
Abstract
Glycoproteins play key roles in various molecular and cellular functions and are among the most difficult to analyze biomolecules on account of their microheterogeneity, non-template-driven synthesis, and low abundances. The stability, serum half-life, immunogenicity, and biological activity of therapeutic glycoproteins, including antibodies, vaccines, and biomarkers, are regulated by their glycosylation profile. Thus, there is increasing demand for the qualitative and quantitative characterization and validation of glycosylation on glycoproteins. One of the most important derivatization processes for the structural characterization of released glycans by mass spectrometry (MS) is permethylation. We have recently developed a permethylation strategy in microscale that allows facile permethylation of glycans and permits the processing of large sample sets in nanogram amounts through high-throughput sample handling. Here, we are reporting the wide potential of micropermethylation-based high-throughput structural analysis of glycans from various sources, including human plasma, mammalian cells, and purified glycoproteins, through an automated tandem electrospray ionization–mass spectrometry (ESI-MSn) platform. The glycans released from the plasma, cells, and glycoproteins are permethylated in microscale in a 96-well plate or microcentrifuge tube and isolated by a C18 tip-based cleanup through a shorter and simple process. We have developed a workflow to accomplish an in-depth automated structural characterization MS program for permethylated N/O-glycans through an automated high-throughput multistage tandem MS acquisition. We have demonstrated the utility of this workflow using the examples of sialic acid linkages and bisecting GlcNAc (N-acetylglucosamine) on the glycans. This approach can automate the high-throughput screening of glycosylation on large sample sets of glycoproteins, including clinical glycan biomarkers and glycoprotein therapeutics.
Introduction
Glycosylation is one of the most common posttranslational modifications on proteins, plays important roles in cell signaling and molecular recognition, and also influences the efficacy of therapeutic molecules.1,2 The study of glycosylated proteins has lagged behind that of nucleic acids and nonglycosylated proteins due to the structural complexity of glycans and their non-template-driven biosynthesis. In the last couple of decades, the number of studies on the biological significance of glycosylation has dramatically increased. Many of these studies have investigated the impact of glycosylation on the efficacy of therapeutics, such as monoclonal antibodies, vaccines, and other glycoprotein-based drugs. 2 However, the analytical characterization of these molecules is difficult and requires highly specialized skill sets, instrumentation, and time-consuming sample handling steps. Routine, automated, and high-throughput workflows, common in genomics and proteomics, are not yet prevalent for glycan profiling. Consequently, the detailed glycan structure analysis is still dependent on a small group of experts, leading to a bottleneck for the progress in biomedical glycomics research. 3
Because of the complex relationships between glycan structures and their functions, the development of highly sensitive, quantitatively and qualitatively accurate and reliable analytical techniques for the characterization of glycosylation on glycoproteins is in high demand.4,5 Considerable progress has been made in the field of glycoprotein characterization due to advancements in mass spectrometry (MS), chromatographic systems, nuclear magnetic resonance, microarray technologies, and improved sample preparation techniques.4–6 Very low sample amounts available from clinical sources, the heterogeneity of glycosylation, and recent discoveries of unusual novel glycoconjugates demand the development of more robust, sensitive, and high-throughput methods for glycoprotein analysis. 7
Several approaches for automated glycan derivatization and analysis have been published, including fluorophore labeling and capillary electrophoresis,8,9 ethyl esterification and matrix-assisted laser desorption/ionization–mass spectrometry (MALDI-MS), 10 permethylation followed by MALDI-MS, 11 fluorophore labeling and ultra-high-performance liquid chromatography (UHPLC), 12 intelligent precursor selection electrospray ionization–mass spectrometry (ESI-MS), 13 and porous graphitized carbon–liquid chromatography–mass spectrometry (PGC-LC-MS). 14 Among these, only MS offers the capability to identify structural isomers without resorting to commercially available standards.15,16 Among MS-based methods, derivatization by permethylation has clearly shown the greatest potential for detailed structure characterization of numerous glycan types.7,15 Permethylation leads to improved ionization efficiency, sensitivity, and stability and enables facile determination of branching, interglycosidic linkages, and the presence of isomers. By making glycans lipophilic, permethylation also allows use of reversed-phase LC for the separation of glycoforms prior to the MS analysis. 17
Several permethylation methods, including solution-phase or solid-phase reactions, can be used for the derivatization of oligosaccharides prior to MS. 7 However, the solution-phase approach is most common and is based on the addition of methyl iodide (MeI) to oligosaccharides dissolved in DMSO containing either powdered 18 or gel-based sodium hydroxide (NaOH). 19 Although this method is successful in various MS-based structural studies of complex oligosaccharides, it is a multistep process involving time-consuming sample handling, sample cleanup steps, and other labor-intensive procedures requiring skilled analysts. Side reactions, such as oxidative degradation and “peeling,” are associated with the high pH resulting from dissolving NaOH powder. 20
These drawbacks were previously addressed by solid-phase permethylation, where a fused-silica capillary or a spin column packed with a solid inorganic base is used. However, solid-phase permethylation procedures require specialized apparatus to pack the solid inorganic base, which is very moisture sensitive. Moreover, to prevent incomplete methylation (“undermethylation”), repeated passage of glycan samples through solid-phase materials is often required.20,21 Recently, an automated and high-throughput glycan preparation and permethylation method in 96-well format was reported as convenient, fast, reliable, and applicable for drug glycan profiling and clinical glycan biomarker studies. 11 The procedure involves N- and O-glycan release, solid-phase extraction (SPE) of glycans using a hydrophilic liquid interaction chromatography (HILIC) resin, permethylation of released glycans, liquid–liquid extraction (LLE), data acquisition, and semiautomated data analysis. This permethylation is performed in a premade plate containing solid NaOH, followed by LLE and multiple transfers of samples between plates at each stage, and has an incubation time of 5 h. 11 Moreover, the specialized 96-well plates are moisture sensitive and expensive.
To address these challenges, we have developed a permethylation strategy that is simple, robust, suitable for large as well as small sample sets, and cost-effective. 22 This efficient and sensitive microplate-based permethylation reaction can be automated and is suitable for the high-throughput, reliable permethylation of glycans.
For the present study, it was our goal to automate as much as possible the detailed structural characterization of large numbers of samples of the same type. The proposed workflow involves the recently published automated, high-throughput permethylation and sample workup, and multistage MSn data acquisition. The latter requires initial setup using experimental results from a single, representative sample, after which the experiment can be conducted automatically on large sample sets, provided the samples are of the same type and the same specific information is sought. Potential examples of such sample sets would include therapeutic antibodies, vaccines, recombinant proteins, patient plasma samples, and cell lines. Here, we report the applicability of this high-throughput glycomics strategy for the characterization of both N- and O-glycans released from human plasma, human cell lines, and purified glycoproteins. We have developed an optimized, automated ESI-MSn direct infusion method using an autosampler for the high-throughput tandem MSn acquisition of permethylated glycans. Thus, the glycoforms and their structural and linkage isomers of 96 glycan samples can be analyzed in automated fashion based on a customized MS acquisition program. We demonstrate a high-throughput ESI-MSn data acquisition program that enabled in-depth structural characterization of glycans, including terminal sialic acid linkages (α-2→3 and α-2→6) on N- and O-glycans and bisecting GlcNAc (N-acetylglucosamine) determination on N-glycans.23,24 We also introduce a way to take advantage of the autoinjector of a widely used LC-MS instrument for the automated direct infusion of a large number of samples.
