
Abstract
The use of bioengineered skin has facilitated fundamental and applied research because it enables the investigation of complex interactions between various cell types as well as the extracellular matrix. The predominantly manual fabrication of these living tissues means, however, that their quality, standardization, and production volume are extremely dependent on the technician’s experience. Simple laboratory automation could facilitate the use of living tissues by a greater number of research groups. We developed and present here an injection molding technique for the fabrication of bilayered skin equivalents. The tissue was formed automatically by two separate injections into a customized mold to produce the dermal and epidermal skin layers. We demonstrated the biocompatibility of this fabrication process and confirmed the resulting bilayered morphology of the bioengineered skin using histology and immunohistochemistry. Our findings highlight the possibility of fabricating multilayered living tissue by injection molding, suggesting that further investigation into this automation method could result in the rapid and low-cost fabrication of standardized bioengineered skin.
Introduction
Bioengineered skin has been extensively used to study the pathogenesis of complex multifactorial pathologies, such as scarring wounds,1–3 inflammatory skin diseases,4,5 and skin cancer. 6 Such skin equivalents typically comprise a biomaterial scaffold and a small number of different cell types that together mimic the dermal and epidermal layers of native skin. 7 When compared with monolayer and two-dimensional co-culture systems, such three-dimensional (3D) living tissues provide a cell environment that replicates human skin more closely, both morphologically and functionally. Many relevant cell reactions and gene expression profiles occur only in a 3D environment; thus, their importance in research and industry has been continually increasing. Furthermore, bioengineered skin is used as an alternative to animal testing in the pharmaceutical8,9 and cosmetic10,11 industries, as well as clinically for the purpose of skin grafting.12,13 Transplantation of skin grafts has been successfully performed in humans in Switzerland, Canada, and the United States.14–17 The demand for skin equivalents is increasing in all fields. Because of the current, predominantly manual, fabrication techniques, however, the quality and quantity of skin equivalents produced are largely dependent on the expertise of the technicians producing the tissues. Increasing the level of automation 18 in the fabrication process could both improve the standardization of bioengineered skin and increase production output, 19 and an automated fabrication technique would ultimately increase the commercial scalability of bioengineered skin.
Thus, to meet the increasing demand for bioengineered skin, fabrication principles that are designed for automation are required. One prominent approach to automating the fabrication of multilayered living tissues is 3D bioprinting.20,21 In this technique, a biomaterial that comprises cells is precisely dispensed at defined positions to create a 3D tissue, and this technique, which has been extensively reviewed, 22 has been repeatedly used to fabricate bilayer skin substitutes.23–28 Industrial 3D printing has thus been successfully used and incorporated in biomedical applications. 29 Besides industrial 3D printing, however, a multitude of other industrial fabrication methods exist, 30 each with specific advantages and strengths that could be of great value for the automation of biomedical fabrication processes.
Injection molding is commonly used in industrial fabrication processes; it offers high standardization, rapid fabrication, and high scalability,31–34 and therefore holds a high potential for the automation of bioengineered skin production. Injection molding has already been successfully used in the biomedical field to fabricate single-component living tissues for facial implants and has been shown to exhibit high geometrical accuracy and mechanical stability. Furthermore, subsequent tissue formation results in high cell viability, and the fabrication is easy, quick, and reproducible.35,36 Injection molding has not yet been applied, however, in the fabrication of multilayered living tissues. The ability to fabricate multilayered living tissues by injection molding could improve fabrication with respect to automation, standardization, and scalability.
In this study, we investigated the fabrication of bilayer bioengineered skin by injection molding and thereby developed a customized mold and an injection prototype. Collagen type I and human dermal fibroblasts were first automatically injected into the mold to produce the dermal layer of skin, and after 1 week of cultivation, the epidermal layer was formed by a second injection of human epidermal keratinocytes. The biocompatibility of the mold and the injection process was confirmed by cell viability and proliferation assays, while the multilayered morphology of the fabricated bioengineered skin was demonstrated by histological and immunohistochemical analyses.
