
Abstract
A diverse range of biochemical and cellular assays are used by medicinal chemists to guide compound optimization. The data collected from these assays influence decisions taken on structure-activity relationship (SAR) campaigns. Therefore, it is paramount that medicinal chemists have a solid understanding of the strengths and limitations of each assay being used to characterize synthesized analogs. For the successful execution of a medicinal chemistry campaign, it is our contention that an early partnership among assay biologists, informaticians, and medicinal chemists must exist. Their combined skill sets are necessary to not only design and develop robust assays but also implement an effective screening cascade in which multiple orthogonal and counter assays are selected to validate the activity and target(s) of the synthesized compounds. We review multiple cases of drug and chemical probe discovery from collaborative National Center for Advancing Translational Sciences/National Institutes of Health projects and published scientific literature in which the evaluation of compounds in secondary or orthogonal assays led to the discovery of unexpected activities, forcing a reconsideration of the original assay design that was used to discover the biological activity of the compound. Using these retrospective case studies, the goal of this Perspective is to hedge toward the development of physiologically relevant assays that are able to capture the true bioactivity of compounds being developed in a medicinal chemistry campaign.
Graphical Abstract
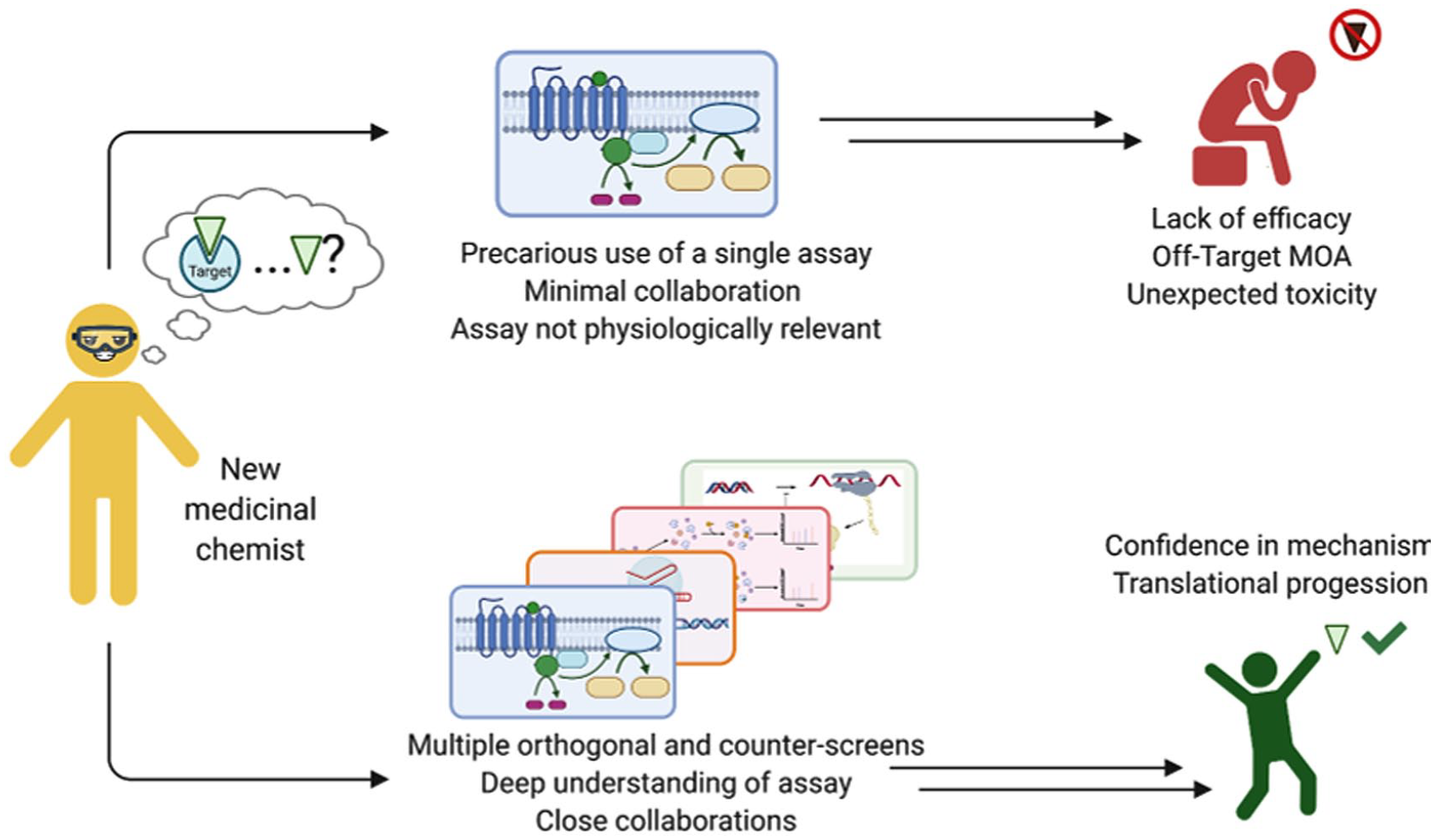
Introduction
Medicinal chemistry is a core component of research conducted at the Division of Preclinical Innovation (DPI) within the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH). Within the DPI, medicinal chemists have the opportunity to collaborate on multiple projects with assay biologists and informaticians. Most projects originate as collaborations with external investigators who possess deep understandings of specific biological targets, pathways, and disease pathologies. Such collaborations often involve high-throughput screening (HTS) followed by medicinal chemistry optimization of hits to discover small-molecule probes that can be used in proof-of-concept experiments. While domain expertise, especially in synthetic organic chemistry, is a prerequisite for medicinal chemists to conduct translational science, we would like chemists to consider another important characteristic of a translational scientist: boundary crossing, especially toward an understanding of assay biology. 1 This Perspective consists of some high-yield case studies from both NCATS DPI and the literature in which evaluation in orthogonal assays produced unexpected activities of compounds. These examples highlight the importance of assay design and establishing a robust screening tree for medicinal chemists to confirm the bioactivity of their compounds.
