
Abstract
SMYD3 (SET and MYND domain-containing protein 3) is a protein lysine methyltransferase that was initially described as an H3K4 methyltransferase involved in transcriptional regulation. SMYD3 has been reported to methylate and regulate several nonhistone proteins relevant to cancer, including mitogen-activated protein kinase kinase kinase 2 (MAP3K2), vascular endothelial growth factor receptor 1 (VEGFR1), and the human epidermal growth factor receptor 2 (HER2). In addition, overexpression of SMYD3 has been linked to poor prognosis in certain cancers, suggesting SMYD3 as a potential oncogene and attractive cancer drug target. Here we report the discovery of a novel SMYD3 inhibitor. We performed a thermal shift assay (TSA)-based high-throughput screening (HTS) with 410,000 compounds and identified a novel benzodiazepine-based SMYD3 inhibitor series. Crystal structures revealed that this series binds to the substrate binding site and occupies the hydrophobic lysine binding pocket via an unprecedented hydrogen bonding pattern. Biochemical assays showed substrate competitive behavior. Following optimization and extensive biophysical validation with surface plasmon resonance (SPR) analysis and isothermal titration calorimetry (ITC), we identified BAY-6035, which shows nanomolar potency and selectivity against kinases and other PKMTs. Furthermore, BAY-6035 specifically inhibits methylation of MAP3K2 by SMYD3 in a cellular mechanistic assay with an IC50 <100 nM. Moreover, we describe a congeneric negative control to BAY-6035. In summary, BAY-6035 is a novel selective and potent SMYD3 inhibitor probe that will foster the exploration of the biological role of SMYD3 in diseased and nondiseased tissues.
Introduction
SMYD3 is a lysine-specific protein methyltransferase reported to methylate lysine residues on histone and nonhistone proteins. It is one of five SMYD family members of methyltransferases that are characterized by an MYND zinc finger domain splitting the catalytic SET domain into two parts. The function of the MYND domain has remained elusive and is thought to be involved in mediating protein or DNA binding. 1 SMYD3 gained a lot of interest as a potential therapeutic cancer target initially based on observed abnormal high expression in cancer tissues. Originally SMYD3 was identified as an overexpressed gene in colorectal and hepatocellular carcinomas,2,3 and a functional oncogenic role in these tissues has been demonstrated in mouse models. 4 Additional studies reported overexpression of SMYD3 in esophageal squamous cell carcinoma, gastric, ovarian, bladder, pancreatic, and non-small cell lung cancer cell line models.5–8 As one potential reason of SMYD3 upregulation, researchers identified activation of the RAS oncogene pathway.9–11 In genetic SMYD3 knockdown experiments in different cancer cell line models, proliferation was significantly reduced, suggesting an essential survival role in these models.5,8,9,12,13
On a mechanistic level, while SMYD3 has been observed to methylate multiple protein targets, the main biological functionality remains elusive. SMYD3 was initially characterized to di/tri-methylate histone 3 at lysine 4 (H3K4), 2 a chromatin mark associated with active gene promotors. In several studies that followed, methylation of H3K4 by SMYD3 has been correlated to aberrantly increased transcription of diverse cancer-promoting genes in different cell line models.5,14–20 More recently, histone 4 at lysine 5 was uncovered to be more efficiently methylated by SMYD3 in vitro. The functional consequences of H4K5 methylation are unknown. 21 Protein localization of SMYD3 was found to be mainly cytosolic,22–24 which suggests that SMYD3 substrates might not be limited to histones. In fact, several studies uncovered an important role of SMYD3 methylation activity on the regulation of nonhistone proteins that affect different and important signaling pathways, thereby promoting cancer phenotypes.9,22,25–28 In contrast to these reported widespread effects on different signaling pathways, organ-specific and whole-body SMYD3 deletion in mice surprisingly does not translate into any obvious phenotype.4,9 Moreover, SMYD3 was not sufficient to either promote tumorigenesis by itself or accelerate tumorigenesis progression in liver-specific SMYD3 overexpressing transgenic mice. 4
Exploration of the mechanistic consequences of a catalytic SMYD3 inhibition is hindered by a lack of specific and potent inhibitors openly available for the research community. The first inhibitors for SMYD3 have been described,
29
particularly
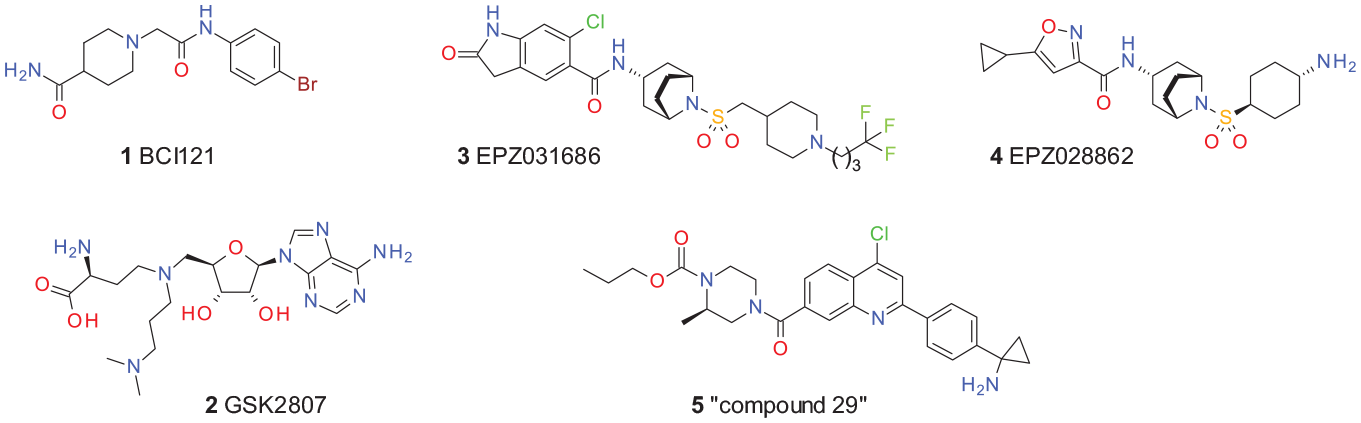
Published SMYD3 inhibitors.
