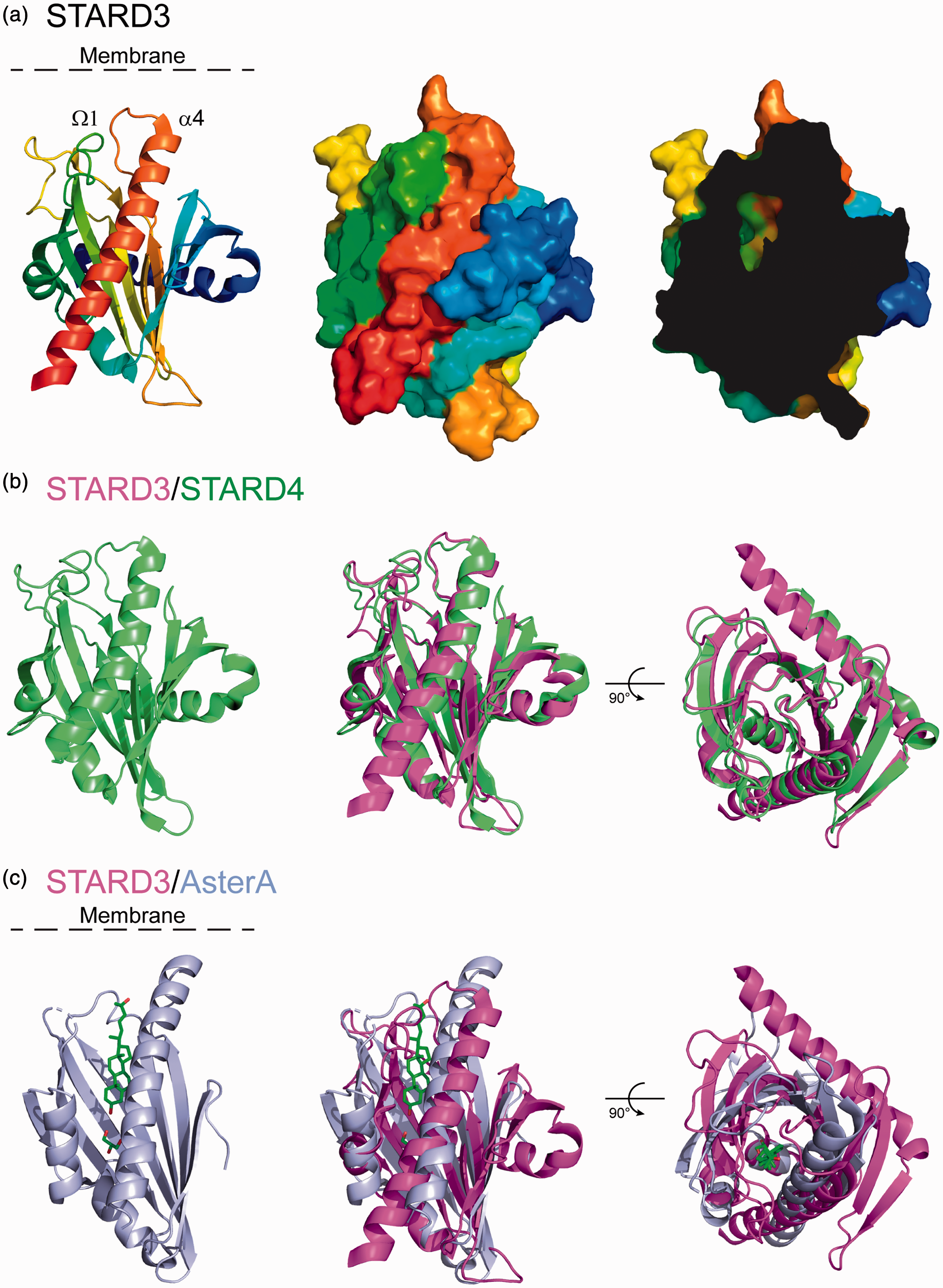
The START domain of STARD3, a cholesterol transfer interface. (a) Three-dimensional (3D) structure of the START domain of STARD3 shown as cartoon (left) or surface (middle) representation. The protein ribbon is rainbow-colored from blue (N-terminus) to red (C-terminus). On the right, a cutaway view of the surface of the domain showing a part of the cholesterol-binding pocket. (b) 3D structure of the START domain of STARD4 alone (green, left) and aligned with STARD3 (magenta, middle). The right-hand panel is rotated 90° about the x-axis. The two structures are highly similar. (c) 3D structure of the START-like domain of Aster-A alone (blue, left) and aligned with STARD3 (magenta, middle). The ligand of Aster-A, 25-hydroxycholesterol, is shown as sticks: green, carbon and red, oxygen. The right-hand panel is rotated 90° about the x-axis. The main difference between the two structures resides in the extended N-terminal α helix present in STARD3 but absent in Aster-A. STARD3: steroidogenic acute regulatory-related lipid transfer domain-3.