Materials and Methods
General Materials
Bovine fetuin, dithiothreitol (DTT), iodoacetamide (IAA), and (MeI were purchased from Sigma Aldrich (St. Louis, MO). Purified human serum IgG was purchased from Innovative Research (Novi, MI) (cat. 7660), and human plasma was purchased from Tennessee Blood Services (Memphis, TN). C18 tips were purchased from Thermo Scientific (Waltham, MA) (cat. 87782). Maltoheptaose was purchased from Carbosynth (Berkshire, England) (cat. OM06868). PNGase F was purchased from New England Biolabs (NEB; Ipswich, MA) (cat. P0704L). All other chemical reagents were purchased from Sigma Aldrich, unless otherwise mentioned. Mass spectrometric data acquisition was performed in positive ion mode on an AB Sciex MALDI TOF/TOF 5800 (Applied Biosystem MDS Analytical Technologies, Waltham, MA) mass spectrometer. ESI-MSn analysis was performed in an Orbitrap Fusion Tribrid mass spectrometer coupled with Dionex Ultimate 3000 nano-LC.
HEK 293f Wild Type Cell Culture
Wild type HEK 293f cells (FreeStyle 293-F cells; Thermo Fisher Scientific) were grown as shake flask cultures in Freestyle media (Freestyle 293 Expression Medium; Thermo Fisher Scientific) at 37 °C under shaking conditions at 120 rpm with 5% CO2. The cells were allowed to grow until reaching a density of 2.5 × 106 cells/mL with viability >95%. Once the desired cell number was achieved, the cells were harvested by centrifugation at 1200 rpm for 10 min at room temperature (20 °C), and the cell pellets were used for further study. Cell number and viability were quantified using a Countess Automated Cell Counter (Invitrogen) using trypan blue dye.
Release of N-Linked Glycans from Cells
About 5 × 106 HEK 293f cells were lysed in a high-salt buffer (2 M NaCl, 5 mM EDTA in 100 mM Tris-HCl, pH 7.5) and centrifuged at 15,000 rcf for 5 min, and the pellet was collected. The protein pellet is subsequently dissolved in a urea lysis buffer (8 M urea, 4% CHAPS, 100 mM DTT, 5 mM EDTA in 100 mM Tris-HCl, pH 8) and heated at 60 °C for 45 min for protein reduction. Further, 300 mM IAA was added to the protein sample and incubated in the dark at room temperature for 45 min. The urea and other salts in the samples were exchanged with NEB glycobuffer 2 (1×) using 10 kDa cutoff filters. The samples in centrifugal filters were first centrifuged to remove the salts and solvents. To the sample in filter 300 µL of NEB glycobuffer 2 (1×) was subsequently added and centrifuged. This process was repeated one more time, and thus the sample buffer was exchanged to NEB glycobuffer 2 (1×) during the process. One microgram of maltoheptaose (internal standard) was added to the protein mixture, followed by 5 µL of PNGase F, and the mixture was incubated at 37 °C for 16 h to release the N-linked glycans. The N-glycans were recovered by passing the mixture through a C18 cartridge, eluting the N-glycans with 5% acetic acid, and lyophilization.
Release of N-Linked Glycans from Human Plasma
About 5 µL of plasma was diluted with 95 µL of water, and the proteins were precipitated by adding 150 µL of chloroform, 500 µL of methanol, and 500 µL of water. The precipitated proteins were washed with 500 µL of methanol and resuspended in 50 µL of 50 mM ammonium bicarbonate buffer, pH 7.8. The proteins were reduced by adding 25 µL of 25 mM DTT (incubated for 45 min at 60 °C), alkylated by adding 25 µL of 90 mM IAA (incubated for 45 min at room temperature in the dark), and desalted using a 10 kDa cutoff centrifugal filter as described previously. The sample was collected in another microcentrifuge tube and 4 µL of PNGase F (NEB) and 1 µg of maltoheptaose (internal standard) were added. The mixture was incubated at 37 °C for 16 h to release the N-linked glycans. To recover the N-glycans, the mixture was passed through a C18 cartridge, and the N-glycans were eluted with 5% acetic acid and lyophilized.
Release of N-Linked Glycans from Purified Glycoproteins
The glycoproteins (1–1000 µg) were dissolved in 50 µL of 50 mM ammonium bicarbonate, pH 7.8, and reduced by adding 25 µL of 25 mM DTT (incubated for 45 min at 60 °C), alkylated by adding 25 µL of 90 mM IAA (incubated for 45 min at room temperature in the dark), and desalted using a 10 kDa cutoff centrifugal filter. The sample buffer was exchanged to NEB glycobuffer 2 (1×) during the process, and the new solution was directly treated with 0.1–10 µL of PNGase F (NEB) and incubated at 37 °C for 16 h to release the N-linked glycans. To recover the N-glycans, the mixture was passed through a C18 cartridge and the N-glycans were eluted with 5% acetic acid and lyophilized.
Preparation of NaOH-DMSO Base
The NaOH-DMSO base preparation is a modification of the one described by Anumula and Taylor. 19 Briefly, 100 µL of 50% NaOH solution taken in a clean stoppered glass tube was mixed vigorously with 200 µL of methanol. About 4 mL of anhydrous DMSO was added to this mixture and mixed vigorously by shaking. This process precipitates the carbonates present in the mixture solution as a white fluffy solid. The tube was centrifuged at 3000g for 5 min; this leads to precipitation of carbonates at the top of the mixture. The precipitated white solid at the top and excess DMSO were removed carefully while retaining the colorless gel at the bottom of the tube. This procedure was repeated four more times until the white solid precipitation ceased. Further, the resulting NaOH-DMSO gel that formed at the bottom of the tube was mixed with 1 mL of anhydrous DMSO and used for the permethylation reaction directly or stored under anhydrous conditions.
Permethylation of Glycans in a 96-Well Plate
The glycans released from the HEK 293f cells, plasma, or purified glycoproteins were dissolved in 50 µL of anhydrous DMSO and transferred to a 500 µL 96-well plate (polypropylene) or a 1.5 mL microcentrifuge tube (polypropylene). To this, 75 µL of the NaOH-DMSO base was added. After mixing by repeated aspiration with the pipette, 25 µL of MeI was added to the well or tube, pipette mixing was repeated, and the sample was incubated on a shaker for 20 min. The permethylation reaction was quenched by adding 100 µL of ddH2O. Excess MeI was bubbled off carefully by immersing the pipette tips into the solution and dispensing air to the reaction mixture. Regular polypropylene tips were used for all addition, mixing, and aspirations. The permethylated glycans were then passed through preequilibrated C18 tips for loading. The C18 tips were washed in 1 mL of ddH2O, and the permethylated glycans were eluted to a fresh plate or tubes containing 100% methanol. The permethylated glycans were dried and stored at −20 °C until MS analysis.