Herein, we describe the identification of a novel, selective, and potent probe inhibitor of SMYD3 from a thermal shift assay (TSA) uHTS campaign using a library of 410,000 compounds. We were able to identify and optimize a series of benzodiazepines resulting in the discovery of BAY-6035. 35 This has excellent properties as a probe that will enable a more comprehensive chemical biology-based target validation of SMYD3.36–38 In addition, we identified a structurally closely related compound BAY-444 lacking SMYD3 activity that may be used as a corresponding negative control. Both compounds are freely available via the Structural Genomics Consortium (SGC).
Materials and Methods
Chemistry
Acetylated 4-oxo-2,3,4,5-tetrahydro-1H-1,5-benzodiazepines have been described in the literature since 1953, and multiple synthetic routes have been published.39–42 In our work, we used commercially available building block
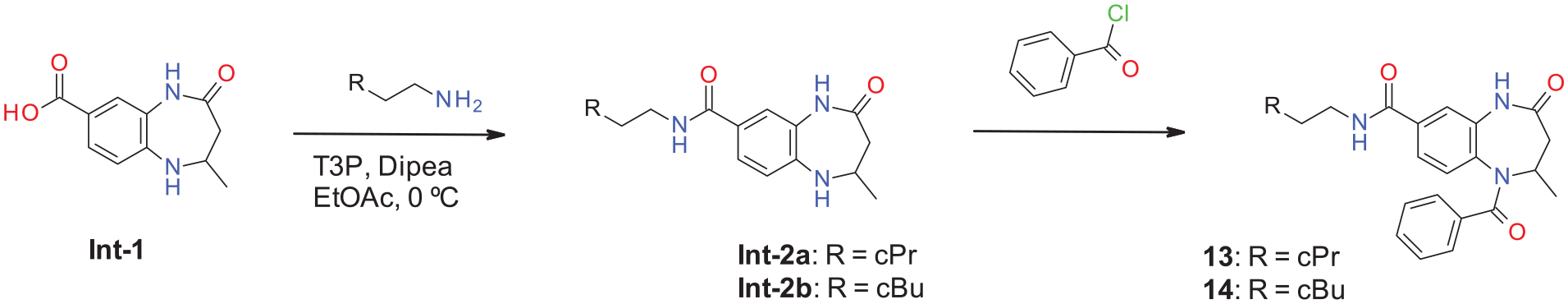
Synthesis of acetylated 4-oxo-2,3,4,5-tetrahydro-1H-1,5-benzodiazepines.
Initially we used chiral separation to obtain both enantiomers, but once it was determined that the (S)-enantiomer was preferred, we set out to synthesize enantiomerically enriched building blocks for further derivatization. Intermediate (S)-Int-7 was synthesized from (S)-β-alanine according to Scheme 2. Coupling acid
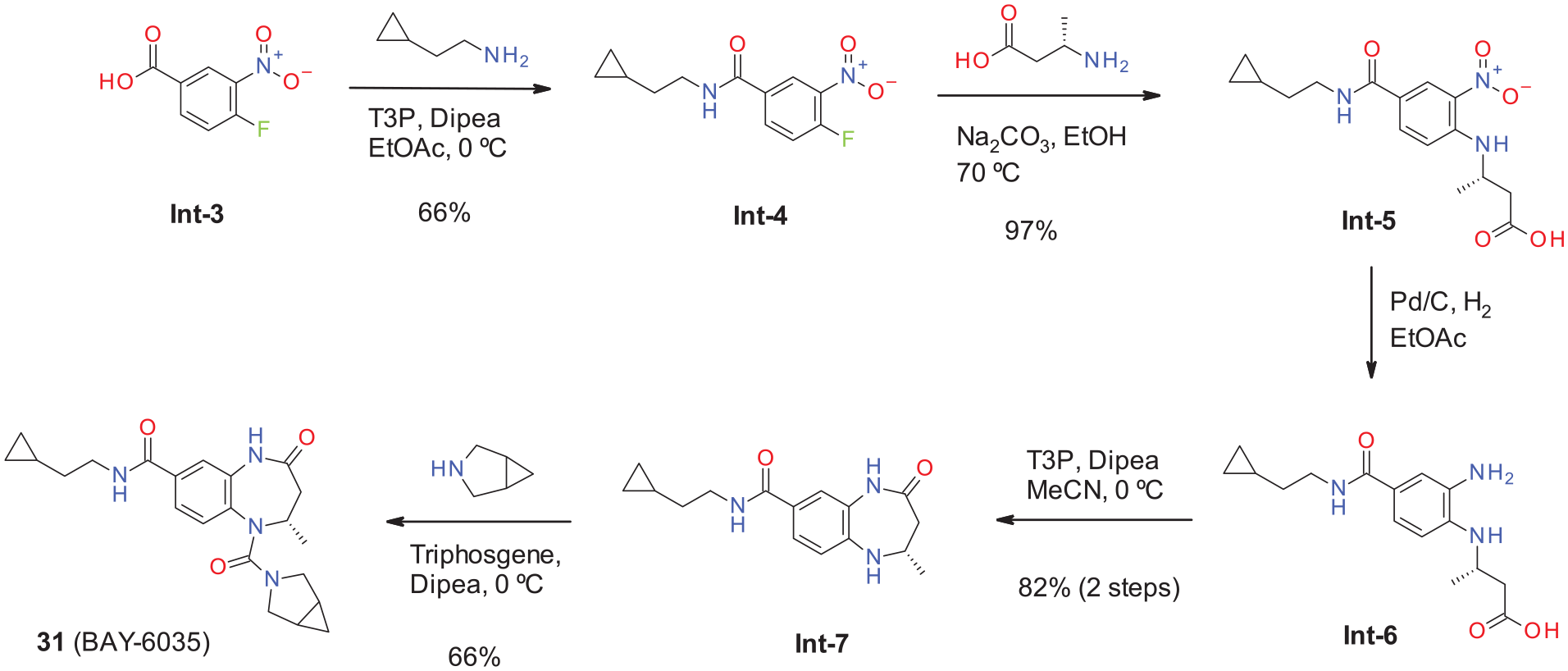
Synthesis of compound
Ureas bearing a second nucleophilic substituent, such as compounds
Detailed syntheses of test compounds used in this study were described 43 and can be found in the supplemental information.
