
Abstract
Triose phosphate isomerase deficiency (TPI Df) is an untreatable, childhood-onset glycolytic enzymopathy. Patients typically present with frequent infections, anemia, and muscle weakness that quickly progresses with severe neuromusclar dysfunction requiring aided mobility and often respiratory support. Life expectancy after diagnosis is typically ~5 years. There are several described pathogenic mutations that encode functional proteins; however, these proteins, which include the protein resulting from the “common” TPIE105D mutation, are unstable due to active degradation by protein quality control (PQC) pathways. Previous work has shown that elevating mutant TPI levels by genetic or pharmacological intervention can ameliorate symptoms of TPI Df in fruit flies. To identify compounds that increase levels of mutant TPI, we have developed a human embryonic kidney (HEK) stable knock-in model expressing the common TPI Df protein fused with green fluorescent protein (HEK TPIE105D-GFP). To directly address the need for lead TPI Df therapeutics, these cells were developed into an optical drug discovery platform that was implemented for high-throughput screening (HTS) and validated in 3-day variability tests, meeting HTS standards. We initially used this assay to screen the 446-member National Institutes of Health (NIH) Clinical Collection and validated two of the hits in dose–response, by limited structure–activity relationship studies with a small number of analogs, and in an orthogonal, non-optical assay in patient fibroblasts. The data form the basis for a large-scale phenotypic screening effort to discover compounds that stabilize TPI as treatments for this devastating childhood disease.
Introduction
Triose phosphate isomerase deficiency (TPI Df) is a rare degenerative disorder that typically affects young children. The disease belongs to a family of diseases called glycolytic enzymopathies, although it is unique from other members in the family because it is characterized by severe neuromuscular impairment and often progressive neurologic symptoms. 1 The typical age of diagnosis for TPI Df is between 1 and 5 years of age, and the most common initial symptom is hemolytic anemia because erythrocytes cannot compensate for defective glycolysis through oxidative metabolism. As the disease progresses, patients experience failure in tissues and organs with high energy requirements, including increased susceptibility to infection, neurological dysfunction, and progressive neuromuscular degeneration. 2 The end stage of this disease constitutes severe paralysis and the inability to breathe without assistance from a respirator. TPI Df leads to a drastically reduced lifespan, with death typically occurring within 5 years of the initial diagnosis. At this time, there are no medical treatments for patients with TPI Df, aside from supportive care, antioxidants, and possibly dietary changes in the form of the ketogenic diet. 3
TPI is a glycolytic enzyme known to interconvert dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (G3P), which allows for increased energy yield from glucose metabolism during glycolysis. The enzyme is known for being a perfect enzyme, meaning that the kinetics of the enzyme are limited only by the speed of diffusion of the substrate. 4 TPI Df is typically caused by specific missense mutations in the TPI1 gene. The most common TPI Df mutation is the TPIE105D (aka E104D in legacy publications); patients with this mutation can be homozygous TPIE105D/E105D or compound heterozygous, typically with TPIE105D and another pathogenic missense mutation resulting in disease. 5 These genetic mutations primarily affect the stability of mutant TPI protein. 6 While the mutant protein dimer remains catalytically active and functional, it has been shown for several pathogenic mutations that the mutant protein is targeted for degradation through the protein quality control (PQC) pathway, leading to very low levels of TPI within the cell. 7 Specifically, both heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) have been demonstrated to play a role in mutant TPI degradation in an animal model of TPI Df. 8 In the absence of an established rodent model of TPI Df, which is currently under development in our laboratory, the animal model most extensively studied has been the fruit fly, Drosophila. The TPI gene is highly conserved in Drosophila, its active site and enzymatic mechanism are identical to those of mammals, and glycolysis is highly conserved across organisms, functioning identically in humans, mice, and flies. Drosophila expressing mutant TPI exhibit phenotypes similar to those observed in humans with TPI Df, such as shortened lifespan, locomotor dysfunction, and neuropathology.7–9