Mechanism of Sterol Transport
The molecular mechanism implicated in the entry and exit of cholesterol from the START domain is a matter of debate. Several models were proposed: a tunnel hypothesis, an intermediate state model implicating movements of the C-terminal alpha 4 helix and the omega 1 loop, and the molten globule hypothesis where the structure of the protein unfolds upon interaction with the acidic environment of the membrane (Figure 1; Bose, Whittal, Baldwin, & Miller, 1999; Iaea, Dikiy, Kiburu, Eliezer, & Maxfield, 2015; Lavigne, Najmanivich, & LeHoux, 2010; Murcia et al., 2006) reviewed in (Letourneau et al., 2015). The tunnel hypothesis is based on the presence of two openings in the structure but given their limited diameters, the authors suggested that additional movements in the alpha 4 helix and omega 1 loop would be necessary to allow sterol entry and exit (Tsujishita & Hurley, 2000; Figures 1(c) and 2(a)). The molten globule hypothesis is based on experimental data showing unfolding of STARD1 at acidic pH (Bose et al., 1999). The model is consistent with the delivery of sterol in the membrane, but it is challenged by the fact that under this unfolded form, the START domain cannot hold a sterol molecule (Roostaee, Barbar, Lehoux, & Lavigne, 2009). The intermediate state model originates from the local unfolding and movement of the C-terminal alpha 4 helix of STARD1 upon interaction with the membrane (Mathieu, Fleury, Ducharme, Lavigne, & LeHoux, 2002; Roostaee, Barbar, Lavigne, & LeHoux, 2009; Roostaee, Barbar, Lehoux, & Lavigne, 2008). This model is completed by other studies showing movements of the omega 1 loop upon cholesterol loading (Murcia et al., 2006). The intermediate-state model including notably conformational changes in the omega 1 loop, and the alpha 4 helix interacting with the membrane is today a good model to explain cholesterol binding and delivery by the START domain (Figure 1(c)).
START Domains Are Not Equal in Terms of Transfer
Early in vivo and in vitro studies using either steroidogenic assays in transfected cells or synthetic donor and acceptor vesicles showed that—in isolation—the START domain of STARD3 had a sterol transfer capacity similar to that of STARD1 (Bose, Whittal, Huang, Baldwin, & Miller, 2000; Watari et al., 1997). Later, a comparative study on the activity and structure of STARD proteins from the sterol binder subgroup (STARD2, D3, D4, D5 and D6) was done by the Bose and Miller’ s groups (Bose et al., 2008). This seminal study using recombinant START domains in isolation for STARD1 and D3 and the whole STARD4, D5 and D6 proteins showed that cholesterol-binding capacity does not predict steroidogenic activity, in other words, the ability for a given START domain to move cholesterol. Bose et al. (2008) showed that although cholesterol binding was quite similar for all the STARD proteins from this subgroup, their capacities to transfer cholesterol were different. In fact STARD1, D6 and D3 were able to rapidly induce pregnenolone production using isolated mitochondria, while STARD4 and D5 had little activity (Bose et al., 2008). This is surprising because the crystal structures of STARD3 and D4 are very similar (Figure 2; Romanowski, Soccio, Breslow, & Burley, 2002). It is also puzzling because STARD4 is an efficient sterol transporter (Mesmin et al., 2011) able to transfer dehydroergosterol (DHE) from donor to acceptor liposome at a rate of seven molecules of DHE per molecule of STARD4 per minute (Iaea et al., 2015). It is difficult to compare transfer activities from different experimental settings, but we showed that under its soluble form (similar to that of STARD4), the START domain of STARD3 had a transfer capacity of 1 DHE molecule per molecule of STARD3 per min (Wilhelm et al., 2017), more modest than STARD4’s. These data indicate that lipid transfer activities between synthetic membranes do not always predict biological activities. Of interest, Iaea et al. (2015) showed that the surface charge of donor and acceptor vesicles modulated STARD4 transfer capacity. Horenkamp et al. (2018) made a similar observation for the isolated START-like domain of Lam2p/Ysp2p and Aster-B/GramD1b. It is then very likely that the donor and acceptor membrane identities in terms of lipid and protein compositions regulate sterol transfer by the START domain. START proteins have a conserved fold, but their primary sequences are different, leaving room for selectivity in their binding with other partner membranes or proteins.
STARD3 Is an Endosome-Bound Sterol Transporter
STARD3 distinguishes itself by the presence of a membrane spanning domain composed of four transmembrane helices (Figure 1(a) and (b); Alpy et al., 2001; Moog-Lutz et al., 1997). The membrane spanning region is critical for the addressing and the proper localization of STARD3 in endosomes (Alpy et al., 2001). We named this region MENTAL domain (Alpy et al., 2005; Alpy, Wendling, Rio, & Tomasetto, 2002) because it is also found in a second human protein called STARD3 N-terminal like (STARD3NL), originally called MLN64 N-terminal domain (MENTAL) homologue (MENTHO) (Figure 1(d); Alpy et al., 2002). The MENTAL domain anchors the protein in endosomes leaving the START domain in the cytosol (Figure 1(b); Alpy et al., 2001). Overexpression of STARD3NL, or of a truncated STARD3 protein containing only the MENTAL domain, results in the enlargement of endosomes (Alpy et al., 2005). In addition, this domain allows the homo- and hetero-oligomerization of both proteins and binds cholesterol (Alpy et al., 2005; Figure 1(d)). Of interest, proteome-wide mapping of cholesterol-interacting proteins using the click chemistry methodology confirmed this finding; several cholesterol-bound peptides were identified in the MENTAL domain (STARD3NL, residues 23–37 and 185–196; Hulce, Cognetta, Niphakis, Tully, & Cravatt, 2013). Altogether, thanks to its ability to dimerize and oligomerize (Alpy et al., 2005), we speculate that the MENTAL domain of STARD3 and STARD3NL build quaternary structures or microdomains at the endosome surface possibly acting as sterol reservoir (Figure 1(d)). In support to this, STARD3 immunolabeling in electron microscopy showed that the protein is not evenly localized on the limiting membrane of endosomes, the labeling appearing as patches, which is compatible with the formation of microdomains (Alpy et al., 2001). Another possibility is that the MENTAL domain acts as a sterol sensing domain to regulate the activity of the START domain.