Compound Activities Can Depend on Enzyme Source and Substrate Choice
Cell-free enzymatic assays that use recombinant protein and fluorogenic substates remain a mainstay of HTS and medicinal chemistry-based optimization. At NCATS, we have adapted and miniaturized a multitude of such assays to discover small-molecule modulators of lysosomal hydrolases implicated in rare disorders such as Gaucher, Pompei, Wolman, and Fabry disease. Gaucher disease (GD) is a recessive monogenic lysosomal disorder caused by the deficiency of glucocerebrosidase (GCase) that results in excessive accumulation of glucocerebrosides or glycosphingolipids, substrates for GCase in lysosomes (
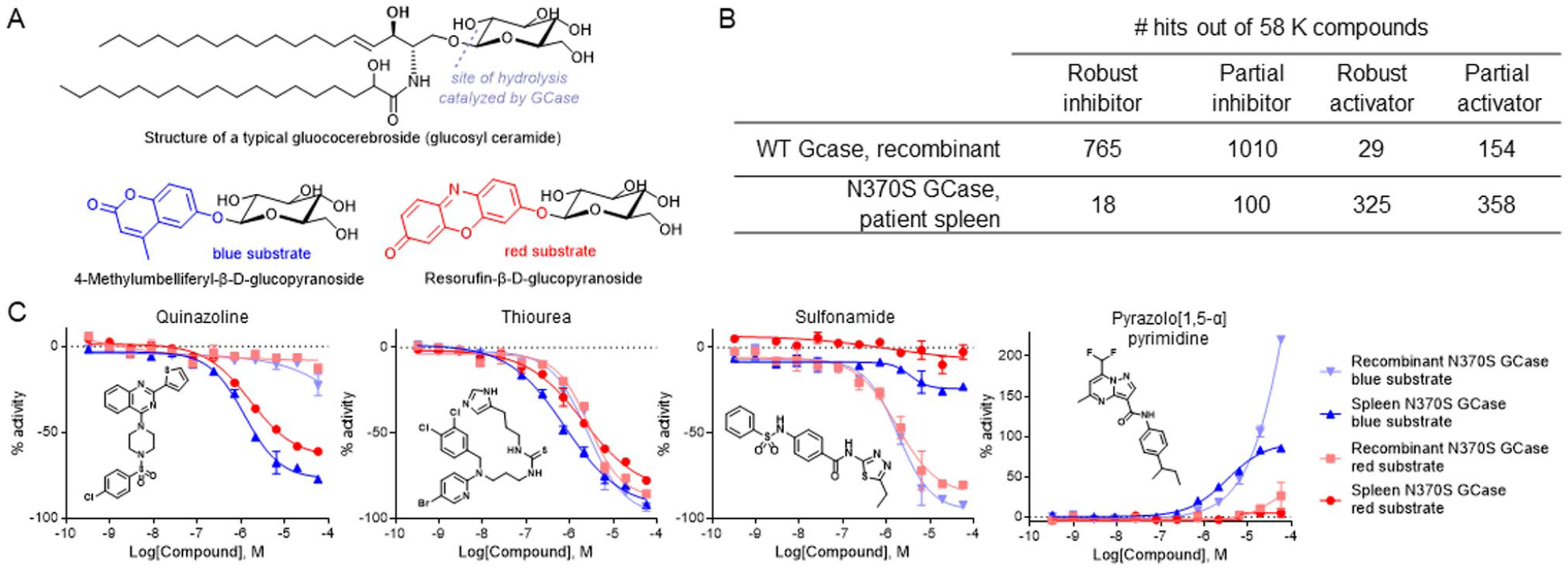
Compound activity dependence on enzyme source and substrate choice. (
Although our initial screening campaign was conducted with WT GCase, we rationalized that it would be more prudent to screen against N370S GCase, the most prevalent mutant found in type 1 GD patients. With access to splenectomy samples from patients, we decided to evaluate spleen homogenates as a natural source of native N370S GCase. To compare screening modalities, we screened a set of 52K compounds versus recombinant WT GCase and splenic N370S GCase using 4-methylumbelliferyl-β-D-glucopyranoside as substrate.
4
Our immediate observation was that we identified more hits with the WT enzyme, and most of these hits were robust (full inhibition curve with asymptotes) and partial inhibitors (
With several active chemotypes in hand, we compared their activities using recombinant and splenic N370S GCase as well as both fluorogenic substrates and discovered some very unusual trends (
Although we have yet to unravel the structural basis of such discordant activities between recombinant enzyme and spleen homogenates, we believe more advanced disease-relevant high-throughout cellular assays are required to discover and verify the most relevant activities of each chemical series. This case highlights the importance of testing several sources of biological targets (recombinant, splenic) and target constructs (WT, mutant) in target-based drug discovery. Unexpected discrepancies should not be dismissed but rather thoroughly investigated.