Materials and Methods
The Principle of Multilayered Injection Molding
Figure 1 illustrates the principle of multilayered injection molding. Three main components are required: the mold, the case, and the injection apparatus (only the nozzle is shown). The mold, which is inserted into the case for the dermal layer injection, consists of a rigid frame and two membranes that together form a circular cavity ( Fig. 1A ). The case consists of top and bottom parts that prevent the membranes from expanding when the material of the dermal layer is injected ( Fig. 1B ). The mold is then removed from the case and transferred into a commercial culture dish for cultivation of the dermal layer. After 1 week of cultivation, the epidermal component is injected ( Fig. 1C ), during which the upper membrane is no longer restricted by the top of the case and bulges upward, resulting in the production of a small additional cavity for the epidermal component. The cells of the epidermal layer are confined to this cavity by the membrane and the dermal layer, and therefore cannot float away. This confinement also facilitates direct transfer into the culture medium with no requirement for a waiting period for cell attachment ( Fig. 1D ).
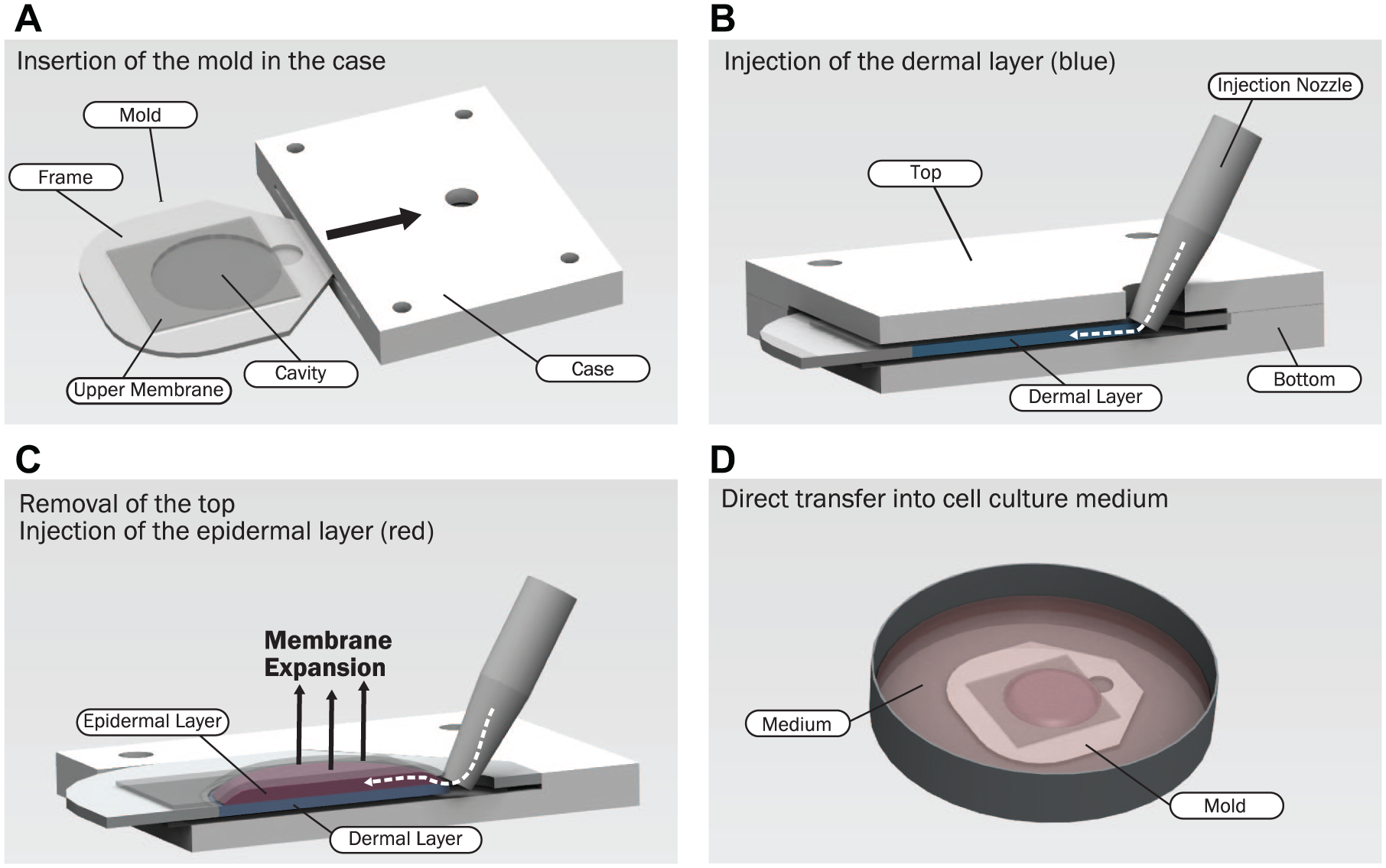
Schematic of the multilayered injection molding process and its components: the mold, the case, and the injection nozzle. (
The injection apparatus was fabricated from an aluminum frame (MakerBeam B.V., the Netherlands) and consisted of three parts, as shown in Figure 2A : the actuation and control unit (I), the dispensing unit (II), and the mold holder setup (III).