One study argued that STARD3 was present on a subpopulation of late endosomes positive for the cholesterol transporter ATP-binding cassette subfamily A member 3 and negative for the oxysterol-binding protein-related protein 1L (ORP1L) and the Niemann–Pick type C1 (NPC1) proteins (van der Kant, Zondervan, Janssen, & Neefjes, 2013). Experiments were done in a malignant melanoma cell line (MelJuSo) using overexpression of fluorescent protein tagged STARD3 at the C-terminus. This study is not consistent with results obtained in other cells including HeLa cells, where untagged STARD3 was localizing with the late endosomal markers LAMP1, CD63, Rab7, and BMP (Alpy et al., 2001; Wilhelm et al., 2017), therefore overlapping with ORP1L- and NPC1-positive endosomes. This discrepancy may reflect cell specific differences in the endosomal compartment; it is known that melanocytes have a cell-type specific endosomal system to generate melanosomes which are lysosome related organelles (Raposo & Marks, 2007). It may also be caused by the presence of the fluorescent protein at the C-terminus that possibly impairs the function of the START domain. Indeed, the START domain is always present at the C-terminus of all STARD proteins, leaving the alpha 4 helix free to interact with acceptor membranes (Alpy & Tomasetto, 2005; Iaea et al., 2015; Tsujishita & Hurley, 2000; Figure 2).
STARD3 Is Ubiquitously Expressed and Regulated in a Cell-Specific Manner
STARD3 is a conserved protein present in the whole animal kingdom as well as in unicellular organisms closely related to animals (Figure 3). STARD3NL is only present in vertebrates evoking a more recent appearance of this protein (Alpy & Tomasetto, 2014b). STARD3 is expressed in most tissues at low levels. Sequence analysis of the gene 5′ region revealed a high GC-content (or guanine-cytosine content) and the absence of canonical promoter elements such as a TATA box. A short promoter of about 200 base pairs upstream of the transcription start sites was sufficient to drive high expression of a reporter gene in vitro (Alpy et al., 2003). In breast cancer cells, there is a strong correlation between STARD3 gene DNA copy number and transcript levels, arguing that amplification of the gene is the main driver for the overexpression (Alpy et al., 2003; Bièche et al., 1996; Tomasetto et al., 1995). In other cells and tissues, STARD3 is likely regulated in a cell-specific manner. A detailed analysis of STARD3 expression in the brain showed a heterogeneous immunostaining of the protein, indicating selective expression in discrete specific cell populations and regions (King et al., 2006). As another example, STARD3 gene expression was shown to be upregulated by 9-cis-retinoic acid in macrophages (Borthwick, Allen, Taylor, & Graham, 2010), and repressed by all-trans-retinoic acid in lymphoma cells (Wang, Cheng, Chuang, Hsu, & Su, 2000). The promoter region of STARD3 gene contains several putative sites for Sterol regulatory element-binding proteins (SREBPs) transcription factors binding; in macrophages, STARD3 mRNA decreases during macrophage differentiation and cholesterol loading and increases following acute cholesterol depletion from the plasma membrane (PM) by methyl beta-cyclodextrin and in response to the release of sequestered SREBPs by LY295427 (Borthwick et al., 2010). Of interest, hepatic expression of STARD3 protein was found to be repressed by genetic obesity in both males and females suggesting that STARD3 might facilitate lipid export, rather than its retention in the liver (Soffientini, Caridis, Dolan, & Graham, 2014). One study recently reported a function of STARD3 in osteoporosis (Lei, Xueming, & Ruihang, 2018). Interestingly, it shows that STARD3 promoted osteoclast differentiation possibly by increasing an inflammatory response (Lei et al., 2018).

Phylogenetic tree of STARD3 orthologs from 115 species. The whole amino acid sequences were aligned by the Clustal Omega program (McWilliam, Li, Uludag, Squizzato, Park, Buso, Cowley & Lopez. , 2013). The phylogenetic tree was drawn with R Studio, R version 3.5.1 (2018-07-02) and the packages ggtree, seqinr, and msa.
Intracellular Functions of STARD3
The presence of a START domain in STARD3 is a strong argument for a role of this protein in intracellular sterol transport. By immobilizing the protein on endosomes, the MENTAL domain restricts its sterol transfer activity to the endocytic compartment territory (Figure 1). However, as the endocytic compartment is highly dynamic and connected with many other cellular organelles, STARD3 may act on many cellular organelles.
Does STARD3 Function in the PM?
Several studies reported that STARD3 was addressed to the PM en route to late endosomes (Holtta-Vuori et al., 2005; Liapis, Chen, Davies, Wang, & Ioannou, 2012; van der Kant et al., 2013; Vassilev et al., 2015; Zhang et al., 2002), supporting the idea that STARD3 might transport sterol at the cell surface. In our laboratory, we occasionally observed STARD3 at the PM in cells expressing very high amounts of the protein, but we interpreted this localization as an overexpression artifact; our opinion is that under physiological conditions, STARD3 is mainly in endosomes (Alpy et al., 2001). However, live cells imaging showed that the protein was in endosomal tubules (our unpublished data; Zhang et al., 2002). It is possible that endosome tubules-containing STARD3 reach the PM bringing STARD3 transiently at PM–endosome contacts. In fact, Vassilev et al. (2015) reported that overexpression of STARD3 tagged with green fluorescent protein (GFP) in a breast cancer cell line, resulted in STARD3 localization in endosomes and for a subset of cells at the PM. The PM staining was attributed to a localization in microdomains in the PM or in vesicles at the proximity of the PM (Vassilev et al., 2015). This observation is consistent with the transient presence of STARD3 in endosomal tubules reaching the PM; whether it is functional is unknown.
Does STARD3 Transport Cholesterol to the Mitochondria?