Biochemical and Cellular Assays Can Show Different Compound Activities
Ever since the discovery of the specific Bcr-Abl inhibitor imatinib, scores of kinase inhibitors have been clinically approved for the treatment of cancers, with hundreds more in clinical trials.11,12 Many kinase inhibitors evolve from HTS and structure-activity relationship (SAR) optimization campaigns that often test for inhibition of recombinant kinases in cell-free formats. However, the following example from NCATS shows how the kinase profiling activity of an inhibitor with cell-free kinases might be in stark contrast with kinase engagement in cells.
Duchenne muscular dystrophy (DMD) is a rare X-linked progressive muscular degenerative disease affecting 1:5000 boys, whereby mutations in the DMD gene lead to complete loss of the dystrophin protein that connects intracellular actin to the extracellular matrix in myofibrils. In one collaboration, NCATS screened for compounds that could upregulate α7β1 integrin, another structural transmembrane protein in myofibers to potentially compensate for the loss of dystrophin.
13
A pilot screen in a murine myogenic-cultured cell line with Itga7+/LacZ (replacement of one opposing α7 integrin allele with a β-galactosidase reporter) identified SU9516 (
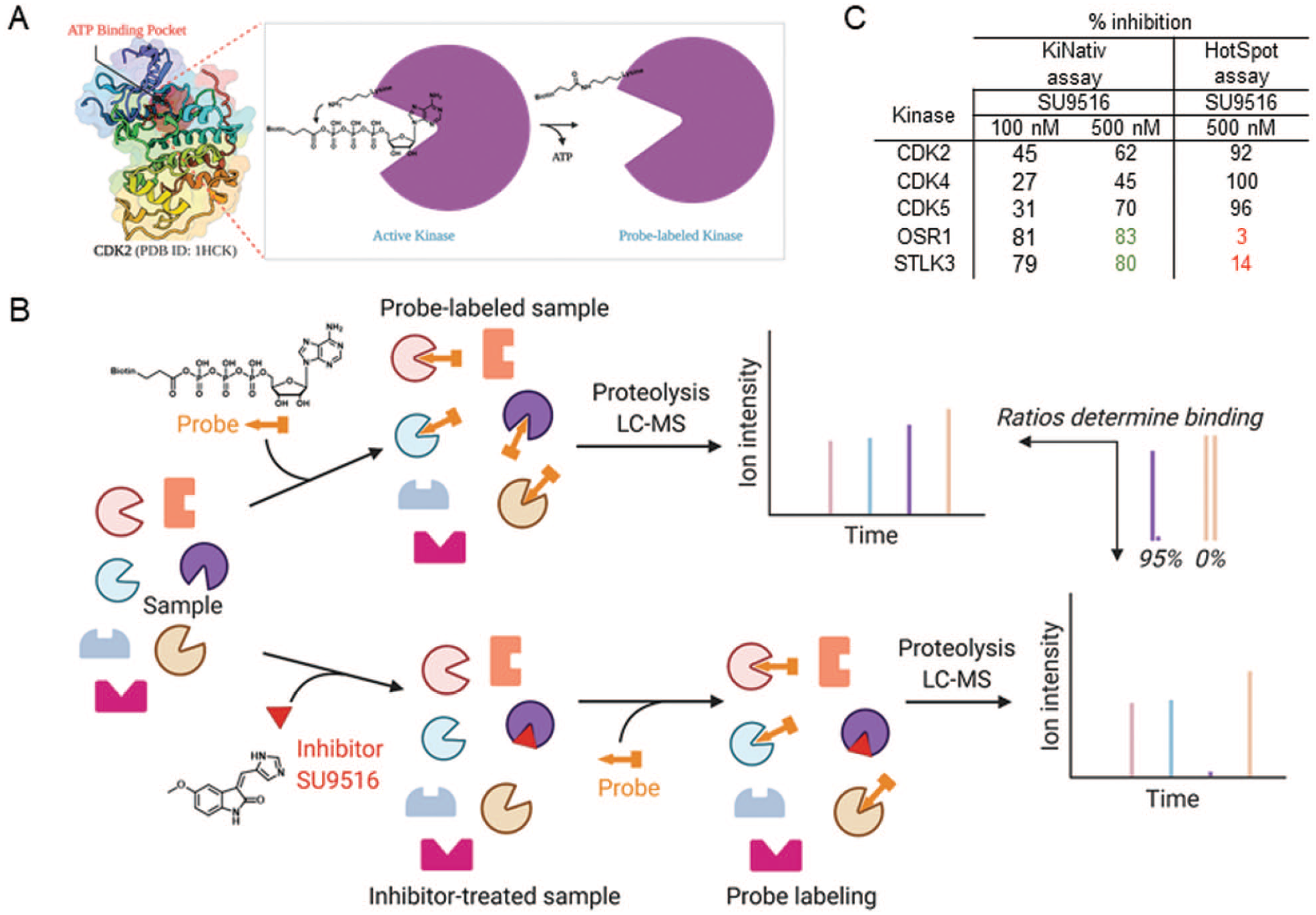
Profiling the kinase activity of SU9516. (
A commercial platform technology (ActiveX Biosciences, San Diego, CA) was used to assess target engagement with the purported target CDK2 and other kinases in myotubes. This particular platform uses a KiNativ probe, a biotinylated adenosine triphosphate (ATP)–mimetic with an acylated biotinylated group at the third phosphate position (
This case reiterates the importance of investigating compound activities in both cell-free and (potentially) the more physiologically relevant cellular assay systems. In certain instances, such as in the case of SU9516, target engagement in cells or tissues might show results discrepant to those obtained under cell-free experimental conditions. Similar observations have been made by others using the KiNativ platform. 16
Multiplex Cellular Kinase Profiling Can Enable Hypotheses of Off-Target Inhibitor Effects
Kinobeads, another chemoproteomic platform to quantify small-molecule kinome engagement in cells, have also exposed disparity in kinome activity between cellular and cell-free assays.