Thermal Shift Assay
We used a TSA to identify compounds that induce a positive shift (stabilization of the protein) of the SMYD3 protein melting temperature (Tm). Thermal melting experiments were performed with a ViiATM Real-Time PCR machine (Thermo Fisher Scientific, Waltham, MA) in a 384-well microliter plate with a final volume of 5 µL. Melting curves were obtained at a protein concentration of 1.5–2.0 µM and 5xSYPRO Orange using buffer containing 20 mM Tris (pH 7.0), 150 mM NaCl, and 1.0 mM DTT. Full-length, untagged SMYD3 protein was used for these measurements. Compounds were diluted in DMSO at a single concentration of 120 μM for primary screening, 100 μM for confirmation test, or for dose–response curves a titration row ranging from 10 up to 200 µM. As a control, 4% DMSO was used. Scans were measured from 25 to 80 °C at a scanning rate of 0.1 °C/s. SMYD1 and SMYD2 proteins were used for selectivity in buffer 20 mM Tris (pH 8.0), 100 mM NaCl, 8.5% glycerol, and 1.0 mM DTT with a protein concentration of 1.4 µM and 8xSYPRO Orange. All TSA data were analyzed using the Genedata Assay Analyzer software.
Purification of SMYD3
Recombinant human SMYD3 (UniProt Q9H7B4, amino acids 1–428) was expressed in Escherichia coli containing an N-terminal TEV-cleavable 6xHis tag. Cell pellets were resuspended in lysis buffer (20 mM Tris [pH 8.0], 500 mM NaCl, 5% glycerol, and 10 mM imidazole) supplemented with complete EDTA-free protease inhibitor tablets. The cell lysate was loaded onto a Ni-NTA (Protino NiNTA) column, eluted with imidazole. The 6xHis tag was subsequently cleaved in solution by TEV. After ion-exchange chromatography (MonoQ10/100GL), SMYD3 was further purified on a Superdex S200 (35/600) column preequilibrated in 20 mM Tris (pH 8.0), 100 mM NaCl, and 1 mM DTT.
Purification of SMYD1 and SMYD2
Recombinant human SMYD1 (UniProt Q8NB12, amino acids 2–490) and SMYD2 (UniProt Q9NRG4, amino acids 2–433) were expressed in insect cells (Sf9) containing an N-terminal TEV-cleavable 6xHis tag. Cell pellets were resuspended in lysis buffer (40 mM Tris [pH 8], 500 mM NaCl, 0.1% IGEPAL, 5 mM imidazole, and 1 mM DTT) supplemented with complete EDTA-free protease inhibitor tablets and 50 U/mL benzonase. The cell lysate was loaded onto a Ni-NTA column, eluted with imidazole, and concentrated using an ultracentrifugal filter unit. Both proteins were further purified on a Superdex S200 column preequilibrated in 20 mM Tris (pH 8), 100 mM NaCl, 5% glycerol, and 1 mM DTT.
SPA for MAP3K2
Inhibitory activities of compounds were evaluated using a scintillation proximity assay (SPA) in which methylation by the enzyme of a biotinylated peptide was measured. The peptide, DYDNPIFEKFGKGGTYPRRYHVSYHGK-Biotin, is derived from MAP3K2 (249–273) and contains the Lys 260 residue previously shown to be methylated by SMYD3. 9 Assays were conducted in 384-well microtiter plates in quadruplicate in a buffer containing 20 mM Tris (pH 9), 2.5 mM DTT, and 0.01% Triton X-100 in a total volume of 10 µL.
The SMYD3 concentration in the assay was 10 nM, while tritiated S-adenosyl-
Isothermal Titration Calorimetry
Eight injections of 4 μL each in 180 s intervals were performed. The heat production appears during the first few injections. In the syringe were 20 mM HEPES (pH 7.4), 150 mM NaCl, and 0.005% Tween 20, 0.3 mM compound concentration and 2.5% DMSO concentration.
SPR Analysis
Surface plasmon resonance (SPR) analysis was performed using a Biacore T200 (GE Health Sciences Inc.) at 25 °C. Approximately 4700 resonance units of biotinylated SMYD3 (amino acids 1–428) was captured onto one flow cell of an streptavidin sensor chip according to the manufacturer’s protocol. A second flow cell was left empty for reference subtraction. SPR analysis was performed in 20 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, and 0.05% Tween-20 supplemented with 2% DMSO. Five concentrations of BAY-6035 (25, 74, 222, 667, and 2000 nM) were prepared by serial dilution. Kinetic determination experiments were performed using single-cycle kinetics with a contact time of 60 s, off time of 300 s, and flow rate of 30 μL/min. To favor complete dissociation of compound for the next cycle, a regeneration step (300 s, 40 μL/min buffer), a stabilization period (120 s), and two blank cycles were run between each cycle. Kinetic curve fittings were done with a 1:1 binding model and the Biacore T200 Evaluation software (GE Health Sciences Inc.). The experiments were performed in triplicate in the presence of 50 μM SAM.