The literature generally presents STARD3 as an LTP able to make contacts between endosomes and mitochondria, implying a role in transferring cholesterol into the mitochondria (Wong, Gatta, & Levine, 2019). The first study by Watari et al. (1997) showed that STARD3 is able to transfer cholesterol to the mitochondria. There, STARD3 was able to induce the first step of steroidogenesis in Chinese hamster ovary (CHO) cells modified to sustain steroidogenesis. It showed that this activity was low when the full-length protein was expressed. In fact, only a construct corresponding to a free START domain (lacking the MENTAL domain and the FFAT motif) was able to sustain steroidogenesis at levels similar to that of STARD1 (Watari et al., 1997). The interpretation of these data was that the sterol transfer function of STARD3 to the mitochondria was not a physiological process. The hypothesis of a proteolytic cleavage of the protein freeing the START domain was then proposed to support the idea of a role of STARD3 in steroidogenesis (Watari et al., 1997). This notion was not confirmed. Actually, STARD3-deficient mice were not impaired for steroid production (Alpy & Tomasetto, 2014b; Kishida et al., 2004). In addition, STARD3 cannot compensate for the lack of STARD1 in lipoid congenital adrenal hyperplasia, a condition where mutations in the StAR gene (aka STARD1) totally prevent steroidogenesis (Stocco, 2001). Nevertheless, correlation between STARD3 expression and either endosome–mitochondria contacts (Zhang et al., 2002) or with cholesterol entry in the mitochondria (Balboa et al., 2017; Charman, Kennedy, Osborne, & Karten, 2010) was reported. Zhang et al. (2002) used live imaging to show that endosomes marked by STARD3-GFP made contacts with mitochondria. However, in this pioneer study, contacts were not quantified and not compared with cells marked with a different endosome marker. Although the authors interpreted the presence of contacts between endosomes and mitochondria as the result of STARD3 expression, it was not confirmed by later studies. A study in liver cells showed that STARD3 overexpression results in severe liver damage and apoptosis (Tichauer et al., 2007). By looking at the molecular mechanisms involved, Balboa et al. (2017) concluded that overexpression of STARD3 was associated with increased mitochondrial cholesterol and with mitochondrial morphologic and metabolic alterations in hepatic cells. Unfortunately, they did not document the changes that are expected to be seen upon STARD3 overexpression, notably cholesterol accumulation in endosomes, and therefore it is difficult to know if the mitochondrial phenotype is due to a direct action of STARD3 on mitochondria or is a result of late endosomal cholesterol accumulation that is deleterious to the cell. Charman et al. (2010) showed that silencing STARD3 reduced steroidogenesis (a read-out for cholesterol transport into the mitochondria) in modified CHO cells and decreased mitochondrial cholesterol content but only in CHO cells deficient for the NPC1 protein. In addition, they showed that some of the mitochondrial metabolic alterations seen in NPC1-deficient-CHO cells can be rescued by STARD3 silencing (Balboa et al., 2017; Kennedy et al., 2014). However, the reduction of STARD3 alone in CHO cells had no effect on mitochondrial metabolism (Kennedy et al., 2014). It is possible that endosome–mitochondria contacts created by other mechanisms, such as endosome enlargement and cholesterol accumulation as in NPC1-deficient cells, may bring STARD3 at the periphery of mitochondria where it can transfer cholesterol to mitochondria by the START domain. This transport appears to play a role in CHO-NPC1-deficient cells, but it is unclear that it is relevant in other cells.
STARD3 Makes Specific Contacts With the ER Through VAPs and MOSPD2
A conserved FFAT-like motif marks the end of the MENTAL domain (Figure 1(a) and (b)). This motif is present in an unstructured region between the MENTAL and the START domain. Canonical FFAT motifs were originally described as linear peptide sequences (EFFDAxE) flanked by an acidic region (Loewen, Roy, & Levine, 2003). FFAT motifs are signals for the interaction with proteins containing a major sperm protein (MSP) domain including vesicle-associated membrane protein-associated proteins (VAP-A and VAP-B), (Furuita, Jee, Fukada, Mishima, & Kojima, 2010; Kaiser et al., 2005; Loewen et al., 2003; Mikitova & Levine, 2012) and motile sperm domain containing 2 (MOSPD2), (Di Mattia et al., 2018). MOSPD2 and VAP proteins contain an MSP domain able to recognize FFAT motifs and to bind to FFAT-containing proteins like STARD3 and STARD3NL (Alpy et al., 2013; Di Mattia et al., 2018). The MSP domain has an immunoglobulin-like fold based on a seven-stranded beta-sandwich (Bullock, Roberts, & Stewart, 1996; Kaiser et al., 2005). Our in vitro analyses revealed that, despite sharing limited sequence homology, the MSP domains of MOSPD2 and VAPs have a conserved fold, the same mode of FFAT motif recognition, and similar affinities (Di Mattia et al., 2018). VAP-A, VAP-B, and MOSPD2 are tail-anchored proteins localized on the ER (Figure 4; Di Mattia et al., 2018; Lev, Ben Halevy, Peretti, & Dahan, 2008). In addition, these MSP domain-containing proteins are able to build ER–endosome contacts with STARD3 (Figure 4). It is unclear whether STARD3 competes with VAPs and MOSPD2 for binding and whether contact sites can contain different tethers. Of note, at the resolution of light microscopy (200–300 nm), we observed both VAPs and MOSPD2 in a given subdomain of the ER engaged in a contact with endosomes (Di Mattia et al., 2018). However, we cannot affirm that the presence of multiple tethers in the same region indicates that all of them are in complex with a partner on the facing membrane as most of these proteins can dimerize. Interestingly, contacts made by VAP and MOSPD2 are likely functionally different (Figure 4). Indeed, MOSPD2 is more than a VAP homolog; it contains an N-terminal cellular retinaldehyde-binding protein and triple functional domain protein (CRAL-TRIO) domain. This lipophilic domain was first identified in the Sec14-protein (Sec14p) of yeast and functions in exchanging phosphatidylinositol and phosphatidylcholine between lipid membrane bilayers (Bankaitis, Aitken, Cleves, & Dowhan, 1990). The function of the CRAL-TRIO domain of MOSPD2 is unknown, but we can speculate that its association with STARD3 might form a molecular machine able to counterexchange sterol with another lipid (Figure 4).