MALDI-TOF MS Analysis of Glycans
The permethylated glycans were dissolved in 10 µL of methanol, and 2 µL of this solution was mixed with 2 µL of 2,5-dihydroxybenzoic acid (DHB) matrix (20 µg in 50% methanol in water), spotted on a MALDI plate, and analyzed on a MALDI-TOF/TOF MS instrument in the reflector mode.
ESI-MSn Analysis of Glycans
The permethylated glycans were dissolved in 20 µL of methanol, and a 5 μL aliquot of the methanolic solution was mixed with 45 μL of ESI-MS infusion buffer (50%; aqueous acetonitrile and 0.1% formic acid). The sample was infused into the mass spectrometer through the autosampler at a rate of 0.5–1 µL/min, and a precursor scan was acquired at 120,000 resolution. Subsequently, collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) MS/MS were acquired by a total ion monitoring (TIM) program, which performs data-independent MS/MS acquisition of all ions from m/z 400 to 2000 with an increment of 1 Da. MSn analysis of the top 20 peaks and targeted MSn based on the inclusion list were also acquired for the detailed structural characterization of glycans.
Structure Interpretation
Assignment of permethylated N- and O-glycan structures was performed manually based on MALDI-MS and ESI-MSn with the aid of software such as GlycoWorkbench 1.1, Data Explorer V4.5, and Xcalibur.
Results and Discussion
We have made modifications to the existing method for the permethylation of glycans and downsized the reaction scale from milliliters to microliters, while improving the sensitivity of the analysis (
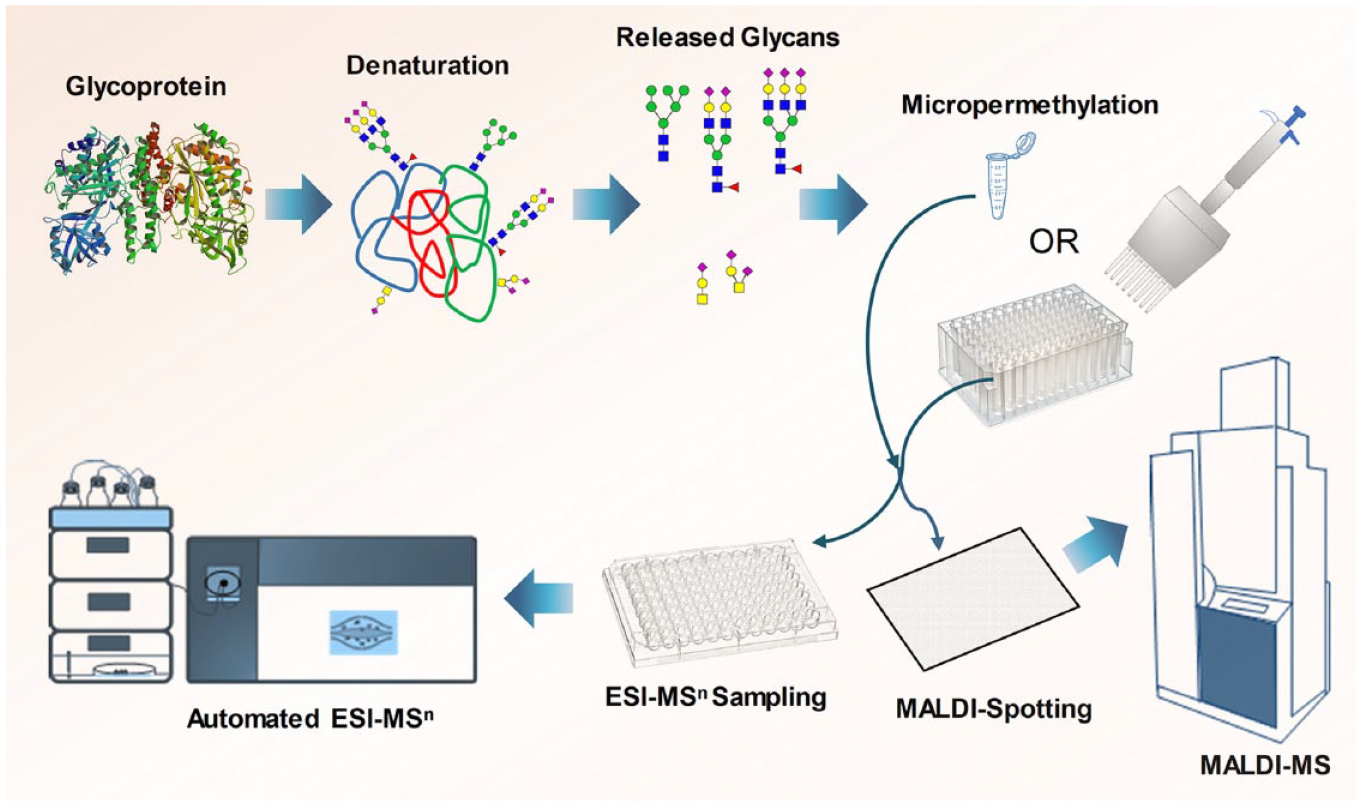
High-throughput and microscale analysis of N- and O-linked glycans through micropermethylation and MALDI-MS/ESI-MSn. The micropermethylation reaction was conducted in a 96-deep-well polypropylene plate (for high throughput) or in a 1.5 mL microcentrifuge tube (for analysis of a small number of samples), reaction was stopped by water, and the permethylated glycans were purified by C18 tips and analyzed by MALDI-MS and ESI-MSn.
Microscale Permethylation of Glycans in Solution Phase
Here we employed a ready-to-use NaOH-DMSO gel as the base for the micropermethylation reaction in 96-well plate format, as we reported previously.