17
Kinobeads work via solid supports that are irreversibly attached to a library of promiscuous kinase inhibitors that bind to kinases present in cell or tissue lysates. After bead washing and proteolytic digestion, bound kinases are quantified based on the release of signature peptide fragments analyzed by LC-MS.
18
In the presence of a competitive free inhibitor, less enzyme binds to the beads, leading to a concentration-dose–dependent reduction of signatures peptides(s) associated with free inhibitor bound kinases (
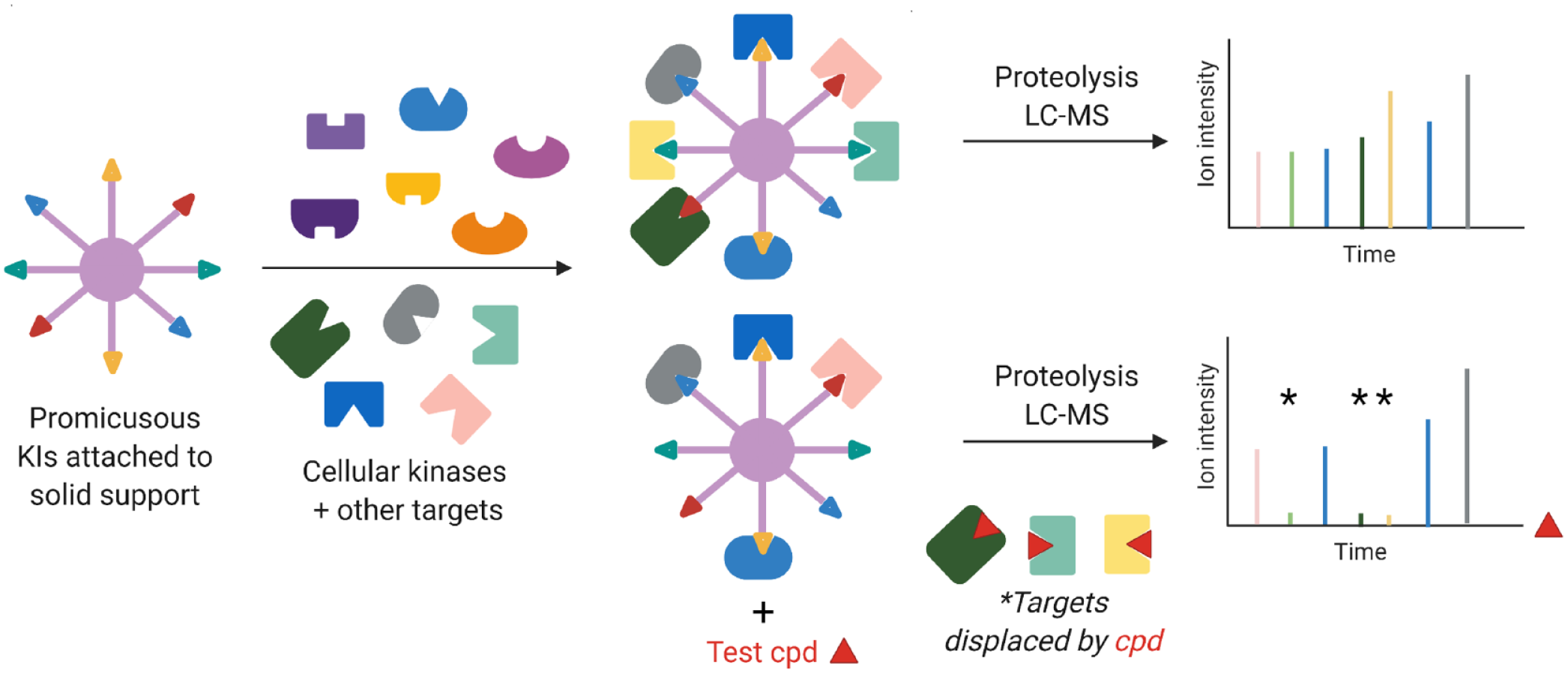
Schematic of Kinobead technology. The assay enabled the characterization of off-target ferrochelatase activity from vemurafenib (test compound) and other kinase inhibitors in clinical development.
Because Kinobeads are affixed with ATP-competitive inhibitors, they also capture other nucleotide-binding proteins such as metabolic kinases and acetyl coenzyme A dehydrogenases via binding to the pyridoxal- or flavin adenine dinucleotide–binding sites, respectively. Using this platform, at least 40 advanced kinase inhibitors (including vermurafenib) were also shown to bind to the heme-synthesizing enzyme ferrochelatase (FECH).