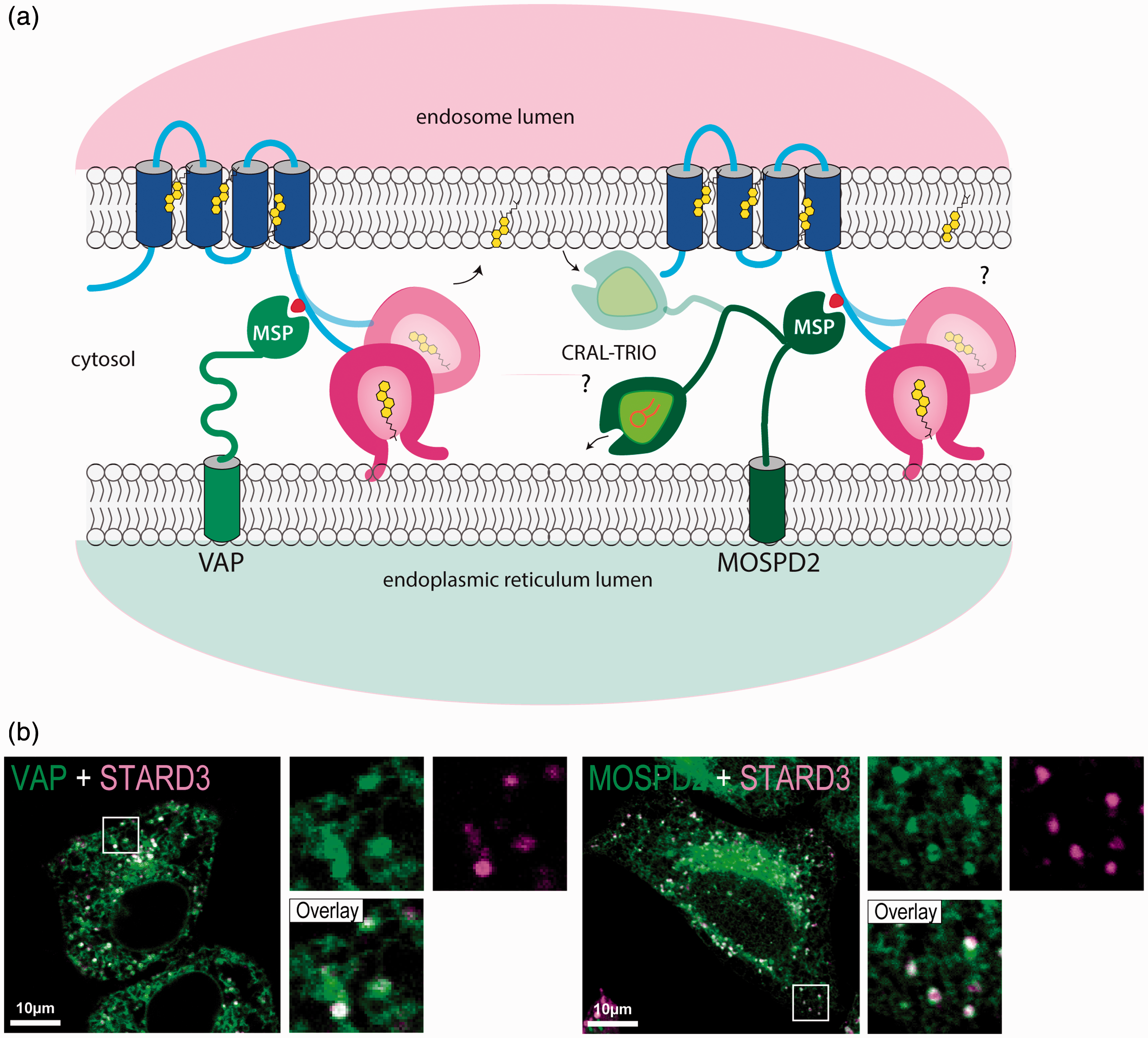
STARD3 creates membrane contacts between endosomes and the ER. (a) Schematic representation of STARD3 positioning in the context of the endocytic compartment and ER–endosome contact via interaction with the ER–proteins VAP and MOSPD2. (b) Localization of STARD3 with VAP and MOSPD2 at ER-endosome contacts. Merge images on the left of STARD3 (magenta) and VAP (green) labeling and on the right of STARD3 (magenta) and MOSPD2 (green) labeling. Insets correspond to a 3.5× magnification. Scale bar: 10 µm. VAP: vesicle-associated membrane protein-associated proteins; MOSPD2: motile sperm domain-containing 2; MSP: major sperm protein domain; CRAL-TRIO: cellular retinaldehyde-binding protein and triple functional domain protein domain; STARD3: steroidogenic acute regulatory-related lipid transfer domain-3.