22
In brief, the glycans in the wells of a polypropylene microplate were dissolved in DMSO and treated with the NaOH-DMSO base, followed by iodomethane. After being sealed with a silica-based lid, the microplates were incubated for 20 min at room temperature to ensure complete methylation. The reaction was quenched by the addition of water, upon which the excess iodomethane was partitioned as a separate lower layer. The iodomethane was removed by repeatedly sparging air through the mixtures by means of a pipette while immersing the tip in the solution. We used the previously optimized reaction conditions for the permethylation of N- and O-glycans released from 1 mg to 1 µg of glycoproteins (human serum IgG), 5 million human cells (N-glycans), and 5 µL of human plasma (N- and O-glycans) (
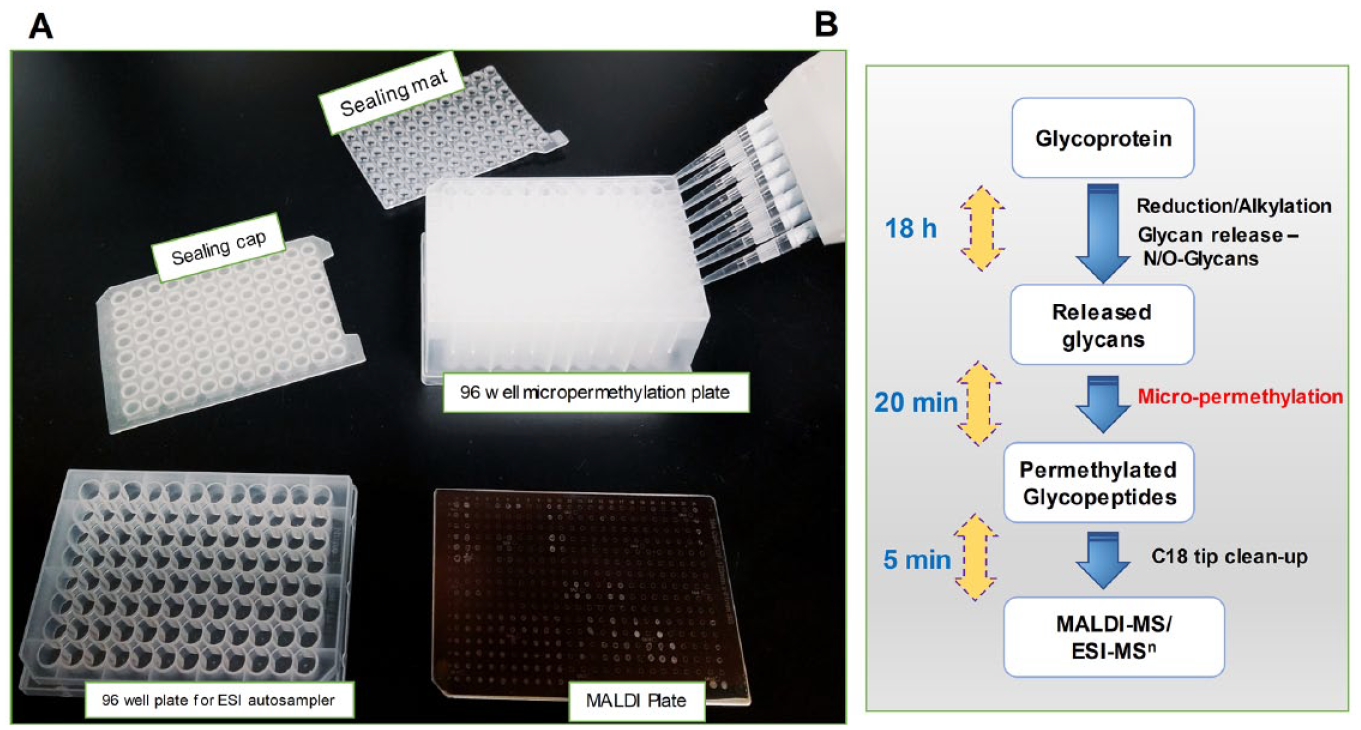
(
Purification of Permethylated Glycans by SPE Using a C18 Tip
LLE has historically been the method of choice for the isolation of permethylated glycans and was also used in a recent report on a high-throughput, microscale variation of the method. 11 The process of LLE requires multiple washes of the organic layer comprising isolated permethylated glycans with water to remove co-extracted DMSO and other reagent contaminants.7,19 This procedure of LLE is labor-intensive and requires high-level skills and precision due to the risks of inefficient separation of contaminants and sample losses. The recently developed automated LLE using robotics helps to a considerable extent, but this involves nonroutine laboratory equipment, making it unavailable to most glycan analysis facilities. 11
To address this limitation of LLE, we explored SPE-based purification of glycans after the permethylation reaction. For this SPE procedure, we used commercially available C18 tips, so that permethylated glycans can be extracted by simple pipetting with single or multichannel pipettes or 96-well semiautomated pipettes. Moreover, the generation of hazardous organic solvent wastes such as CH2Cl2 and CHCl3 can be avoided. Additionally, the SPE method is also suitable for the extraction of permethylated negatively charged glycans that cannot be recovered through LLE. 25 After preconditioning with methanol, the C18 tips were equilibrated with water, and further, the quenched, aqueous permethylation reaction mixture was passed through the tips. The tips were subsequently washed with water onto a separate microplate, and the permethylated glycans were eluted on another plate by passing methanol through the tips. To ensure complete transfer, each extraction step was carried out in five passes through the C18 tips. After the final step, the methanol solvent was evaporated using the pipette, redissolved in methanol, and spotted directly to the MALDI plate after premixing with matrix or used for ESI-MSn profiling.
Performing the reaction in microscale allowed us to achieve improved sensitivity and throughput while using the permethylation reagents in microliter quantities. In comparison with the only commercially available permethylation kit in 96-well format, our method has remarkable advantages, such as shorter reaction and sample purification times, and sample handling efforts. The commercial permethylation kit involves a permethylation reaction time of 1 h and sample purification by LLE involving sophisticated robotics that requires 5 h of sample processing in total. Our method is very cost-effective and consistent, involves a reaction time of only 20 min, and can be used for the analysis of glycans released from 1 mg to 1 µg of glycoproteins. In addition, the C18-based SPE (C18 tips) is employed for the sample cleanup instead of cumbersome LLE, and thus generates less harmful solvent wastes.
Our preliminary studies on oligomannose and glycans released from bovine RNase B, bovine fetuin, bovine κ-casein, human transferrin, and human IgG demonstrated that the solid-phase separation using C18 tips is effective for the permethylation reaction in microscale.