18
Vermurafenib (
Proteomics Enables Investigation of Ocular Toxicity in Alzheimer’s Disease
Activity-based protein profiling (ABPP) has been shown to be a versatile approach to gauge small-molecule target engagement, especially with proteins bearing nucleophilic residues needed for catalysis. 25 ABPP typically uses chemical probes that have promiscuous electrophilic warheads that covalently bind to the nucleophilic residue (e.g., serine hydroxyl in serine hydrolases or sulfhydryl groups in cysteine side chains). The warhead is attached to a reporter, often a fluorescent group or biotin, that helps in detection and identification of the protein. Such probes can also be designed from active small-molecule leads by decorating them strategically with functional groups such as diazirines, which generate light-activated reactive species for irreversible binding to their target. The following example showcases such a probe that was able to reveal off-target toxicity of several β-secretase 1 (BACE1) inhibitors due to cathepsin D (CatD) inhibition, a target that these compounds had previously been optimized against in biochemical assays with purified protein.
BACE1 (part of the pepsin aspartyl protease super family) has been regarded as a promising target for Alzheimer’s disease (AD) due to its role in catalyzing the breakdown of amyloid precursor protein (APP) to amyloid beta (Aβ), which accumulates as cerebral Aβ plaques in the brains of AD patients.
26
Although BACE1 inhibitors are yet to be approved by the U.S. Food and Drug Administration (FDA), several generations have been developed (e.g., LY2811376, AMG-8718, PF-9283;
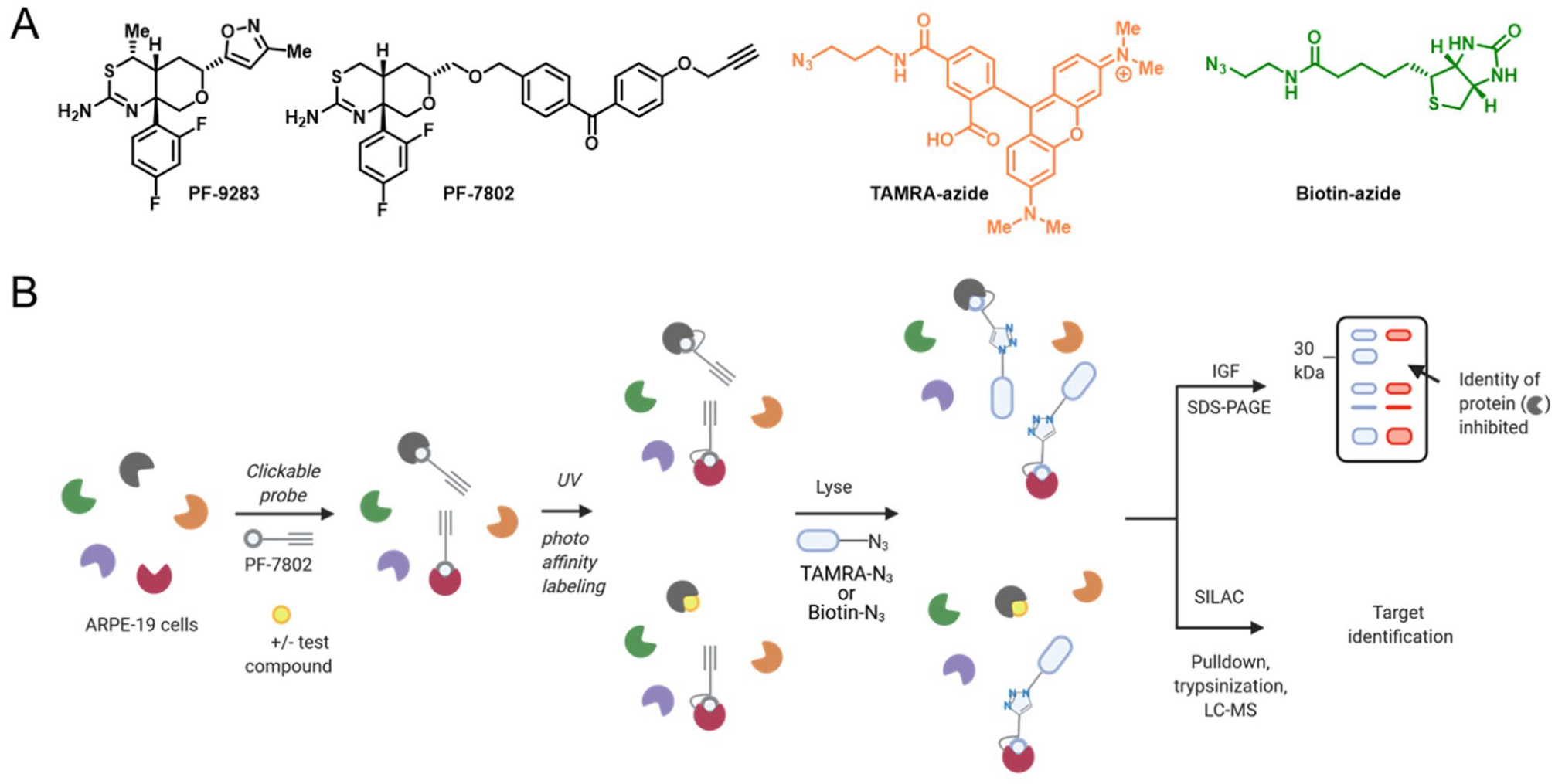
Characterization of ocular toxicity of β-secretase (BACE) inhibitors by proteomics. (
This case study shows the benefit of cellular on-target validation using a complementary photoaffinity-labeling chemoproteomics approach. Although this case led to drugs being pulled from clinical development, it led to new assay development and toxicology standards that can be applied to future drug candidates operating in the same pathway.