The Interaction Between STARD3, STARDNL, and VAP/MOSPD2 Proteins Has a Strong Impact on the Subcellular Architecture and on Endosome Dynamics
In cells overexpressing either STARD3 or STARD3NL, endosomes are covered by the ER. This ER–endosome tethering results in the formation of extended membrane contact sites where the distance between the ER and endosome membranes is less than 10 nm (Alpy et al., 2013). Of interest, extended ER–endosome contacts mediated by STARD3NL alter the dynamics of the endosomal compartment by preventing vesicle-to-tubule transitions (Alpy et al., 2013). Membrane contacts between the ER and endosomes have been previously described in the context of intracellular cholesterol sensing. They involved another sterol transfer protein named ORP1L (Johansson, Lehto, Tanhuanpää, Cover, & Olkkonen, 2005; Rocha et al., 2009). Of note, both STARD3 and ORP1L interact with VAPs and MOSPD2 proteins in the ER (Alpy et al., 2013; Di Mattia et al., 2018). However, at contacts between endosomes and the ER, ORP1L mediates cholesterol transfer from endosomes to the ER (Ridgway & Zhao, 2018; Zhao & Ridgway, 2017), while STARD3 mediates cholesterol transfer from the ER to endosomes (Wilhelm et al., 2017). In addition, in cells grown in the absence of low-density lipoprotein (LDL) cholesterol condition, VAPs and ORP1L are required for transport of ER-derived cholesterol to a subpopulation of endosomes (Eden et al., 2016). These results support the idea that ER–endosome contacts are built by distinct protein complexes and lipid transport at these sites might be flexible.
STARD3 and VAP Form an Autonomous Sterol Transfer Machine
STARD3 builds physical contacts between endosomes and the ER. How these contacts function in sterol exchange was recently uncovered using in vitro and in vivo approaches.
STARD3 Favors Cholesterol Transport in Endosomes and Reduces PM Cholesterol
Cholesterol in situ labeling in cells expressing high amount of STARD3 showed that STARD3 modulates the intracellular repartition of cholesterol (Wilhelm et al., 2017). Consistent with earlier studies, STARD3 expression is associated with increased cholesterol in late endosomes (Alpy et al., 2001). To better understand the mechanism of this accumulation, we used STARD3 mutants impaired in their ability either to transfer cholesterol or to make contact sites. Mutants lacking the START domain or containing a two amino acid substitution in the START domain lipid-binding cavity did not accumulate cholesterol in endosomes. Likewise, mutation in the FFAT motif inactivating binding with VAP prevented cholesterol accumulation in endosome (Wilhelm et al., 2017). As the PM is enriched in cholesterol, we studied its cholesterol content in the context of STARD3 expression. The PM contains different pools of cholesterol (Das, Brown, Anderson, Goldstein, & Radhakrishnan, 2014). Cholesterol-enriched domains are accessible to the domain 4 (D4) of Perfringolysin O (Θ toxin), a cholesterol-binding toxin (Ohno-Iwashita et al., 2004). Using a fluorescent protein tagged GFP-D4 as a biosensor for membrane cholesterol in living cells, we showed that STARD3 expression was associated with a reduction of signal at the PM that we assumed reflects a reduction of PM cholesterol. Of note, Vassilev et al. (2015) reported that STARD3-GFP was implicated in cholesterol delivery in the PM at the expense of the ER in MCF7 cells. As we observed an opposite results in HeLa cells, where STARD3 expression is associated with cholesterol accumulation in endosome and cholesterol reduction at the PM (Wilhelm et al., 2017), we overexpressed an untagged version of STARD3 in MCF7 cells and looked at cholesterol distribution. We could not confirm Vassilev et al. (2015) observations; in MCF7 cells, STARD3 expression results in cholesterol accumulation in late endosomes and a reduction of PM cholesterol (our unpublished data).
Membrane Tethering Boosts Sterol Transport by the START Domain
In vivo studies support the model in which STARD3 and VAP-made late endosome–ER contacts allow cholesterol transport. To have direct evidence of this transport, we reconstituted the tethering complex in vitro using recombinant proteins and synthetic liposomes. As mentioned earlier, compared with STARD4, and indirectly with Lamp proteins (Iaea et al., 2015; Jentsch et al., 2018), the sterol transfer function of the soluble START domain of STARD3 is modest. But in contrast to STARD4, which is free, STARD3 is hooked on endosomes by the MENTAL domain and attached to the ER by a FFAT-MSP interaction (Figure 4). To test sterol transport in this defined context, contact sites were mimicked in vitro, by STARD3-decorated endosome-like liposomes and VAP-decorated ER-like liposomes, and sterol transport was measured in real time using a fluorescence resonance energy transfer-based assay (Mesmin et al., 2011; Moser von Filseck, Vanni, Mesmin, Antonny, & Drin, 2015). In this in vitro setting, within the first second, endosome–ER contact formation occurred and was associated with a fast sterol transport; the equilibrium in sterol between both populations of liposomes was reached within few minutes. The initial transport rate was about 24 sterol molecules/min transported by one STARD3 molecule, an order of magnitude higher than unattached STARD3 (Wilhelm et al., 2017). This was not observed for START-like proteins; curiously, the sterol transfer capacity of Lam2p/Ysp2p and Aster B/GramD1b is not favored by tethering (Horenkamp et al., 2018). In the case of STARD3, sterol transport assays are consistent with in vivo studies (Wilhelm et al., 2017), showing that the complex formed by STARD3 and VAP in membrane contacts functions as an independent molecular machine transferring sterols in a rapid manner.