22
In the present study, we have applied the micropermethylation strategy to characterize the N- and O-glycans released from low quantities of more complex samples such as human plasma and cell lines. We have used maltoheptaose with permethylated m/z 1497.6 for N-glycans in nonreduced form and m/z 1513.8 for O-glycans in reduced form, as an internal standard for the quantitation of glycans. Sensitive detection of N- and O-glycoforms was achieved from 5 µL of human plasma and 5 million HEK 293f cells (
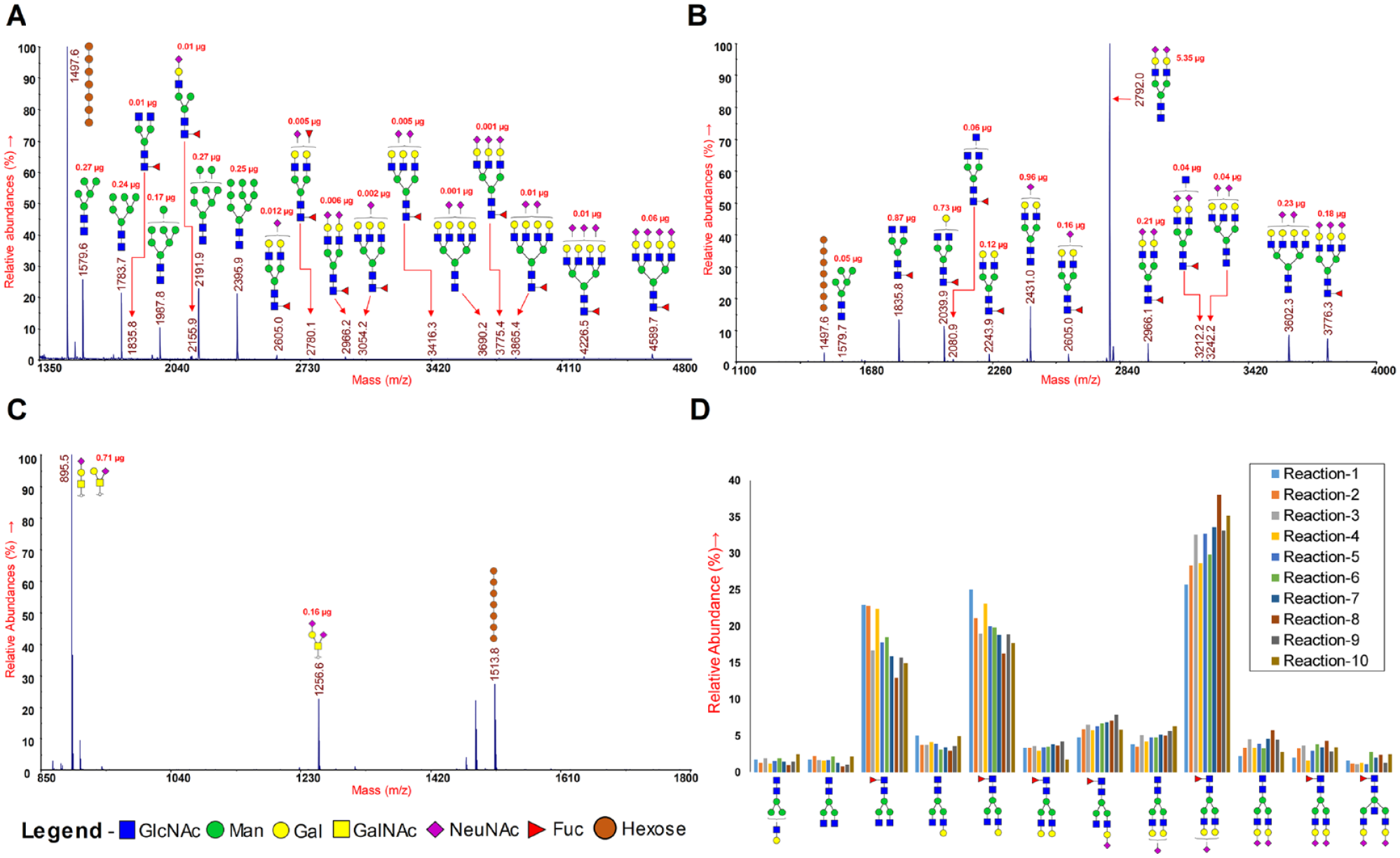
Glycans were released from glycoproteins by PNGase F (N-glycans) or β-elimination (O-glycans), micropermethylated, cleaned up by C18 tips, and analyzed by MALDI-MS/ESI-MSn. MALDI-MS spectra of released and permethylated glycans: (
Evaluation of Micropermethylation Reaction Consistency
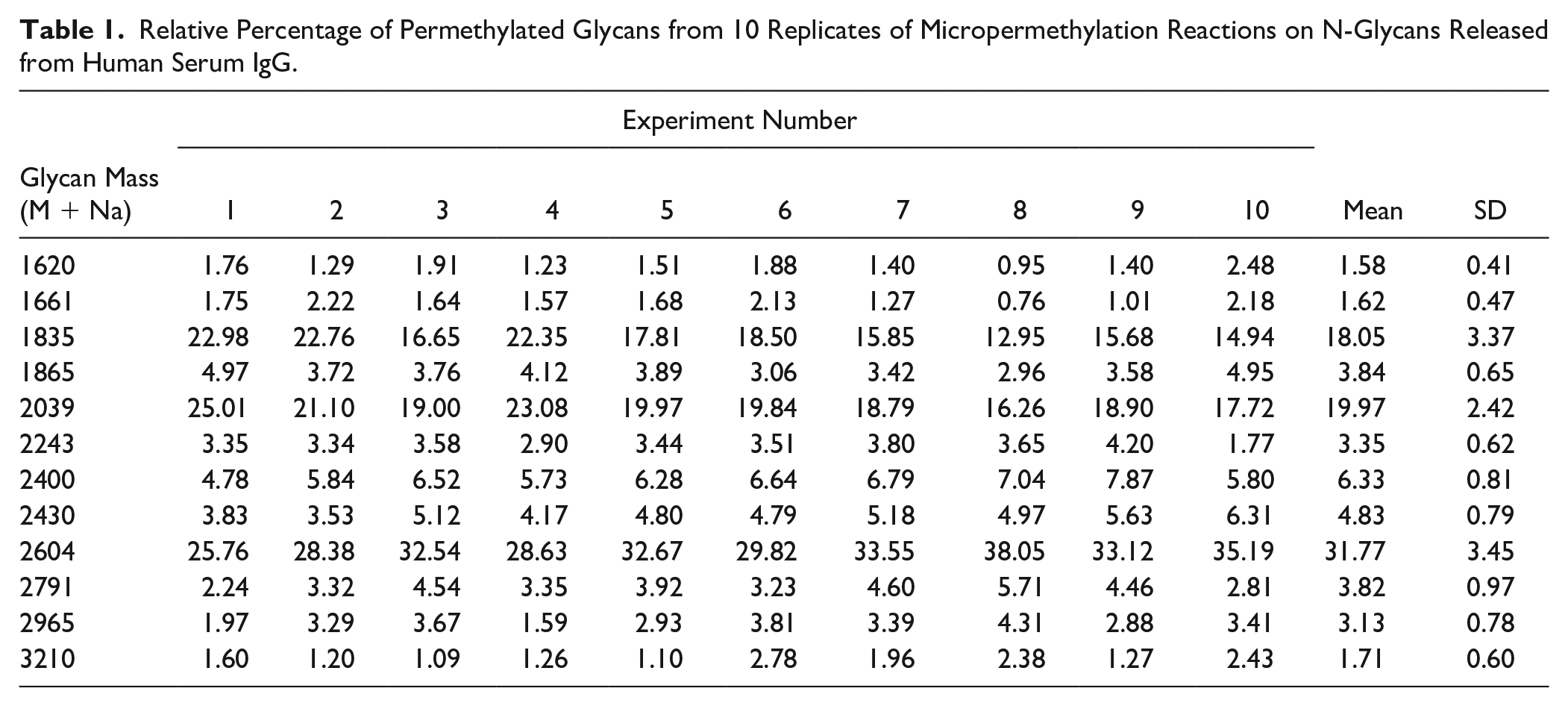
We performed 10 micropermethylation reactions on N-glycans released from the same human serum IgG and acquired MALDI-MS of these 10 reactions in duplicate. Relative abundances of glycoforms were calculated from the average peak area obtained from each of the duplicate spots of 10 micropermethylation reactions (
Table 1
). Notably, the relative abundances of glycoforms obtained from 10 reactions are consistent and comparable to those of regular permethylation and MALDI-MS. No statistically significant difference was observed between the reactions (ANOVA, p > 0.05) (
Relative Percentage of Permethylated Glycans from 10 Replicates of Micropermethylation Reactions on N-Glycans Released from Human Serum IgG.