OTS167 and Its Oncologic MELK-Independent Activity
Although not absolutely necessary for FDA approval, target deconvolution and a thorough understanding of a drug candidate mechanism of action can guide therapeutic use and predict potential toxicities.41,42 In preclinical discovery programs, when a compound shows an expected phenotype in disease-relevant cells, a simple check for target validation would be to eliminate the putative target and check whether the compound still elicits the phenotype. In a retrospective study, Sheltzer et al.
43
sought to establish the role of maternal embryonic leucine zipper kinase (MELK) as a prognostic biomarker for cancers addicted to MELK. They chose the triple-negative breast cancer cell lines Cal51 and MDA-MB-231 and the melanoma cell line A375, which had been shown, via RNA interference experiments, to be dependent on MELK for their growth. Using CRISPR-Cas9 technology, they induced frameshift mutations in various regions of MELK to knock out the protein. After verifying by Western blot that MELK expression had been ablated, they were surprised to see that the rate of proliferation in these cells remained unaffected. Furthermore, the MELK inhibitor OST167 (
Sheltzer et al.
46
expanded such mechanistic reexamination to 10 additional cancer drugs targeting six different targets that are in preclinical development and clinical trials. Among key considerations in choosing the targets was that they had no reported mutations that rendered the compounds ineffective. Unfortunately, their observations were the same: the compounds inhibited cancer cell proliferation with the same potencies in the absence of their target. They honed in on a T-LAK cell-originated protein kinase/PDZ binding kinase inhibitor OTS964 (
These case studies stand as cautionary tales for the uncovering of a target and how even when the recognized pathway may prove noncanonical, phenotypic responses may remain. They highlight the importance of target investigation using modern genomic methods such as CRISPR/Cas9 and that RNA interference alone cannot be reliable validate a target. They show that target associations with phenotypes that are consistent with chemical and genetic perturbations are further strengthened by the identification of compound-resistant mutants.
Reporter Assays: PTC124 as a Potent Inhibitor of Firefly Luciferase
Because of tolerable expression and high sensitivity, luciferase and green fluorescent protein reporters are often used in high-throughput assays to tag proteins and measure their concentration, localization, and study signal transduction. Reporter genes that code for luminescent proteins can be inserted after the promoter region (the region of DNA where the transcription machinery binds before start of transcription of a gene) of a protein of interest or next to it. Luciferase enzymes need a substrate that consumes molecular oxygen and ATP to produce a product, adenosine diphosphate, and light emission, which is the output that is measured. For firefly luciferase (FLuc), the substrate and products are D-luciferin and oxyluciferin, respectively (
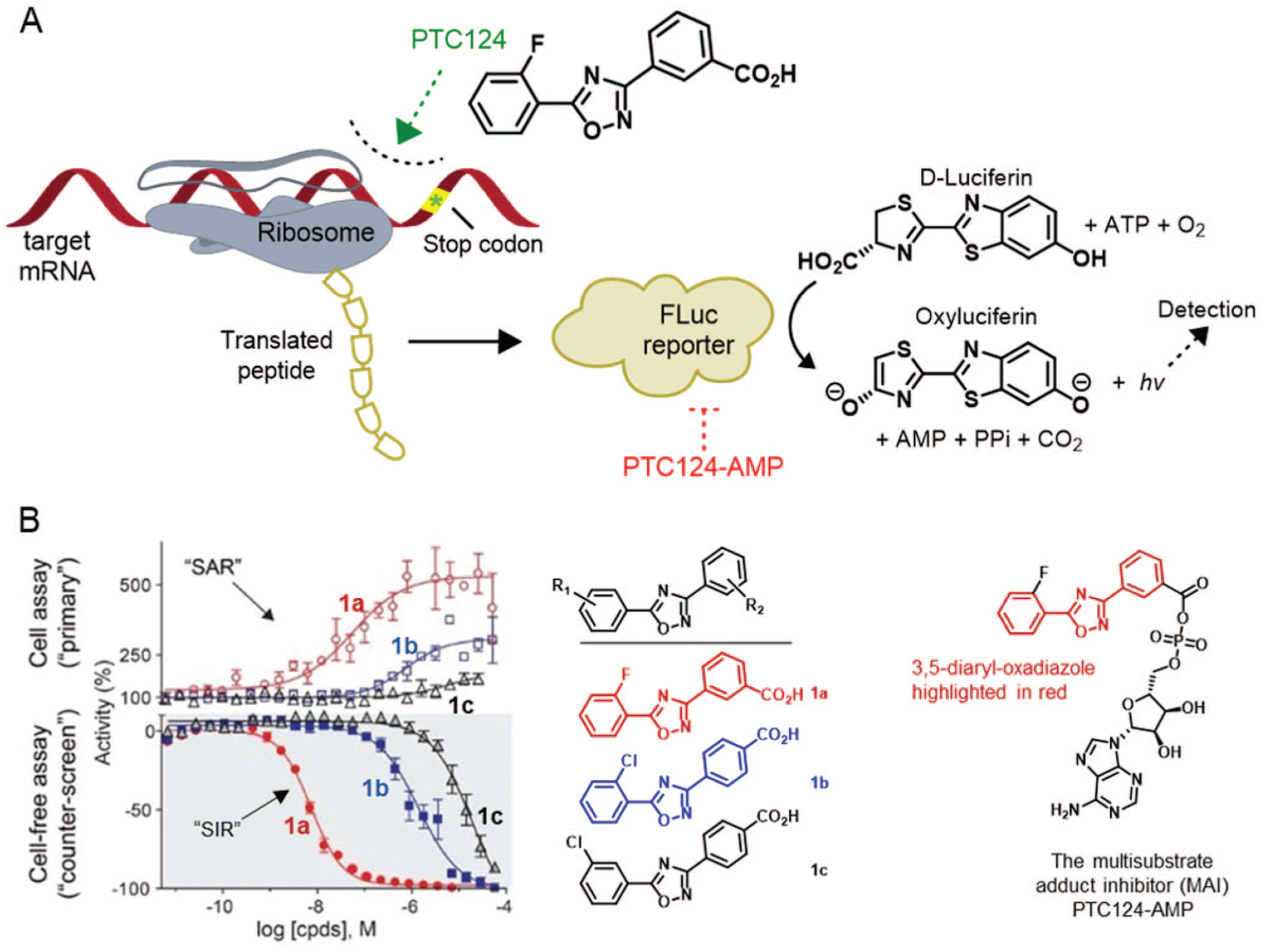
Compound-mediated interferences in luciferase-based assays. (