STARD3/VAP Contacts Move Cholesterol From the ER to Endosomes
It was first thought that STARD3 could export LDL-derived cholesterol from endosome to the ER where it is esterified. Many lines of evidence are against this idea: the expression of STARD3 in different cell lines including CHO, HeLa, and cells being CV-1 (simian) in Origin (COS) results in cholesterol accumulation in endosomes (Alpy et al., 2001; Holtta-Vuori et al., 2005; Liapis et al., 2012). In addition, STARD3 overexpression did not cause a significant change in cholesterol esterification (Liapis et al., 2012). Finally, STARD3 expression in NPC1- and NPC2-deficient fibroblasts does not alleviate cholesterol accumulation in endosomes, it rather increases sterol load (Alpy et al., 2001, 2002; Holtta-Vuori et al., 2005). To have a better insight on the source of cholesterol mobilized by STARD3/VAP contacts, we labeled cholesterol in situ in cells expressing STARD3, in the absence of LDL cholesterol, and in the absence of de novo synthetized cholesterol. Of interest, while the absence of LDL-derived cholesterol had no significant effect on cholesterol accumulation driven by STARD3, in the absence of de novo synthesis, cholesterol was not accumulated in endosomes. These results support the notion that STARD3/VAP contacts are involved in the delivery of neosynthetized cholesterol in endosomes (Wilhelm et al., 2017). The cellular processes involved in sterol uptake and synthesis are tightly regulated by the transcription factors from the SREBPs family (Horton, Goldstein, & Brown, 2002). Notably, SREBP-2 is a transcriptional regulator of genes participating in the synthesis and uptake of cholesterol. We were surprised to note that the endosomal–cholesterol accumulation and PM depletion driven by STARD3/VAP contacts were imperceptible to the SREBP2 transcription factor despite the fact that the pathway was still functional (Wilhelm et al., 2017). All these results suggest that different pools of cholesterol coexist in endosomes, notably the LDL-cholesterol en route to the ER, and a pool of newly formed cholesterol moving to endosomes.
STARD3/VAP Contacts Modulate the Inner Membrane Composition of Endosomes
Endosomes are cholesterol rich organelles, and therefore bringing more cholesterol from the ER to endosomes appears counterintuitive. One recent study showed that cholesterol can move from the ER to endosomes. Eden et al. (2016) reported that ER–endosome contacts made by Annexin A1 and S100A11 support cholesterol transport by ORP1L from the ER to endosome in the context of Epidermal Growth Factor signaling. This ER-to-endosome transport made by ORP1L and VAP is involved in the formation of endosome intraluminal vesicles (Eden et al., 2016). Actually, the morphology of endosomes under conditions of VAP/STARD3 contacts is changed. Endosomes with homogenous intraluminal vesicles were rarer in these cells compared with control cells. The endosomes were generally pleomorphic and enriched in internal membranes including multilamellar and multivesicular structures (Wilhelm et al., 2017). Given that the complex formed by STARD3 and VAP fluxes cholesterol toward endosomes, this pool of cholesterol might be preferentially used to make inner membranes in endosomes.
STARD3, More Than a Cholesterol Transporter
A surprising function for STARD3 was discovered in silkworms (Bombyx mori). Silkworm larvae build cocoons in silk which naturally displays a yellow color. This color results from the presence of carotenoids in the silk, a chemical which is not synthetized by these animal but taken up from mulberry leaves through the alimentation (Sakudoh et al., 2007; Sakudoh & Tsuchida, 2009). Genetic analyses showed that in silkworms, the STARD3 gene has evolved to produce two different proteins by alternative promoter usage and splicing: A START-only protein named carotinoid-binding protein which is involved in carotenoid transport in the silk glands, and Bombyx mori Start1, which has the same structure as STARD3 found in other animals, and is likely involved in cholesterol transport (Sakudoh, Tsuchida, & Kataoka, 2005; Tabunoki et al., 2002). Of note, in humans, two carotenoids, lutein and zeaxanthin, accumulate in the central region of the retina, the macula lutea, that is responsible for central vision. The name macula lutea originates from the yellow color of this structure, which results from the presence of carotenoids. Interestingly, a carotenoid-binding protein was purified from human retina (Bhosale et al., 2009). This protein binds lutein with a Kd of 0.45 µM. Based on the molecular weight of this protein and its detection by anticarotinoid-binding protein antibodies, STARD3 was proposed to be the carotenoid-binding protein in human eyes (Bhosale et al., 2009). Accordingly, the START domain of STARD3 was shown to bind lutein with a Kd similar to that of cholesterol of 0.45 µM (Li, Vachali, Frederick, & Bernstein, 2011). These data suggest that STARD3 might be responsible for carotenoid accumulation in the human retina, thus implying that STARD3 could bind several ligands, and therefore have a general function in cholesterol transport and a specialized role in the retina.
STARD3 has been recently incriminated in viral infection and replication. Actually, factors implicated in endocytosis and intracellular cholesterol homeostasis are generally needed for viral infection. The NPC1 protein has been shown to be essential for filovirus infection notably Ebola virus infection (Carette et al., 2011; Cote et al., 2011; Herbert et al., 2015). In that case, the protein itself acts as a viral receptor. However, for many other viruses, endosomal cholesterol contributes to their entry and replication: notably Chikungunya, Zika, West Nile, and Dengue viruses (Wichit et al., 2017). Of interest, a recent publication reported that NPC1 and STARD3 were implicated in the entry and replication of Hepatitis C virus, and not of the Dengue virus (Stoeck et al., 2018). While the molecular mechanism implicating STARD3 is not known, it might bring cholesterol to the replication organelle, a structure composed of single and double membrane vesicles (Romero-Brey et al., 2012). This study supports the idea that individual LTPs like STARD3 may represent new vulnerabilities for many viruses.
Conclusion
STARD3 is an LTP able to transport cholesterol; the different structural and functional regions present in the protein cooperate to ensure a fast and efficient intracellular transport of cholesterol between endosomes and the ER (Figure 5). The fact that STARD3 can partner with three different ER receptors (VAPs and MOSPD2) suggests that it might be involved in the transport of different pools of cholesterol, coming and leaving endosomes, involving the ER and perhaps other organelles and LTPs. Along this line, the physiological relevance of a complex made of STARD3 and MOSPD2 on intracellular lipid transport needs to be addressed. The isolation, purification, and molecular characterization of the contacts made by STARD3 under different conditions will clarify its role in pathophysiological pathways. In addition, structure function experiments with the complete protein in complex with either VAP or MOSPD2 will help to capture the mechanism of sterol transport. The role of STARDNL, the START domain-less protein, remains enigmatic and should be clarified. We speculate that STARD3NL assists STARD3 by the creation of microdomains at the surface of late endosomes ready to assemble into extended membrane contact sites and to store a pool of cholesterol to be transferred by the START domain. The role of STARD3 in binding and trafficking other ligands has been established in silkworm and in the human retina; more studies are needed to understand the function of this transport. STARD3/VAP and STARD3/MOSPD2 complexes are examples of a growing number of molecular complexes implicated in inter-organelle communication and exchange, and their characterization will provide new insights about intracellular communication and lipid distribution.