Fabrication prototype. (
The actuation and control unit was located in the upper part of the prototype and is denoted as I in Figure 2A . The setup was based on the open-source project OpenSyringePump 37 and adapted for injection molding. The adapted part files and code are published to supplement this article. 38 The system was controlled by an Arduino Uno microcontroller board with an ATmega328P chip (Arduino SRL, Italy). The user interface was an LCD Keypad shield with a matrix of 16×2 characters (RGB LCD I2C Shield Kit 16x2; Adafruit, New York, NY, USA) to display the set parameters and five buttons to receive inputs from the user, allowing them to set the dispense volume and injection rate. The total dispensed volume was shown on the display. Optionally, the injection and its respective parameters could also be controlled by a computer via a USB connection. The microcontroller code was based on the OpenSyringeProject for Arduino 39 and modified for this project. The microcontroller drives a standard triple-stack NEMA 17 stepper motor via a motor driver (Big Easy Driver; Sparkfun Electronics, Niwot, CO, USA) at 24 V (power supply unit; Alpha Elettronica, Collecchio, Italy). To increase precision and continuity during the injection process, the stepper was actuated by microstepping with a factor of 16. Depending on the syringe configuration, this resulted in 3200 steps per revolution and a theoretical resolution of between 0.1 and 0.3 µl per microstep. A trapezoidal treaded spindle with a pitch of 1.5 driven by the motor was used to move a sled, which was guided by linear ball bearings and connected to the syringe attachment.
The dispensing unit is denoted as II in Figure 2A , and it is shown in detail in Figure 2B for dermis injection and Figure 2C for epidermis injection. The unit was designed to hold a standard 10 ml syringe (B. Braun, Melsungen, Germany) and was printed from polylactic acid (PLA; Prusa Research, Prague, Czech Republic) with a fused deposition molding (FDM) 3D printer (i3 MK3; Prusa Research). A 1 ml pipette tip (Rainin, Dübendorf, Switzerland) was attached to the syringe with a silicone seal and used as injection nozzle for the dermis injection ( Fig. 2B ), while the same adapter with a 10 ml syringe equipped with a flexible Western blot pipette tip (Corning, Corning, NY, USA) was used for the epidermis injection ( Fig. 2C ).
The mold holder setup is denoted as III in Figure 2A and comprised a mold that was inserted into a case and mounted in a case holder. The case holder was a flexible piece of polycarbonate (PC) that lifted the case into position and facilitated the rapid and easy exchange of cases. The mold is shown in Figure 2E . The frame was cut from a 1 mm polystyrene (PS) sheet with a Trotec Speedy 100 laser cutter (Trotec AG, Sankt Gallen, Switzerland). The membrane (Unique-Mem Track-Etched Membrane; Oxyphen AG, Wetzikon, Switzerland) was cut to size and manually welded to the upper and lower surfaces of the frame. The membrane had a pore size of 5 µm, which allowed the exchange of nutrients and waste products between the tissue and the culture medium. The final product was a mold with a disk-shaped cavity with a height of 1 mm and a 23 mm diameter, with an opening on one side with a diameter of 5 mm ( Fig. 2E ). The mold was sterilized by oxygen plasma treatment for 20 min (Femto plasma cleaner; Diener Electronic GmbH, Ebhausen, Germany). The case was cut from 5 mm PC sheets and autoclaved. The mold was inserted into the case under sterile conditions in a laminar flow hood and fixed with autoclaved M3 screws ( Fig. 2D ). For the injection of the keratinocytes, a top is used that has an opening above the cavity to allow for membrane expansion ( Fig. 2C ). To promote the differentiation of the keratinocytes in the epidermal layer, simple stands were designed to lift the mold to the air–liquid interface ( Fig. 2F ). The stands were cut from 1 mm polystyrene sheets and sterilized by autoclaving. The setup was placed in a sterile laminar flow hood for experimental manipulations, and syringes, nozzles, molds, and stands were used as sterile disposables and replaced after each experiment.