In orthogonal assays in which the FLuc reporter was replaced with Renilla luciferase (RLuc), PTC124 and analogs failed to inhibit RLuc biochemically and to activate RLuc in cells, further supporting the hypothesis of FLuc inhibition. Introducing PTC124 to purified FLuc or RLuc increased the trypsinization half-life of FLuc but not RLuc, suggesting a stabilized E.I complex for PTC124 and FLuc that was further supported by thermal denaturation assays.
49
Although counterarguments were made in favor of PTC124 not acting in a FLuc-dependent manner, confirmation of the E.I complex was found through x-ray crystallography.49,50 LC-MS experiments found that the reversible inhibitor responsible for the complexation was actually PTC124-AMP, a high-affinity multisubstrate inhibitor with a specific meta-carboxylate motif needed to bring about the potent activity (
The search for luciferase reporters with reduced promiscuity toward chemical libraries, smaller size, and brighter signal has led to the discovery of RLuc, NanoLuciferase (NanoLuc), and the turbo luciferase (TurboLuc). Although a class of arylsulfonamides inhibit RLuc,
55
the most recent 16 kDa TurboLuc shows reduced frequencies of interference by low-molecular-weight compounds.
56
At NCATS, the NanoLuc reporter is widely used as a split luciferase in high-throughput cellular thermal shift assay (CETSA) experiments to investigate cellular target engagement. A small fragment of NanoLuc (11 amino acid peptide HiBiT, or a 15 amino acid variant 86b) is fused to proteins of interest and allowed to interact with small molecules. This is followed by the addition of the larger fragment of NanoLuc (11S or LgBiT) whose surface complements the surface formed by the smaller fragment; the reconstituted enzyme can then emit light on the addition of the substrate furimazine after heating, to quantify the amount of soluble protein (
G-Protein–Coupled Receptor: Ligand Bias and Activity Dependency on Assay Format
G-protein–coupled receptors (GPCRs) are attractive therapeutic targets for various diseases covering about 30% of all FDA-approved drugs.
59
These seven-transmembrane receptors bind to extracellular ligands, undergo conformational changes to activate a GTP-to-GDP exchange in an intracellular heterotrimeric G-protein (Gα, Gn, Gγ;
The pain and addiction field has also been influenced by the desire to develop µ-opioid receptor (MOR) agonists that selectively activate the G-protein (analgesia) and not the β-arrestin (abuse, constipation, and respiratory suppression) pathway, a concept that has been challenged by the failure of the G-protein–biased MOR agonists TRV130 to reduce abuse-related effects in rats. Recent work published at the NIH has shown that the galanin receptor forms heteromers with MOR and negatively regulates the adverse dopaminergic effects of opioids.63 These observations hint at the complex pharmacology associated in the discovery of small molecules to modulate GPCR function, the significance of which is further corroborated by the HEAL Initiative (Helping to End Addiction Long-term Initiative), a nationwide effort initiated by NIH to tackle the opioid crisis and develop nonaddictive pain medications.