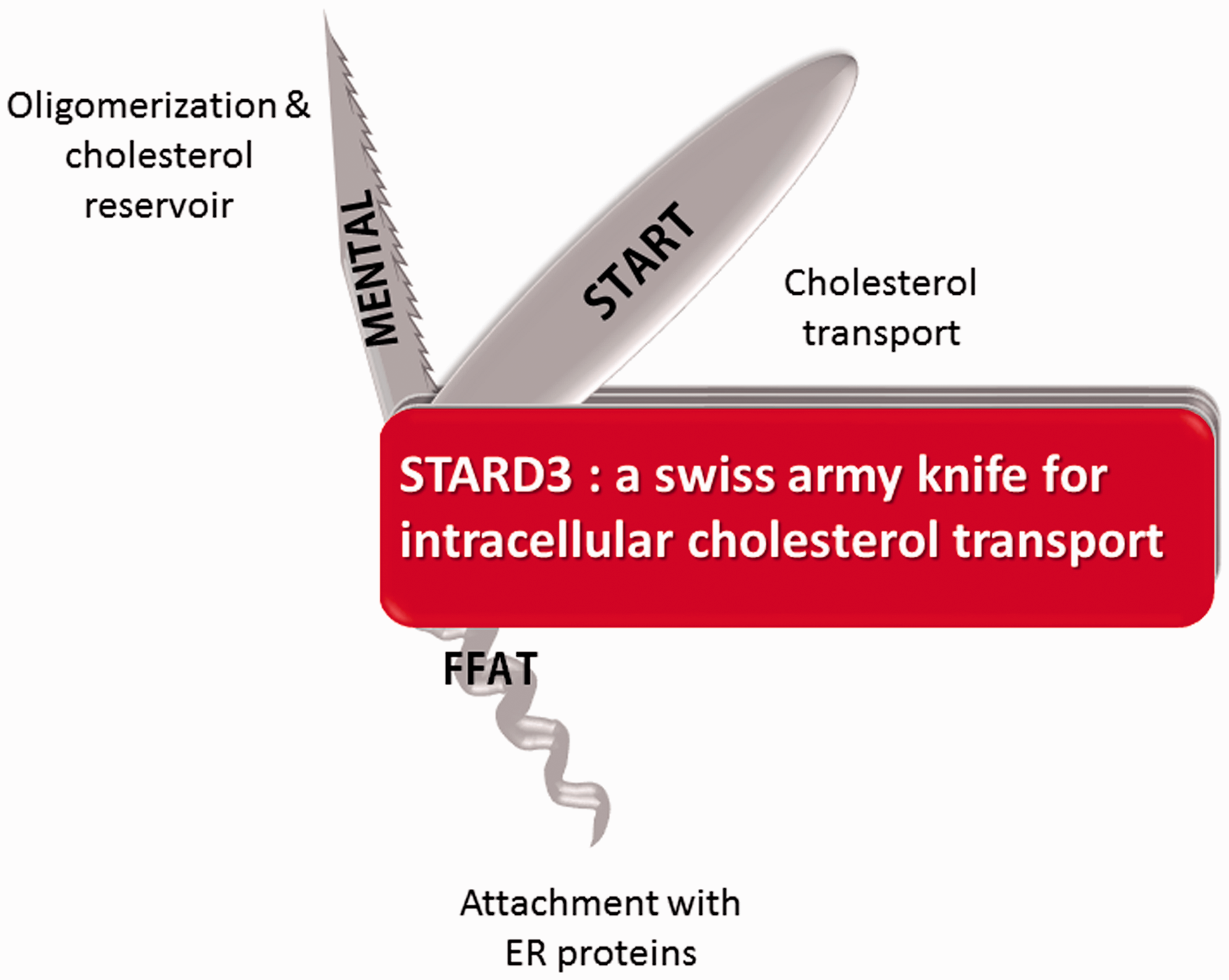
Title visual narrative. MENTAL: MLN64 N-terminal domain; FFAT: two phenylalanines in an acidic tract motif; START: steroidogenic acute regulatory-related lipid transfer domain; ER: endoplasmic reticulum; STARD3: steroidogenic acute regulatory-related lipid transfer domain-3.
Footnotes
Acknowledgments
The authors acknowledge core funds from the Institut National de Santé et de Recherche Médicale (http://www.inserm.fr/), the Centre National de la Recherche Scientifique (http://www.cnrs.fr/), the Université de Strasbourg (![]() ), and the ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir ANR-10-IDEX-0002-02.
), and the ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir ANR-10-IDEX-0002-02.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: LV is supported by an allocation from the Ministère de l’Enseignement Supérieur et de la Recherche (France; http://www.enseignementsup-recherche.gouv.fr/). TDM is supported by a Fondation pour la Recherche Médicale fellowship. M. L. is supported by a fellowship from the Faculty of Medicine, University of Strasbourg. This work was supported by grants from the Institut National Du Cancer INCA (INCA_9269; www.e-cancer.fr) and the Ligue Contre le Cancer (Conférence de Coordination Interrégionale du Grand Est; ![]() ).
).