Cell Culture
Neonatal human dermal fibroblasts were acquired from Thermo Fisher (C0045C; Gibco Media, Thermo Fisher Scientific, Carlsbad, CA, USA). Human primary keratinocytes were provided by the Department of Dermatology of the University Hospital Zürich under the assistance of the SKINTEGRITY Biobank. Cells were extracted from surplus skin biopsies from consenting patients who had signed an informed consent that was approved by the local Institutional Review Board (EK647 and EK800). Fibroblasts were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 1% penicillin/streptomycin, 1% HEPES, and 10% fetal bovine serum. Keratinocytes were cultured in keratinocyte serum-free medium (SFM, cat. no. 17005042; Gibco) supplemented with 1% penicillin/streptomycin. Cells were cultured in standard conditions at 37 °C and 5% CO2, and the culture medium was changed three times a week. Fibroblasts for experiments were used until passage 10, and keratinocytes until passage 5. All materials were purchased from Thermo Fisher and Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated.
The dermal layer’s composition was based on recipes described by L. Mazzone et al. 40 and Braziulis et al. 41 Therefore, an acidic solution of bovine collagen type I (Symathese) was dropwise neutralized with a buffer (containing 200 mM HEPES, 0.32 M NaHCO3, and 0.15 M NaOH) and mixed with fibroblasts suspended in DMEM. The mixture was prepared on ice to reduce the gelation rate, facilitating injection. A total volume of 0.75 ml containing 75,000 fibroblasts was injected into each mold at a rate of 300 µl/s. After 15 min at 37 °C, the collagen had become crosslinked, and the molds were released from the case and transferred into DMEM supplemented with 1% penicillin/streptomycin, 1% HEPES, and 10% fetal bovine serum. After 7 days in culture, the mold containing the dermal layer was placed into the injection prototype, and a flexible Western blot tip was inserted into the mold ( Fig. 2C ). One million keratinocytes were injected between the dermal layer and the membrane of the mold in a volume of 100 µl keratinocyte–SFM and at a speed of 125 µl/s, using the injection setup described above and as illustrated in Figure 1C . The dermo-epidermal equivalents were directly transferred into keratinocyte–SFM supplemented with 1% penicillin/streptomycin culture for 9 days. The molds were then lifted to the air–liquid interface and cultured for an additional 10 days in keratinocyte differentiation medium consisting of three parts DMEM and one part Ham’s F12 media (cat. no. 11765054; Gibco) containing 5% fetal bovine serum, hydrocortisone (0.4 µg/mL), cholera toxin (1 × 10−10 M), transferrin (5 µg/mL), insulin (5 µg/mL), triiodothyronine (2 × 10−11 M), and 1% penicillin/streptomycin. The medium was added beneath the skin equivalents to facilitate keratinocyte maturation. Medium was changed five times a week.
Cell Viability Assay and Proliferation Assay
A cell viability assay was performed after both the dermis and epidermis injection processes. The dermal component was prepared as described above in a volume of 50 µl containing 50,000 viable fibroblasts, and it was either automatically injected at a speed of 300 µl/s or manually pipetted into a 96-well plate (TPP, Trasadingen, Switzerland; n = 30). Manually prepared samples served as a control group. Fibroblast viability was analyzed 3 h after the production of the dermal layer. The gels were digested with collagenase in DMEM (4 mg/ml) for 45 min, and cell viability was measured with a trypan blue exclusion assay and with an automated cell counter (Countess-II Invitrogen, Thermo Fisher).