X-Ray Crystallography
Purified SMYD3 was crystallized at 293 K using the hanging-drop vapor diffusion method. One microliter of the protein solution at ~10 mg/mL was mixed with 1 µL of the reservoir solution containing 8% PEG 3350, 50 mM magnesium formate, and 100 mM Tris (pH 7.5). No additional SAM was added. Needle-shaped crystals were soaked in mother liquor with compound concentrations of 3–6 mM. Soaked crystals were flash-frozen in liquid nitrogen. Crystallographic data were collected at Bessy Synchrotron (Berlin, Germany). Data reduction was performed with XDS, phasing was performed by molecular replacement using PHASER, and refinement was carried out using COOT and REFMAC5 as implemented in the CCP4 software package. The coordinates of the Smyd3 complexes of compounds
Cellular SMYD3 Assay
HeLa cells (ATCC CCL-2) were seeded in 12-well plates (1 × 105 cells/mL) in DMEM (Wisent) supplemented with 10% fetal bovine serum (Wisent), penicillin (100 U/mL), and streptomycin (100 µg/mL). On the next day, cells were co-transfected with 0.1 µg/well untagged SMYD3 wild-type or catalytic inactive SMYD3 mutant (deletion in amino acids 205–211 in the catalytic SET domain) and 0.9 µg/well HA-tagged MAP3K2 using Xtreme gene HP transfection reagent (Roche, Basel, Switzerland), following the manufacturer’s instructions. After 4 h, media was removed and the cells were treated with inhibitors or a DMSO control. After 20 h, media was removed and the cells were lysed in 100 µL of total lysis buffer (20 mM Tris-HCl [pH 8], 150 mM NaCl, 1 mM EDTA, 10 mM MgCl2, 0.5% TritonX-100, 12.5 U/mL benzonase [Sigma-Aldrich (St. Louis, MO)], and complete EDTA-free protease inhibitor cocktail [Roche]). After 1 min of incubation at room temperature, sodium dodecyl sulfate (SDS) was added at a final concentration of 1%. Total cell lysates were analyzed on 4%–12% Bis-Tris Protein Gels (Invitrogen) with MOPS buffer (Invitrogen) and blotted for 1.5 h (80 V) onto a polyvinylidene fluoride membrane (GE Healthcare Amersham Hybond, Fisher Scientific) in Tris-glycine transfer buffer containing 20% MeOH and 0.05% SDS. Blots were blocked for 1 h in blocking buffer (5% milk in phosphate-buffered saline with Tween [PBST]–0.1% Tween 20 PBS) and then incubated