Automated Direct Infusion of Permethylated Glycans for ESI-MSn Analysis
Currently, detailed structural characterization of glycans, including branching, linkages, and topology, is performed by manual direct infusion of permethylated glycans into the mass spectrometer. This manual infusion process is tedious, allows only a few sample acquisitions per day, and is prone to operator error. We envisaged that by employing the autosampler and sample loading system of a regular nano-LC system, the direct infusion of samples can be achieved automatically. Toward this goal, we infused the permethylated glycans into the ESI-MS system through an autosampler using the loading pump of a Dionex Ultimate 3000 nano-LC (
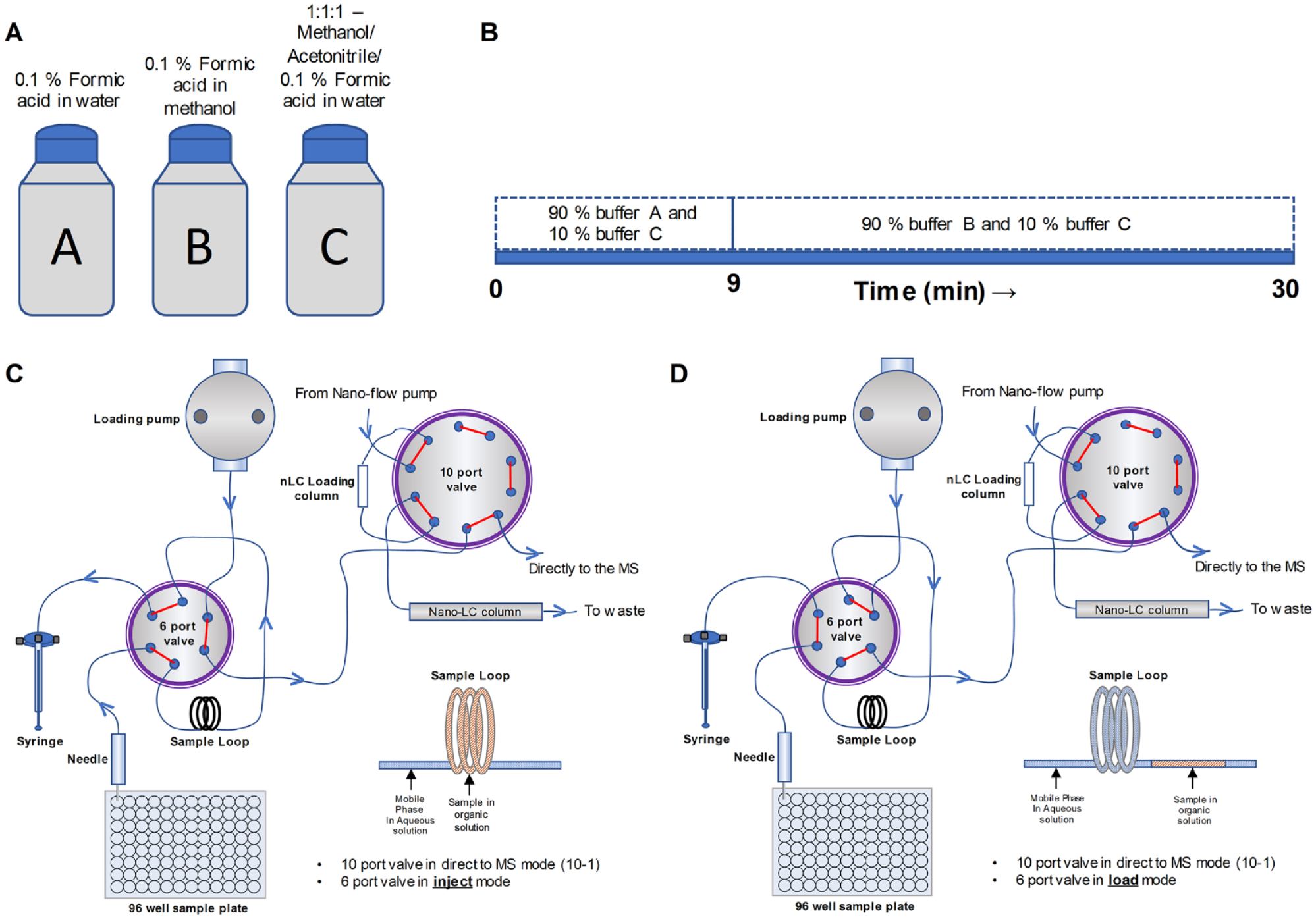
Diagram showing the connections for the automated ESI-MSn through direct infusion via an autosampler. The sample in the 96-well plate is injected and infused directly to the MS through the loading pump bypassing the nano-LC column. (
Optimizing the Buffer Constituents to Obtain Optimum Sample Elution, ESI Spray, and Introduction of Adduct Ions
Bypassing the loading column meant that we could not take advantage of its ability to prefocus the sample and initially led to unacceptable peak broadening and loss of sensitivity. To address this problem, we reconstituted the sample in an organic buffer, which prevented fast mixing of the sample plug with the much more polar mobile phase during autosampler pickup, transfer to the sample loop, and subsequent transport to the MS. Thus, we optimized the buffer constitution by changing the ratio of organic solvents and water in such a way that the sample picked up from the vial does not get mixed immediately with the mobile phase during the transport. Potential sample mixing with the mobile phase would affect the MS detection sensitivity as the sample would get diluted by the higher volume of the mobile phase. We tried several different combinations of buffer and solvents, monitoring spray stability, peak elution, and peak detection, and observed optimal performance with a mixture of 90% buffer A (0.1% formic acid in water) and 10% buffer C (1:1:1 0.1% aqueous formic acid/acetonitrile/methanol) as the mobile phase for the transport of sample reconstituted in 50% acetonitrile (
We next optimized the introduction of metal ions to the sample to form adducts that have been shown to enhance diagnostic fragmentations during the ESI-MS experiment. Obtaining these adducts through the addition of salts of either sodium (Na+) through 1 mM NaOH or lithium (Li2+) through 1 mM Li2CO3 is commonly practiced during ESI-MSn experiments via direct infusion. 26 For this purpose, we initially introduced the ions to the mobile phase by adding the salts to buffer C and changing its composition with buffer A. However, we found that this practice does not impart sufficient adducts to the analyte. We concluded that this is the result of the lack of mixing between the mobile phase and the sample during injection. Minimal mixing was the designed outcome of using a high organic percentage of the sample buffer to avoid peak broadening, and we reasoned that this could also be used advantageously to introduce salts through the sample. Thus, we added the respective salts to the sample in the vials instead of to any of the mobile phase components. Using this approach, we observed the efficient formation of adducts during the ESI-MSn acquisition. This provided remarkable flexibility in the process because different salts can be introduced conveniently and quickly without the need for solvent exchange and excessive flushing of the LC system between the use of different salts.