To analyze keratinocyte viability after the epidermal layer injection process, 58,000 viable keratinocytes suspended in 100 µl of keratinocyte–SFM were either automatically injected at a speed of 125 µl/s or manually pipetted into a 96-well plate (n = 30). Manually pipetted keratinocytes served as a control group. Keratinocyte viability was measured after 3 h with a commercially available live/dead viability assay (L3224; Thermo Fisher). Keratinocytes were incubated for 1 h in a solution of 6 µM calcein AM and 10 µM ethidium homodimer-1. Fluorescent readings were acquired with a plate reader (BioTec Synergy HT, Winooski, VT, USA), and cell viability was calculated from the fluorescent readings relative to controls, which were set to 100%.
The biocompatibility of the customized mold was analyzed with a commercially available viability/proliferation assay (Celltiter Blue, similar to Alamar Blue; Promega, Dübendorf, Switzerland). The metabolic activity of cells results in the reduction of the nonfluorescent resazuring into the highly fluorescent resorufin. The measured fluorescence relates to metabolic activity and is an indicator of cell viability and proliferation. Dermal layers were injected into the customized molds as well as into commercially available cell culture wells (TPP, Trasadingen, Switzerland; n = 33). After 24 h of cultivation, the dermal tissues were incubated with the dye for 3 h, and cellular metabolic activity was measured with a plate reader (BioTec Synergy HT). The statistical analysis of both viability and proliferation tests was performed as described by Lee et al., 28 using a two-tailed t-test and Excel software (Microsoft, Seattle, WA, USA). Statistical significance was set at p < 0.05.
Histology and Immunofluorescence Microscopy
Twenty-six-day-old bilayered skin equivalents were fixed in 4% formalin, embedded in paraffin, sectioned at 5 µm, stained with hematoxylin and eosin (H&E), and imaged by light microscopy (Zeiss Axiovert A1; Carl Zeiss AG, Feldbach, Switzerland).
Sections were also used for immunofluorescence. The antibodies used to visualize components of the epidermal layer were against keratin 14 (1:5000, BioLegend, San Diego, CA, USA) and cytokeratin 10 (1:100; Dako, Glostrup, Denmark). Secondary antibodies were anti-rabbit AF488 (1:200; Jackson ImmunoResearch, West Grove, PA, USA) and anti-mouse Cy3 (1:500; Jackson ImmunoResearch), and Hoechst 33342 (1:500; BioLegend) was used for staining of nuclei. Sections were deparaffinized, rehydrated, and washed three times in Tris buffered saline (TBS; 1×). Antigen retrieval was performed by incubating the sections for 1 h at 90 °C in sodium citrate buffer (1×). After incubation, slides were cooled down to room temperature (RT) and washed twice in TBS. Sections were permeabilized in TBS containing 0.1% Triton-X for 5 min at RT. After blocking for 30 min in 12% bovine serum albumin (PAN-Biotech, Aidenbach, Germany) and 0.1% Tween 20 (9127.2; Carl Roth, Karlsruhe, Germany) in TBS, the primary antibody mixtures were added in the blocking buffer and incubated at 4 °C overnight. The next day, sections were washed in TBS and incubated with the secondary antibody mixtures for 30 min at RT. Sections were washed twice in TBS containing 0.1% Tween 20. After a final washing step in TBS, the samples were mounted with Mowiol (81381; Sigma-Aldrich) and dried at RT overnight. Images were acquired using an AxioCam MRm connected to an Axio Imager.A1 microscope (Carl Zeiss AG). Images were processed with Apple Preview (Apple; Cupertino, CA, USA) and Adobe Illustrator (Adobe; San Jose, CA, USA) for contrast/brightness and layout, respectively.
Results
Biocompatibility of the Fabrication Process
Fibroblast viability in the dermal layers was measured 3 h after their production. A trypan blue exclusion assay showed a fibroblast viability of 97.9% ± 2.1% for the automatically injected cells (n = 30), and one of 97.8% ± 2.0% for the manually produced dermal layers (n = 30).
A live/dead viability assay was performed on keratinocytes 3 h after injection and showed a viability of 87.7% ± 5.3% for the automatically injected keratinocytes (n = 30) and of 86.9% ± 6.6% for their manually pipetted counterparts (n = 30). There was no significant difference in cell viability of either fibroblasts or keratinocytes in comparison to their respective control group. Results of the cell viability test are shown in Table 1 .