with primary antibodies: anti-HA tag (1:500; Santa Cruz Biotechnologies, Dallas, TX, cat. sc-7392), anti-SMYD3 (1:2000; Abcam, Cambridge, UK, cat. ab183498), anti-MAP3K2 (1:1000; CST, Danvers, MA, cat. 19607), and anti-MAP3K2-K260me3 (1:2000) in blocking buffer overnight at 4 °C. The antibody anti-MAP3K2-K260me3 was produced in-house with rabbit antisera generated using the DNPIFEKFGK(me3)GGTYPRRYHV KLH-conjugated peptide at the University of Toronto, Division of Comparative Medicine facility and affinity purified (
Results
Screening for SMYD3 Inhibitors Using a High-Throughput TSA
The proposed link between SMYD3 function and cancer motivated us to screen for small-molecule inhibitors of the enzyme. To this end, we developed an SMDY3 TSA to identify compounds that induce a positive shift (stabilization of the protein) of the protein melting temperature (Tm). Based on our experiences with SMYD2, a closely related family member of SMYD3, and the identification of BAY-598,
44
we focused on the development of a TSA uHTS for lead identification. With the inhibitor series leading finally to BAY-598, we observed a very good correlation between TSA stabilization and catalytic inhibition. This prompted us to change the screening strategy for SMYD3 from an enzymatic assay toward a binding/stabilization-based TSA. We screened a core library of ~410000 small molecules to identify compounds that stabilize SMYD3. For the primary screen, compounds were measured at a single concentration of 120 µM. Compounds stabilizing more than 1.2 K were subsequently reconfirmed in triplicate retests. This approach identified more than 3900 confirmed SMYD3 hits. To further characterize the primary hits, we also tested for stabilization of the closely related SMYD1 and SMYD2 proteins. This resulted in 1238 confirmed and specific stabilizers of SMYD3 (
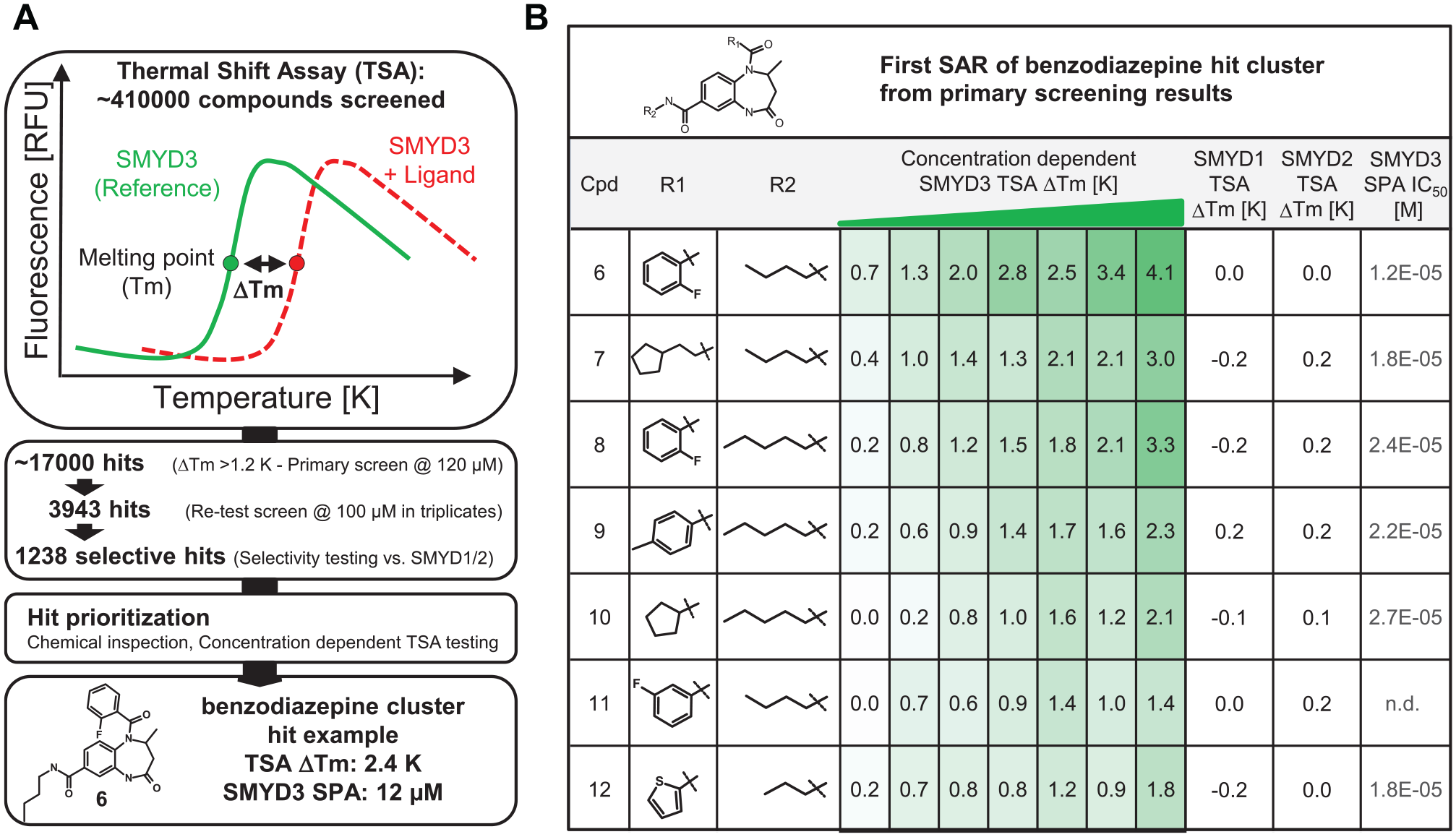
TSA screening. (
Based on the dose-dependent TSA results, our attention was drawn to a cluster of benzodiazepine-based compounds, which showed a first structure–activity relationship (SAR) (
X-Ray Crystallography of Compound 6 (Hit)
In order to guide further optimization, we selected
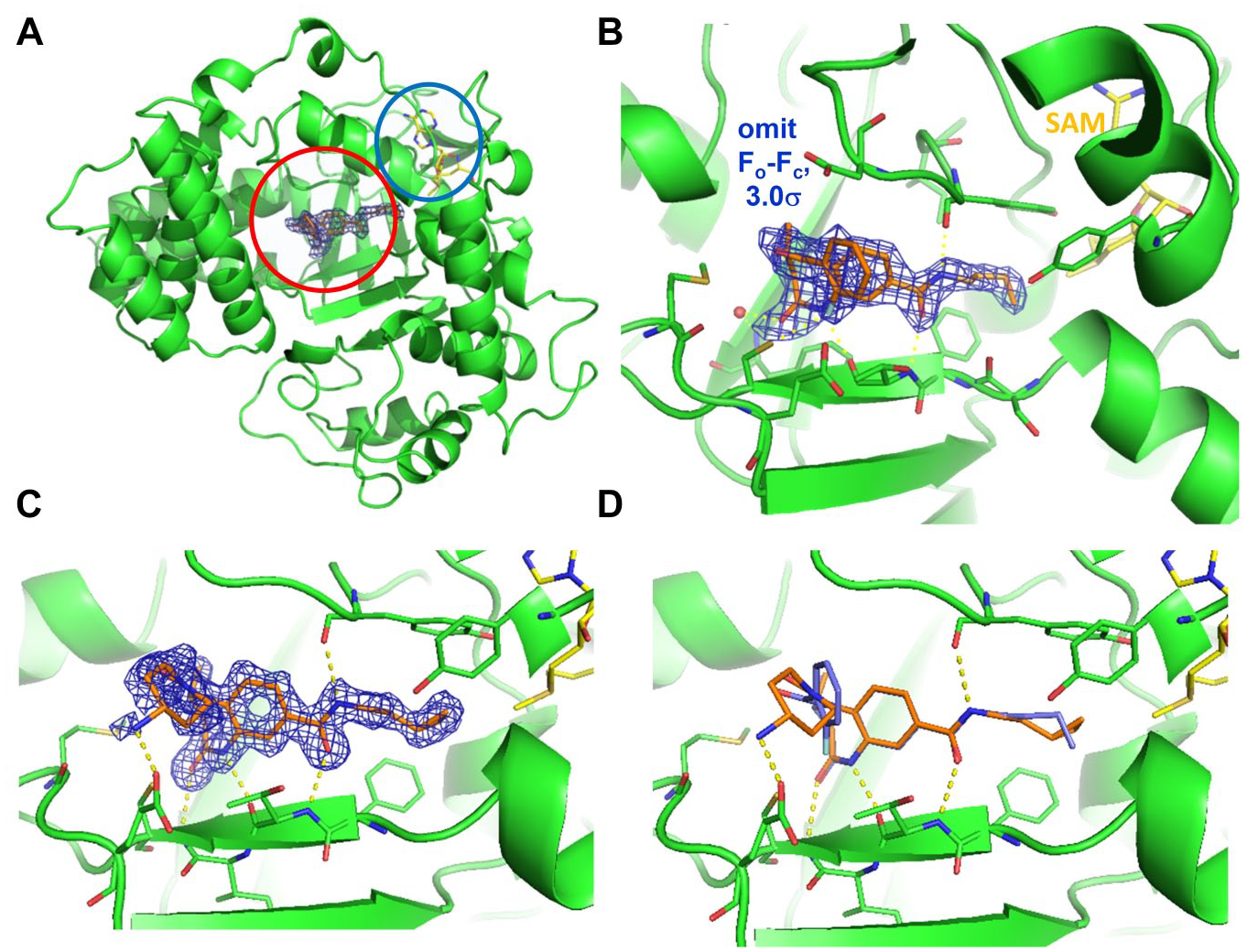
Crystal structures of