Configuration of LC System for Automated Direct Infusion
For automated direct infusion, the microplate containing the samples (dissolved in 50% aqueous acetonitrile with sodium or lithium ions) was placed in the autosampler, and the sample was introduced into the sample loop by the autosampler syringe (
N-glycans were released from bovine fetuin, permethylated by micropermethylation, and analyzed by ESI-MSn through direct infusion via an autosampler. (
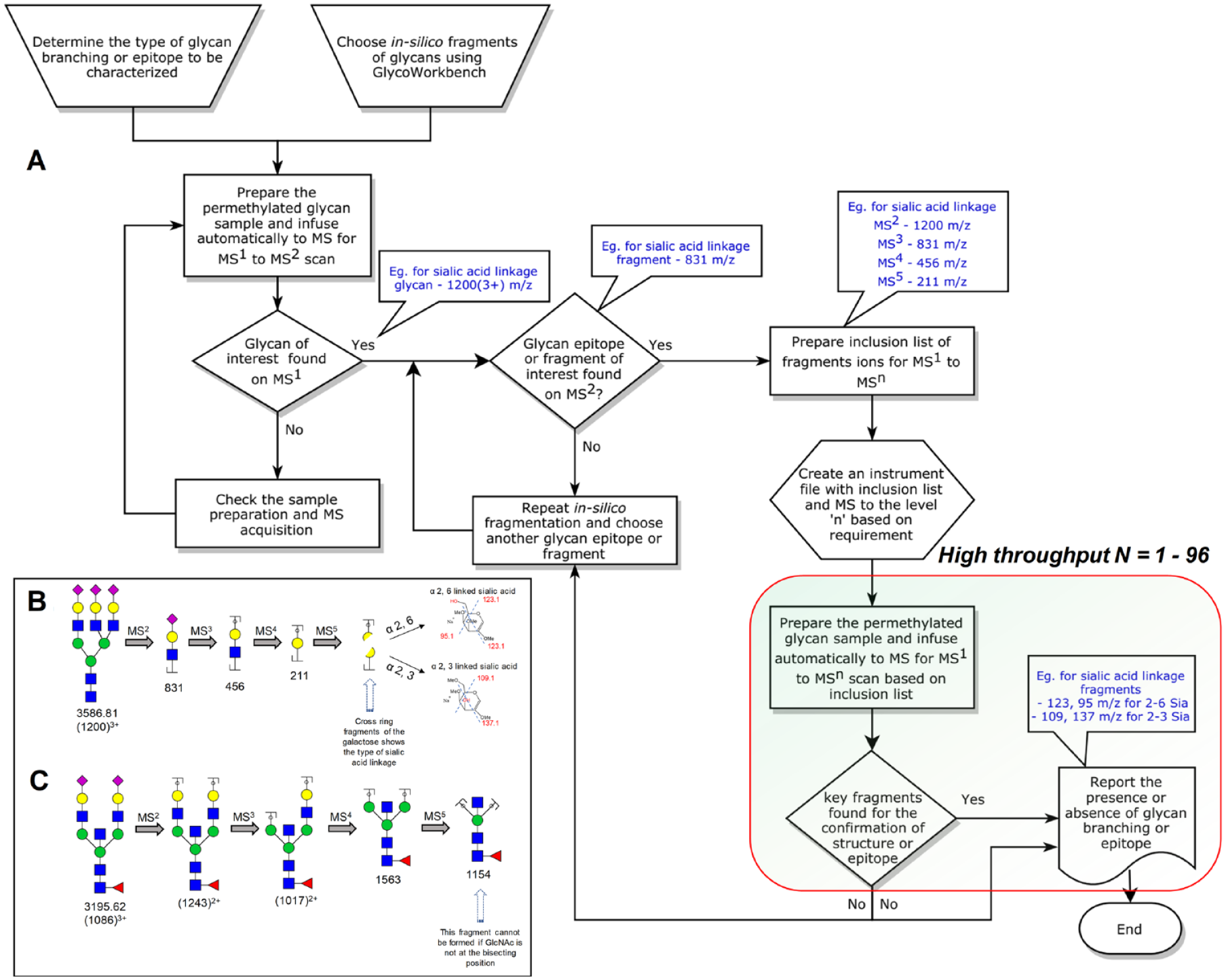
Programming MSn Experiments for Automated Structural and Linkage Isomer Identification
Permethylation allows the effective elucidation of glycan structural details by tagging the hydroxyls that are not part of glycosidic linkages. However, retrieving this type of information requires rather complex, multistage MSn experiments that resist automation. We have developed a process by which an MS instrument method file can be constructed that enables fully automated multistage MSn experiments for the structural characterization of certain biologically or pharmaceutically important glycan epitopes for large numbers of samples of the same type. The proposed workflow requires a one-time initial setup using one representative from a large number of samples. Subsequently, the multistage MSn data acquisition can be performed under full automation. A survey MS
1
and data-dependent MS
2
within the top 3 s was conducted on the released and permethylated N-glycans from bovine fetuin (
(
Sialic Acid Linkage Determination by Automated ESI-MSn
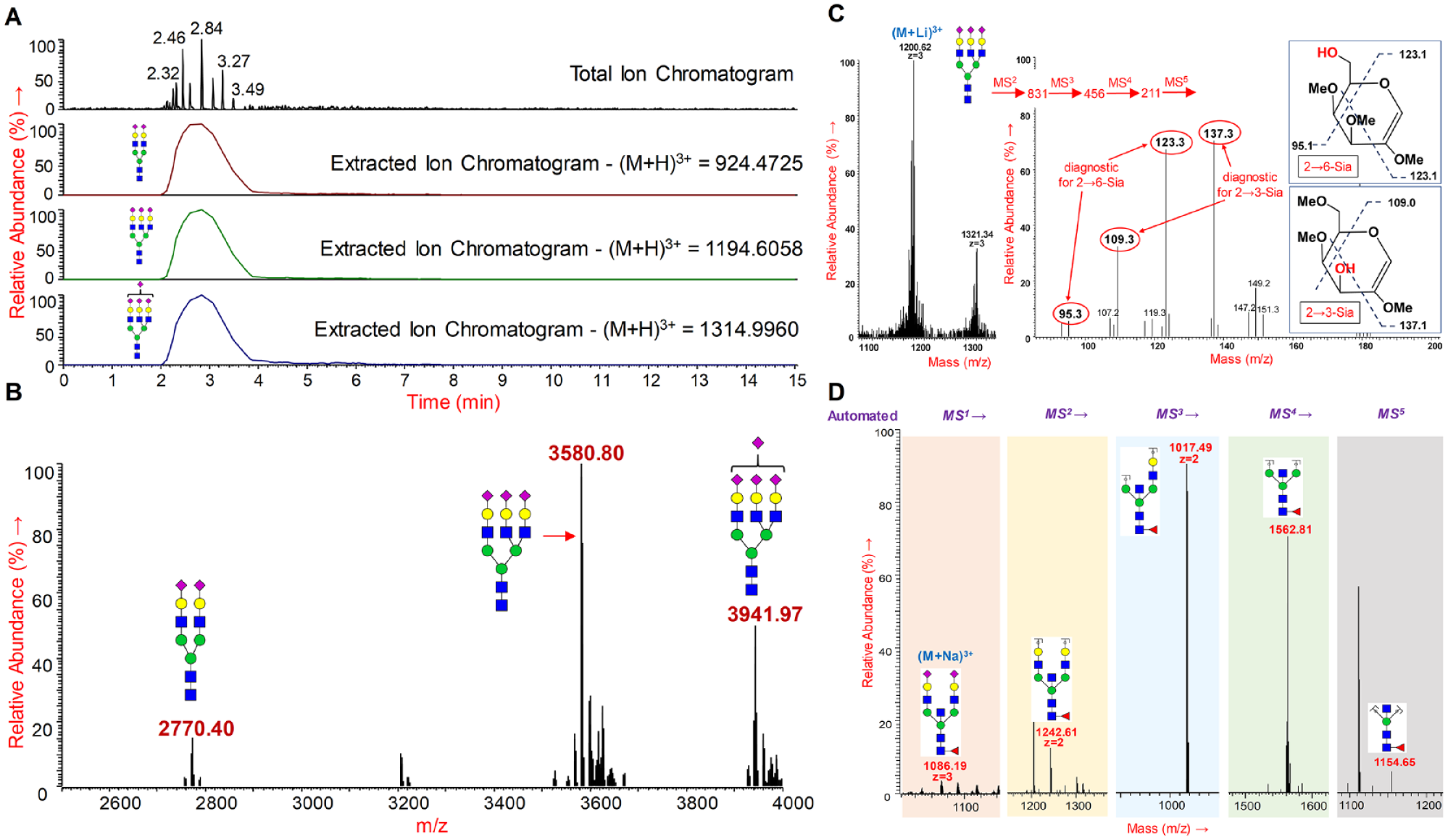
A typical example of a biologically significant epitope variation is the regioisomerism involved in the linkage of sialic acid to galactose in sialylated glycoproteins. Sialic acids are linked to the preceding galactose through either α-2→3 or α-2→6 linkages. Glycan-mediated cell signaling and ligand binding processes frequently rely on sialic acid residues at the glycan nonreducing termini, and the terminal linkages of sialic acid play a critical role in such processes. The expression of sialyltransferases and the sialylation linkages they create on the surface of T cells change during their maturation in the thymus. 27 Another study demonstrated that the anti-inflammatory activity of IgG Fc is influenced by the type of sialic acid linkages on the N-glycans. 23 Human parainfluenza virus type 1 (hPIV1) binds only to α-2→3-linked sialic acid residues, whereas type 3 (hPIV3) binds only to α-2→6-linked sialic acid residues. 28 This underscores the importance of sialic acid linkage determination, which is commonly done by employing either lectins or tandem sequential MSn fragmentation methods.23,27
Our proposed process allowed the automated determination of the sialic acid linkages on the N-glycan termini. To this end, the released and micropermethylated N-glycans from bovine fetuin were infused into the mass spectrometer with our automated injection program and analyzed using the customized and automated MSn program containing the MS 1 to MS 5 fragment mass inclusion list. The glycans were infused with lithium salts for increased cross-ring fragmentation. 23 The cross-ring fragments of the partially methylated galactose to which the sialic acids are either 2→3- or 2→6-linked are different and allow the identification of sialic acid linkages.23,26
The sialic acid linkage (α-2→3 and α-2→6) was determined on bovine fetuin N-glycans using the ESI-MS
5
experiment through the direct infusion of micropermethylated N-glycans via autosampler. The triantennary trisialylated N-glycan (M+Li; 1200.283+) precursor was automatically isolated and fragmented by MS
2
to form the sialylated LacNAc (N-acetyl lactosamine) fragment 831.431+. Further, the sialylated LacNAc fragment was de-sialylated by MS
3
fragmentation to form the LacNAc fragment 456.241+. MS
4
on the LacNAc fragment 456.241+ formed the fragment 211.111+ of galactose to which the sialic acids are linked. Subsequent MS
5