Cell Viability Results, Obtained 3 h after Processing of Fibroblasts (Trypan Blue Exclusion Assay) and Keratinocytes (Live-Dead Staining).

No significant difference between manual pipetting and automated injection molding was found.
Cell proliferation was also analyzed after 24 h using a commercial proliferation assay on bilayered skin equivalents cultured in either the customized molds or commercially available culture wells. There was no significant difference in the results from the skin equivalents cultured in the customized molds and the control groups (n = 33).
Bilayered Skin Equivalents Fabricated with Injection Molding
Fabrication of bilayered skin equivalents by injection molding was rapid because the injection time of both the dermal and epidermal layers was less than 1 min. In-mold cultivation also facilitated easy handling of the fragile tissue. Skin equivalents were cultivated for a total of 26 d, and H&E staining revealed a continuous and multilayered (stratified) epidermal layer with no stratum corneum ( Fig. 3A ). H&E staining further revealed the presence of vital fibroblasts in the dermal layer. Differentiation of the epidermal layer was evaluated by immunofluorescence staining using two keratinocyte markers: keratin 14 for basal keratinocytes and cytokeratin 10 for differentiating keratinocytes. Both markers were found in the epidermal layers of the fabricated skin equivalents ( Fig. 3B–3E ). Figure 3F shows a homogeneous dermal layer after 7 days of cultivation. Only minimal radial shrinkage was observed after 26 d of culture in all of the bilayered skin equivalents produced. Figure 3G shows a representative macroscopic image of a bilayered skin equivalent after 17 d of cultivation.
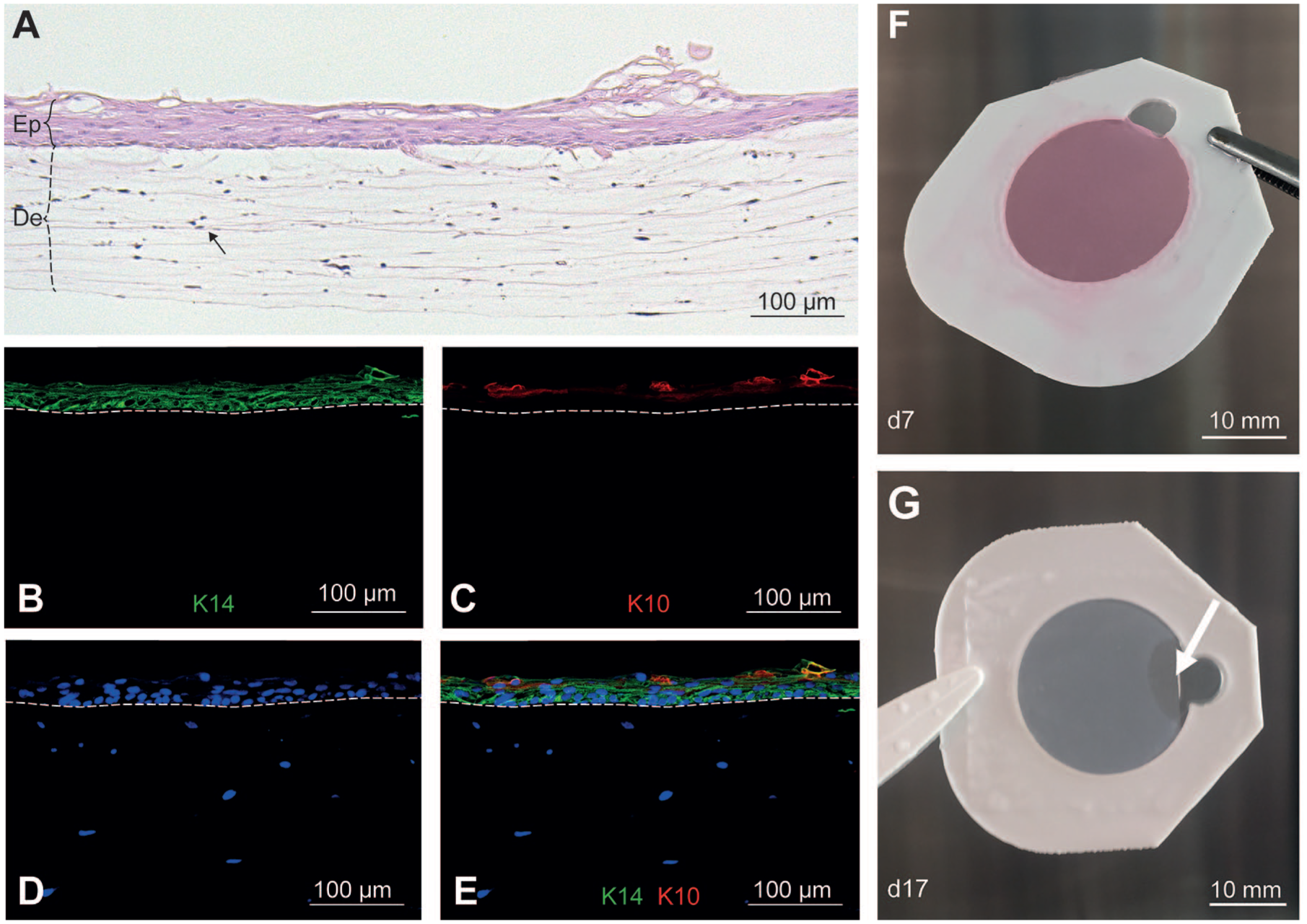
Bilayered skin equivalents fabricated by injection molding. (
Discussion
Fabrication techniques are needed that are specifically designed for automation to efficiently produce bioengineered skin. Injection molding is known to be fully automatable, is scalable in industrial applications, and can create standardized high-quality products. 32 Therefore, the use of injection molding for biomedical applications offers great potential for the automation of the fabrication of bioengineered skin.
Here, we have presented the results of our feasibility study of the use of injection molding principles for the fabrication of bilayered bioengineered skin. For this study, we developed a device and a customized mold that facilitate the successive injection of two cell-containing materials that form the dermal and epidermal layers of the skin. This study supplements previous reports of injection molding for the production of other living tissues,35,36 and it also extends them by using more than one cellularized component. The resulting bilayered skin equivalents produced a stratified epidermal layer on top of a dermal layer containing vital fibroblasts, which was verified with histology and immunohistochemistry.