Structure−Activity Relationships
We first turned our attention to the variation of the amine side chain, exploring interactions in the lysine binding channel. Replacing the n-butyl chain with shorter or longer chains did not increase potency. Small cycloalkyl rings were the most beneficial for potency, and we explored compounds bearing the cyclopropylethyl and cyclobutylethyl side chains in more detail. In comparison with the original n-butyl-substituted racemic hit compounds, replacement by cyclopropylethyl or cyclobutylethyl substituents to address the lysine channel yielded a potency improvement by one order of magnitude. The benzoic acid analogs
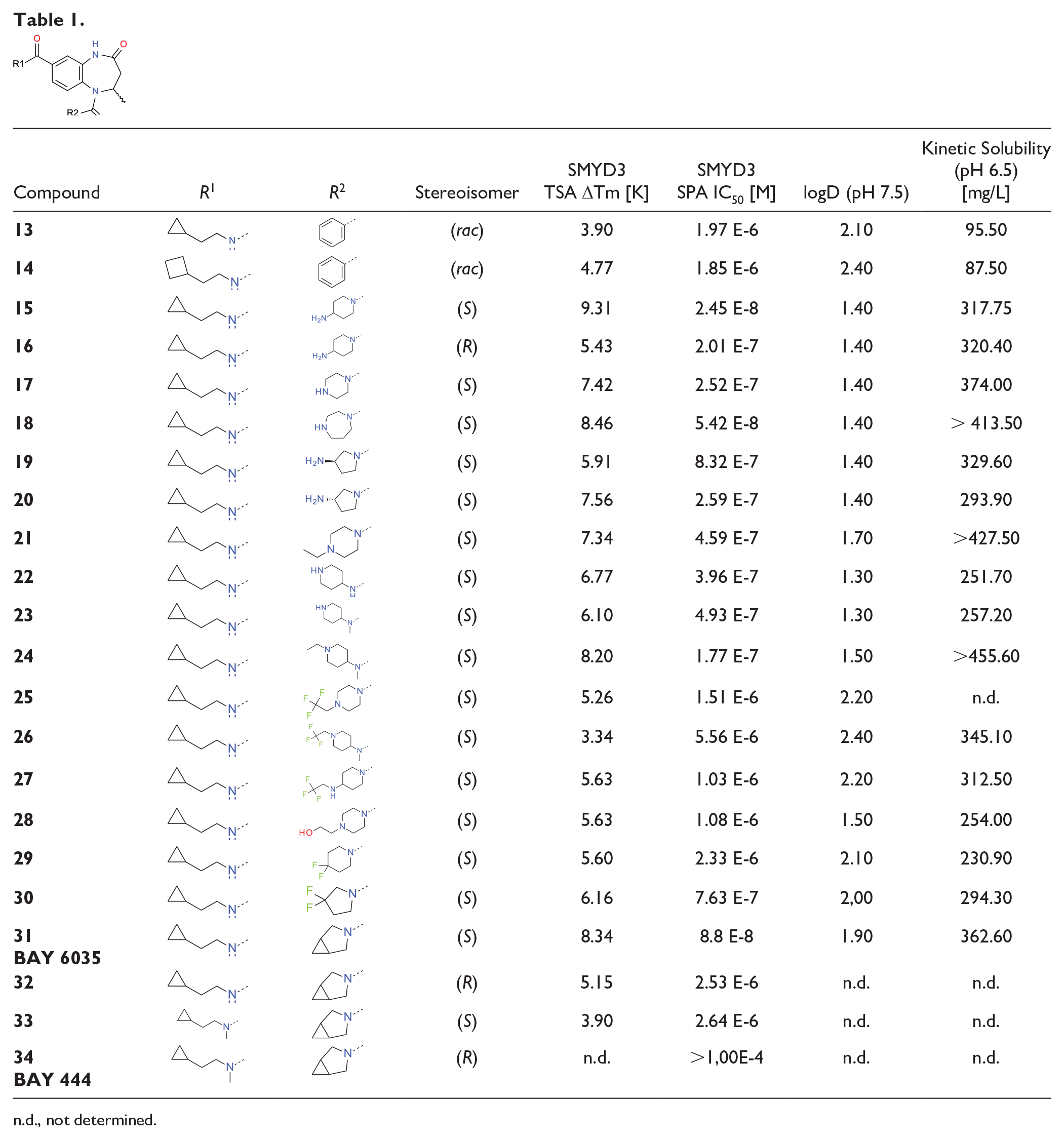
n.d., not determined.
The presence of three negatively charged amino acids (Glu 192, Asp 241, and Glu 294) in close neighborhood to the benzoic acid side chain in the crystal structure of
Despite using a racemic mixture for crystallization experiments, only the (S)-enantiomer
We synthesized a range of urea-containing compounds. The replacement of the benzoic acid amide by a urea-linked aliphatic heterocycle was in general associated with a potency improvement, but all replacements of the aminopiperidine moiety were less potent than compound
We hypothesized that the low permeability and strong efflux could be attributed to the presence of the basic amine introduced for the sake of potency optimization, so we subsequently searched for alternative options. We focused on small aliphatic ring systems and found that while difluoropiperidine (
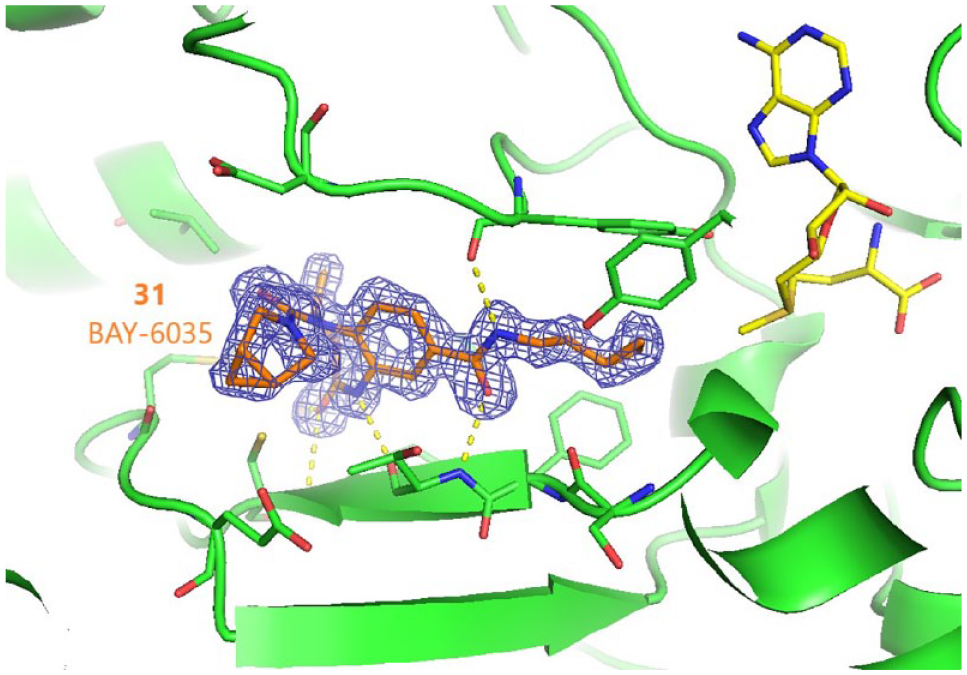
X-ray structure of
Cellular Activity of SMYD3 Inhibitors
In order to characterize whether the activity of our series in the biochemical assay is translated to inhibition of SMYD3-dependent methylation in cells, we transiently overexpressed HA-tagged MAP3K2 and untagged SMYD3 in HeLa cells and determined the levels of MAP3K2 trimethylation at lysine 260 (
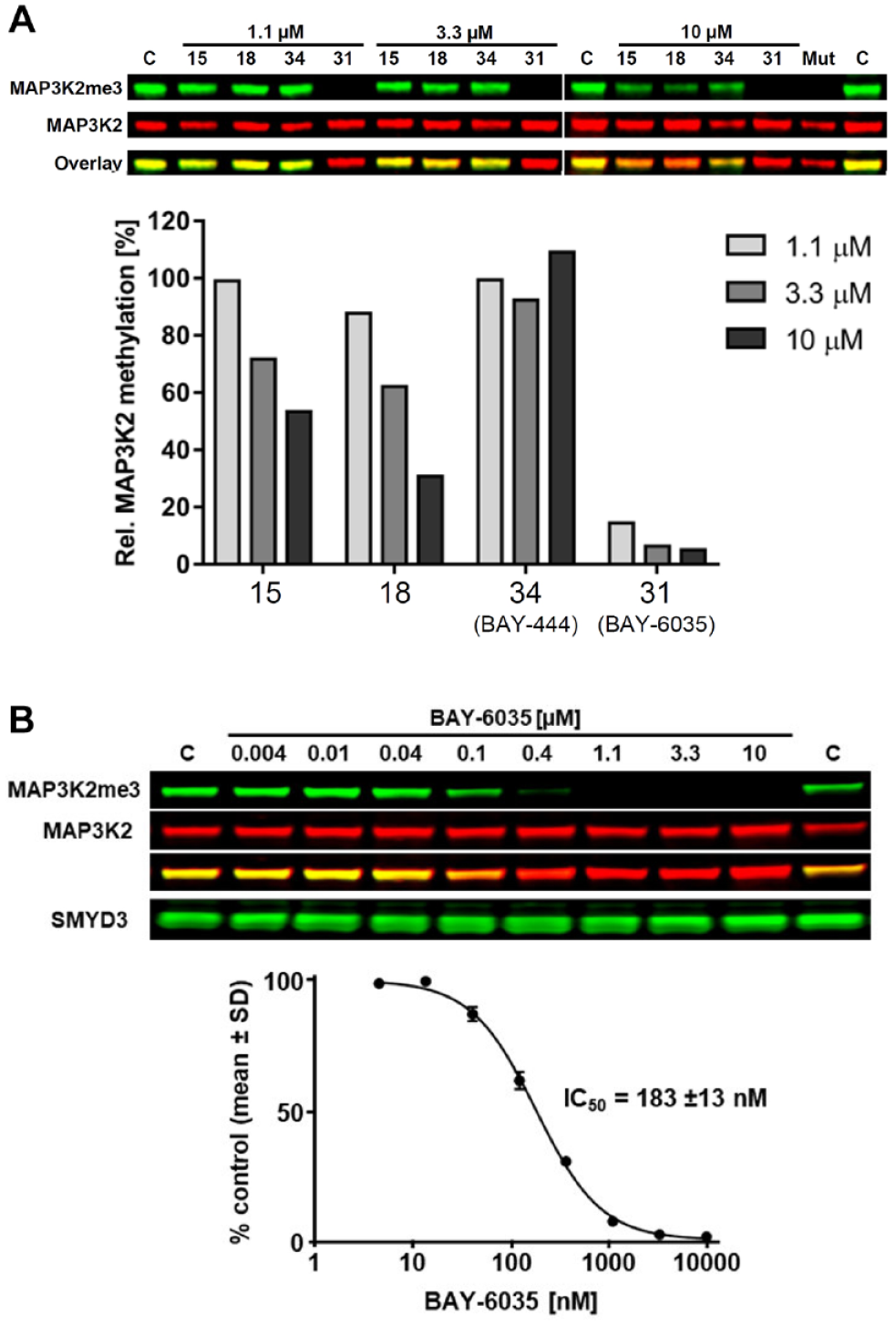
SMYD3 inhibitor compounds reduce MAP3K2 methylation in cells. HeLa cells were transfected with SMYD3 (wild-type or catalytic mutant) and HA-tagged MAP3K2 and treated with the indicated compounds for 20 h. MAP3K2 K260 trimethylation levels were determined by Western blot and normalized to total HA-MAP3K2 levels. Mut, catalytic inactive SMYD3 mutant (deletion in amino acids 205–211 in the catalytic SET domain). (