64
The paucity of predictive animal models in pain has also thwarted the clinical translation of therapies. However, at times, a simple verification of compound activity in cells that express a rodent receptor may be pragmatic before advancing to a rodent model (
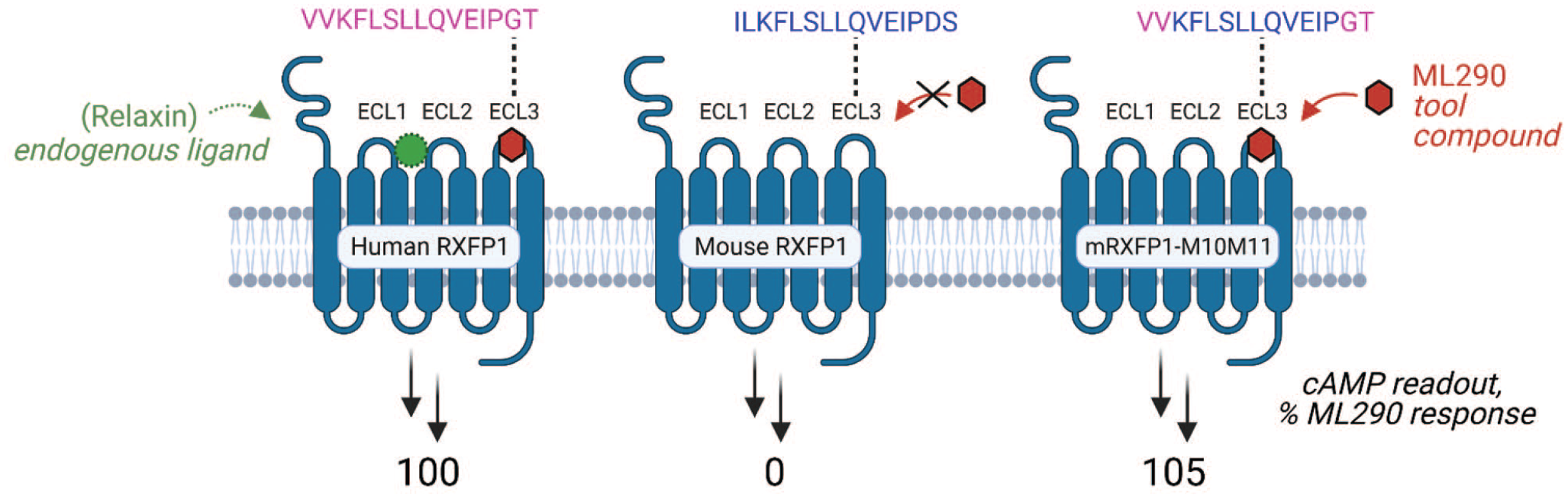
Influence of G-protein–coupled receptors target construct on probe activity. In a project targeting the relaxin hormone receptor, the activity of ML290 is dependent on the sequence of the ECL3 portion of human/mouse RXFP1 receptors. To create a more translationally relevant mouse model, a partially humanized mouse construct (right) was necessary to restore ML290 activity.
These examples highlight the complexities in predicting the therapeutic efficacy of GPCR-bound small molecules a priori based on their pharmacologic activity. Thus, it may be prudent to advance molecules with distinct pharmacologic profiles and adopt assays that use physiologically relevant cells with verified receptor expression and a disease phenotype that can be reversed.
Perspectives
In this Perspective, we summarize several case examples of an assay design that significantly affects medicinal chemistry efforts. There are several important, high-yield take-away messages from these series of vignettes for medicinal chemists and biologists alike. First, seemingly innocuous factors such as the substrate choice, enzyme source, and cell-free versus cellular assays can have significant impacts on the observed biological activity, and these choices can dramatically guide hit selection and SAR. Second, compounds can interfere with the assay readout and can confound data interpretation. This interference is less predictable in more complex assays and can even produce an apparent SAR that reflects compound-dependent interference (SIR). Third, these case studies demonstrate the importance of establishing multiple lines of evidence for informing hit picking and medicinal chemistry optimization. This can include confirmatory, orthogonal, and interference counterscreens to de-risk for interferences, lack of selectivity, and irreproducibility. Furthermore, multiple types of experiments are usually needed to support meaningful (potent, selective) cellular target engagement. Techniques such as proteomics (including ABPP), targeted genetic manipulation (CRISPR), and CETSA can be vital in this regard.
Scientific translation, including preclinical drug and probe discovery, is a team sport that is crucially dependent on crosstalk between multiple disciplines. Results, whether assay testing from biologists or new chemical analogs from chemists, should not be simply passed over the transom. On the contrary, the aforementioned vignettes should encourage medicinal chemists to engage with their biologist colleagues who characterize new analogs. Many of these cases studies are retrospective in nature—reexaminations that ultimately led to the recognition of the unexpected results. This is expected because translation of a basic discovery to a therapeutic is not linear, as often depicted. On the contrary, translation requires constant interactions of neighborhoods of disciplines and expertise, followed by revisitation and reevaluation of the approaches that are being used in a dynamic process. 67 Indeed, these cases demonstrate that scientific approaches (and assays) evolve as a project progresses.
To better recapitulate disease pathophysiology, in vitro assays are getting more complex. This is evident by the current focus on assays that use multiple types of cells, including patient-derived pluripotent stem cells and complex in vitro models such as organoids and tumor spheroids.68–72 Advances in rapid high-throughout mass spectrometric analysis are also leading to label-free analysis.73,74 However, we believe simple biochemical and cellular assays stand their ground in medicinal chemistry. Their reductionist approach is powerful for making decisions about SAR. The goal of this Perspective is to promote partnership and shared understanding about the advantages and limitations of the assays and, more importantly, to encourage scientists to share their learnings about success and failures in their approaches.
Supplemental Material
sj-pdf-1-jbx-10.1177_24725552211026238 – Supplemental material for The Impact of Assay Design on Medicinal Chemistry: Case Studies
Supplemental material, sj-pdf-1-jbx-10.1177_24725552211026238 for The Impact of Assay Design on Medicinal Chemistry: Case Studies by Joshua R. Born, Vinoth Kumar Chenniappan, Danielle P. Davis, Jayme L. Dahlin, Juan J. Marugan and Samarjit Patnaik in SLAS Discovery
Footnotes
Acknowledgements
Authors acknowledge the contributions from the Early Translational Branch of DPI NCATS and extramural collaborators that formed the basis for many of these case studies. Figures were created with BioRender.com and ChemDraw.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.