The fabrication setup was shown to be both rapid and easy to use, with the injection time for both the dermal and epidermal layers being less than 1 min. The fabricated skin, cultured during a period of 26 days, has shown the suitability of the process and the materials used. This finding has been backed by the cell viability and proliferation assays. For further in-depth analysis, cell tests should be performed for longer periods of time. Furthermore, maintaining the fragile bioengineered skin in the mold during cultivation notably improved the ease of handling of the tissue and therefore reduced the risk of damage during handling.
This automated process produced bilayered bioengineered skin that had a continuous dermal layer containing vital fibroblasts, and a continuous epidermal layer comprising multiple layers of keratinocytes. Initial differentiation was observed in the epidermal layer, as evidenced by keratin 14 and cytokeratin 10 expression in basal and differentiating keratinocytes, respectively. No fully differentiated stratum corneum was produced, however. This final differentiation step may require an extended cultivation time or additional stimulators of epidermal differentiation.5,42 Precultivation of the fibroblasts in the dermal equivalent before keratinocyte seeding was performed for 7 days, since this has been shown to be crucial for the equilibrated maturation of the bilayered skin. 43 In recent experiments using 3D bioprinting, a fibrinogen matrix was used without the need for precultivation of fibroblasts. 27 Under the premise that this material creates comparable results, the injection molding process could be adapted to inject both layers in direct succession and without precultivation. In future studies, more cell types could be introduced to increase the resemblance of the tissue to natural skin (e.g., endothelial cells for blood vessels or melanocytes for skin pigmentation).
The injection device developed here is of low complexity and can be built from readily available components for less than US$300. The prototype is based on an open-source syringe pump and can be easily recreated from the information provided by its developers 39 and in this article. The device has only an A4 paper-sized footprint and can therefore be used under a laminar flow hood. This allows for its use under sterile conditions in standard laboratory infrastructure. The open-source availability, low production costs, and low complexity of the system could enable many researchers to automatically produce multilayered living tissue using biomaterials and cells of specific interest to them.