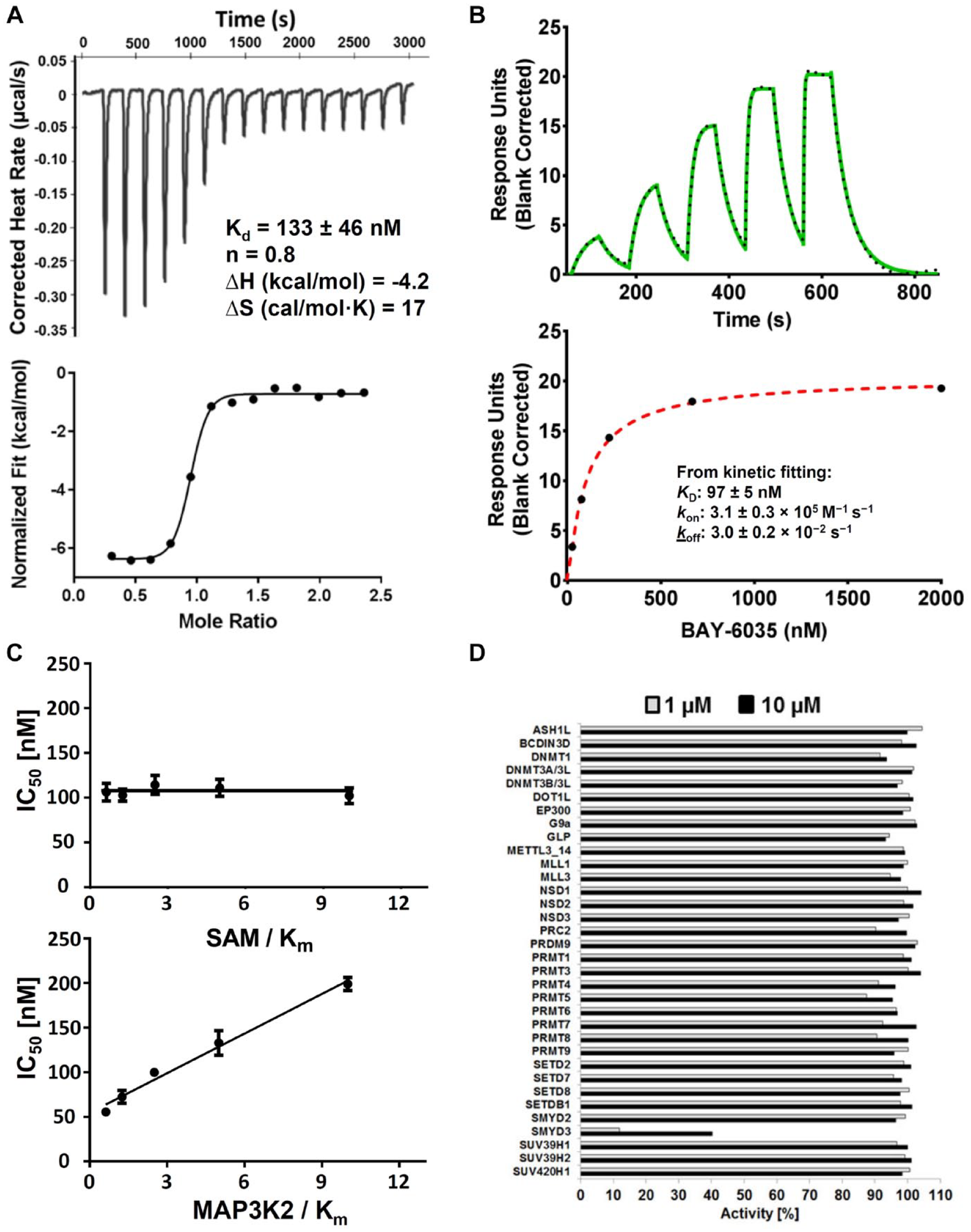
Characterization of BAY-6035. Confirmation of binding activity by (
Full Characterization of BAY-6035 as a Novel Chemical Probe for SMYD3
The good combination of the biochemical and cellular potencies of BAY-6035 prompted us to further characterize this compound. We analyzed the binding of BAY-6035 to SMYD3 in isothermal titration calorimetry (ITC) (
To further characterize BAY-6035, we tested the selectivity against a set of 34 protein methyltransferases, including several that contained a SET domain and three DNA methyltransferases. BAY-6035 showed highly selective inhibition of SMYD3 with no activity on other methyltransferases (
Discussion
SMYD3 and other protein methyltransferases have recently emerged as potential therapeutic cancer targets. Validation of SMYD3-associated cancer phenotypes has so far been mainly based on correlative studies of increased protein level in cancer tissues or knockdown/knockout experiments. For a more unbiased interpretation of observed effects and comprehensive validation as a therapeutic drug target, fully profiled chemical probes are highly desirable.
38
In this study, our aim was to generate a novel SMYD3 probe inhibitor, which should be openly accessible to the research community. This probe may enable more cross-validation studies based on catalytical inhibition of the enzymatic function(s) of SMYD3. We decided to switch our screening approach to a binding/stabilization-based TSA for this target protein, based on previous experiences with the closely related SMYD2 enzyme.
44
Using a TSA had several advantages in comparison with assays based on catalytical activity, including being low cost, easy to establish, and fast and having relatively low protein requirements.
47
During the hit validation phase, the TSA was crucial for the further selection process. Dose-dependent TSA stabilization was used to further narrow down our primary hit list toward more specific hits. We finally prioritized the benzodiazepine hit cluster, which showed clear concentration-dependent stabilization effects in the TSA. Primary hit