fragmentation of this galactose fragment 211.111+ formed signature crosslink fragments supporting the presence of both 2→3- and 2→6-linked sialic acids.23,26 We prepared an inclusion list with all of the above-mentioned key fragments for the confirmation of 2→3- and 2→6-linked sialic acids and programmed an instrument file with MS to level 5. The presence of key MS
5
fragments validating sialic acid linkages was monitored on the MS data file (
Identification of Bisecting GlcNAc on Human Plasma by Automated ESI-MSn
Another example of a biologically important glycan structural detail is bisecting GlcNAc. This feature is important for a variety of biological functions, such as cell–cell and cell–matrix interactions, cell growth control, and tumor progression.29,30 A β1,4-linked GlcNAc residue is termed “bisecting” if it is attached to the 4-position of a β-mannose of the N-glycan core, between the antennae linked to the 3- and 6-positions of that same β-mannose residue. Biosynthetic attachment of this residue is catalyzed by N-acetylglucosaminyltransferase-III (GnT-III/Mgat-III). The presence of bisecting GlcNAc on N-glycans can be unambiguously determined by MSn sequential fragmentation of permethylated glycans. 24
Taking an approach similar to sialic acid linkage determination, we used automated MSn fragmentation for the detection of bisecting GlcNAc in human plasma N-glycans. Serum N-glycan bisecting GlcNAc is indicated as a potential determinant for longevity, changes in cellular physiology, and biomarkers for various diseases, including cancer.23,29,31,32
The bisecting GlcNAc determination on human plasma N-glycans was determined by an ESI-MS
5
experiment through the direct infusion of micropermethylated N-glycans via an autosampler. The MS data acquisition method was programmed to perform MS to level 5 by choosing fragments at each MS level in such a way as to confirm the bisecting GlcNAc structure. The biantennary, disialylated, bisecting GlcNAc-bearing N-glycan glycoform with a precursor mass of 1085.853+ (M + Na) was fragmented by CID, and the resulting de-sialylated MS
2
fragment 1242.102+ was further fragmented by CID MS
3
. Subsequently, the two LacNAc units on each arm were removed by sequential MS
4
(1017.492+) and MS
5
(1562.771+) fragmentations. The presence of fragment 1154.571+ on the MS
5
spectrum confirmed the bisecting GlcNAc as such a fragment would not be possible if the GlcNAc were attached to one of the antennae. We made an inclusion list with all of the above-mentioned key fragments for the bisecting GlcNAc confirmation and programmed an instrument file with MS to level 5. The presence of the key MS
5
fragment was monitored on the data file to validate the presence of bisecting GlcNAc (
The automated method described here will help in screening the large sample set of plasma for the structural variation in key glycan epitopes such as sialic acid linkages, bisecting GlcNAc, alpha-galactose, and core fucosylation.
We have used the micropermethylation process for the analysis of both N- and O-glycans released from human cell lines, human plasma, and purified glycoproteins. The micropermethylation of glycans coupled with automated ESI-MSn profiling would enable convenient, rapid, and reliable high-throughput screening of glycosylation in detail and can be used for the analysis of large sample sets of glycoprotein therapeutics and glycosylated biomarkers.
Supplemental Material
Supplemental_Material_Micropermethylation_Shajahan_et_al – Supplemental material for Simplifying Glycan Profiling through a High-Throughput Micropermethylation Strategy
Supplemental material, Supplemental_Material_Micropermethylation_Shajahan_et_al for Simplifying Glycan Profiling through a High-Throughput Micropermethylation Strategy by Asif Shajahan, Nitin T. Supekar, Digantkumar Chapla, Christian Heiss, Kelley W. Moremen and Parastoo Azadi in SLAS Technology
Footnotes
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received the following financial support for the research, authorship, and/or publication of this article: a grant from the U.S. National Institutes of Health (S10OD018530).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.