Because the presented experiment was a feasibility study, the injected material was kept on ice to reduce the viscosity, thus minimizing the shear stress exposed to the cells. A moderate injection speed was chosen to avoid high pressure. Shear stress and injection pressure both have a negative influence on cell viability. 44 To further improve injection speed and cell viability, different viscosities can be investigated to find important shear stress, process time, and process pressure thresholds.
In traditional injection molding, the materials are injected into costly, reusable metal molds. 32 To reduce cost and the inconvenience of cleaning and sterilization, a simple single-use plastic mold was developed. The mold we present here is built from a laser-cut plastic frame on which two pieces of membrane are welded to both sides; the membrane on the mold enables the exchange of nutrients and waste products between the tissue and the culture medium. The skin equivalent can therefore be directly formed and cultivated in the mold, and the tissue is stabilized by the membranes and therefore protected from ripping, folding, and accidental damage during transfer and medium changes, as well as during fixation for histological analysis. If robotic manipulation for full process automation is later required, this protection with respect to the handling of the tissue could be additionally beneficial. The production of the molds with the laser cutter enables the user to customize the shape and size of the created tissue for their specific applications with respect to both the two planar dimensions and tissue height. In cases when the number of donor cells or the amount of biomaterial is restricted, the mold size could be minimized. With skin grafts, in contrast, it could be favorable to maximize the size and adapt it to the shape of the wound.
Cell-induced shrinking is a common problem in research models of bioengineered skin because of the contractive nature of fibroblasts.45,46 Shrinkage can lead to unwanted densification, loss of permeability, surface corrugation, or even folding of the tissue, which can hinder subsequent experiments. 46 Previous reports have demonstrated high shape accuracy when producing living tissues by injection molding.35,36 Using the present technique with customized molds, we too qualitatively observed geometrical accuracy in the radial direction, meaning the circumference of the tissue stayed true throughout several weeks of culturing, with very little radial shrinkage of the tissues during cultivation and occurring only at the injection point ( Fig. 3G ). Reducing shrinkage could be a significant and important feature of the fabrication technique described here, but it requires further in-depth investigation to confirm this effect.
Injection molding and extrusion-based 3D bioprinting have different advantages. Bioprinting is well suited for complex 3D geometries in which a precise positioning of certain features in the 3D volume is required. 22 A small nozzle with high spatial accuracy is inefficient for filling a homogeneous volume, however, and increasing the nozzle diameter reduces shape fidelity and surface quality.
Injection molding, by comparison, is well suited for rapidly fabricating homogeneous volumes with high shape accuracy and surface quality. 32 Our results suggest that injection molding is very well suited for the fabrication of simplified bilayered tissues, because it allows standardized fabrication of tissues with high shape and surface quality in large numbers and in a cost- and time-efficient manner, independent of the required tissue size.
We have demonstrated the feasibility of fabricating a multilayered living tissue with an injection molding approach. We developed an automated system and used it to fabricate bilayered skin, comprising human dermal fibroblasts and keratinocytes, by injecting the components stepwise into a customized mold to form the dermal and epidermal layers. After further research and development, this technique may result in a rapid, standardized, and cost-effective fabrication method for high-yield multilayered living tissues for clinically applicable skin grafts and for research purposes.
Footnotes
Acknowledgements
We would like to acknowledge the contributions of Jimmy Hu and Samuel Arpagaus to this research with their student theses. We would like to thank Prof. Dr. Sabine Werner and Dr. Paul Hiebert of ETH Zürich for their support with immunohistochemistry; Prof. Michael Detmar, Dr. Carlotta Tacconi, and Jihye Kim of ETH Zürich for their support and advice; the ETH Zürich teaching laboratory for access to the very same; Andrea Gubler for her technical support; Dominik Siallagan for support with Figure 1; Gaetana Restivo and the SKINTEGRITY Biobank for the keratinocytes; the ScopeM Facility for their support with histology; and Enago (![]() ) for the English language review.
) for the English language review.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received the following financial support for the research, authorship, and/or publication of this article: The authors thank the SKINTEGRITY project of University Medicine Zürich for the financial support and for providing the framework for this project. The authors are further thankful for financial support by the vonTobel Foundation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.