BAY-6035 represents a novel structurally orthogonal chemical probe, which will be valuable for further target validation and exploration of additional undiscovered functional roles of SMYD3. In a first report using a chemical probe for SMYD3 target validation,
Supplemental Material
sj-pdf-1-jbx-10.1177_24725552211019409 – Supplemental material for Discovery of the SMYD3 Inhibitor BAY-6035 Using Thermal Shift Assay (TSA)-Based High-Throughput Screening
Supplemental material, sj-pdf-1-jbx-10.1177_24725552211019409 for Discovery of the SMYD3 Inhibitor BAY-6035 Using Thermal Shift Assay (TSA)-Based High-Throughput Screening by Stefan Gradl, Holger Steuber, Joerg Weiske, M. Szewczyk, Norbert Schmees, Stephan Siegel, Detlef Stoeckigt, Clara D. Christ, Fengling Li, Shawna Organ, Megha Abbey, Steven Kennedy, Irene Chau, Viacheslav Trush, Dalia Barsyte-Lovejoy, Peter J. Brown, Masoud Vedadi, Cheryl Arrowsmith, Manfred Husemann, Volker Badock, Marcus Bauser, Andrea Haegebarth, Ingo V. Hartung and Carlo Stresemann in SLAS Discovery
Footnotes
Acknowledgements
We thank Elisa Gibson and Taraneh Hajian for the support of SMYD3 protein purification. In addition, we thank Tina Stromeyer for her assistance in the crystallization experiments and Albine Bolotokova for her help in managing the compounds. Simon Holton is acknowledged for helpful discussions and critical reading of the manuscript.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: S.G., H.S., J.W., N.S., S.S., D.S., C.D.C., M.H., V.B., M.B., A.H., I.V.H., and C.S. were or are employees and stockholders of Bayer AG. S.G., H.S., J.W., N.S., S.S., C.D.C., M.H., V.B., and C.S. are listed as inventors of a patent application related to SMYD3 inhibitors. 43
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Structural Genomics Consortium is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer AG, Boehringer Ingelheim, Canada Foundation for Innovation, Genentech, Genome Canada through Ontario Genomics Institute (OGI-196), EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking (EUbOPEN grant 875510), Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer, Takeda, and Wellcome (106169/ZZ14/Z).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.