Seminal studies in Drosophila have documented that genetic [by RNA interference (RNAi)] and pharmacological interventions (by inhibitors of the proteasome and Hsp70 and Hsp90) result in elevated TPI levels and reverse the detrimental effects of mutant TPI. 8 Hsp70 and Hsp90 are, however, ubiquitously expressed and interact with a wide variety of client proteins and co-chaperones to fulfill their cellular roles. 10 Although numerous drugs modulate Hsp70, Hsp90, and the proteasome, these drugs are all exceptionally toxic, and currently their clinical significance centers on their intrinsic toxicity and ability to sensitize cells to chemotherapies. We have recently used mutant TPI in a powerful Drosophila genetic screen to elucidate all (or at least most) of the PQC components that regulate TPI turnover. 11 We performed a genome-wide RNAi screen of all known and predicted components of the ubiquitin–proteasome system (UPS) and PQC pathways and numerous unknown proteins with domains similar to those commonly found in UPS and PQC components. Importantly, the screen identified ~27 proteins that are critical to TPI stability (including many that were not previously known to be involved in its turnover), novel proteins with no known or associated PQC function for any substrate, including some that could contribute to protein folding or ubiquitination, or are likely to be involved in co-translational PQC. 11 The majority of these potential targets are known to be much less promiscuous than Hsp70 and Hsp90, which are known to modulate >75% of the proteome, suggesting that discovery of less toxic agents that stabilize mutant TPI should be feasible. 11
In an effort to develop effective and nontoxic treatments for TPI Df, we developed a high-throughput phenotypic screening assay (which captures all mechanisms that govern TPI stability) to identify novel pharmacological regulators of mutant TPI degradation. We developed a modified HEK-293T cell line that expresses mutant TPIE105D protein fused to a green fluorescent protein reporter (TPIE105D-GFP). The assay is a phenotypic screen enabling the unbiased assessment of novel compounds as modulators of TPI stability based on a cellular fluorescent readout, which would include compounds that slow the degradation of mutant TPI in human cells. The assay was initially tested by administration of PQC inhibitors that targeted known regulators of mutant TPI and were thus predicted to increase mutant TPI levels, resulting in fluorescence intensity changes in HEK cells stably expressing TPIE105D-GFP. Once the assay was validated, the National Institutes of Health (NIH) Clinical Collection of compounds was screened in the mutant TPIE105D-GFP HEK cells to investigate any potential pharmacological regulation of mutant TPI levels, focusing on drugs that have a history of use in human clinical trials. Some of the hits from this pilot screen were further investigated using human patient cell lines and showed increases of endogenous mutant TPI. The assay performed to accepted HTS standards in multiday variability studies and is ready to be used in large-scale HTS. Insights gained from this high-throughput screen may allow for the development of new treatment options for patients with TPI Df.
Materials and Methods
Generating and Culturing HEK293 TPIE105D-GFP Cells
HEK293 cells expressing a mutant (E105D) TPI-GFP fusion protein were created using the Flp-In System (cat. nos. K6010-01 and K6010-02, Invitrogen, Waltham, MA) following the manufacturer’s protocol. Briefly, a host cell line expressing the transfected Flp-In target site vector, pFRT/lacZeo, was obtained following zeocin-resistant selection. TPIE105D-GFP fusion construct was cloned into the pcDNA 5/FRT expression vector. Co-transfection of 0.1 µg of the pcDNA 5/FRT construct (390 ng/µL) and 0.9 µg of the Flp recombinase expression vector (pOG44, 127 ng/µL) into the Flp-In host cell line was performed with 3.75 µL Lipofectamine 2000 reagent (Invitrogen). A hygromycin dose–response was performed to determine the minimum concentration of hygromycin B required to kill untransfected host cells. Clones of cells were selected that exhibited hygromycin B resistance. Expression of the TPIE105D-GFP fusion protein was verified via Western blot. The cells were cultured using standard methods (37 °C, 5% CO2) in normal media [Dulbecco’s modified Eagle medium (DMEM) with 10% serum, 100 U penicillin/100 µg streptomycin/ml; Lonza, Basel, Switzerland]. To document subcellular localization of mutant GFP-tagged TPI, nuclei were stained with Hoechst 33342 and imaged on a PerkinElmer Opera Phenix confocal high-content reader (PerkinElmer, Waltham, MA) in the ultraviolet (UV) and GFP channels, respectively. A z-series of images (63× water immersion objective, 90 planes, 0.3 µm z-steps) was acquired in both channels, and the color combined stack was subjected to three-dimensional (3D) reconstruction in MetaXpress (Molecular Devices, San Jose, CA). A single slice of the reconstructed image was chosen to illustrate the phenotype of mutant TPI-GFP expression. In addition, a 3D rendering was created in Harmony 5.0 (PerkinElmer).
NIH Clinical Collection
The NIH Clinical Collection is a plated array of 446 small molecules that encompasses US Food and Drug Administration (FDA)-approved drugs and agents that have a history of use in human clinical trials. The collection was assembled by the NIH through the Molecular Libraries Roadmap Initiative as part of its mission to enable the use of compound screens in biomedical research and is maintained as assay-ready daughter plates at the University of Pittsburgh Drug Discovery Institute (UPDDI). The library is maintained under temperature- and humidity-controlled conditions as recommended.12,13 A full list of compounds in the library is presented in
Additional Compounds
A full list of compounds used for the prescreen and for follow-up studies is presented in
Assay Development and Prescreen with Selected PQC Inhibitors
HEK293 TPIE105D-GFP cells were plated (50,000 cells/well in collagen-coated 96-well thin-bottom microplates; cat. no. 655956, Greiner Bio-One, Monroe, NC) and allowed to attach overnight. Cells were treated with four-point, twofold gradients of 5× treatment solutions of test agents (six replicates per condition). The final concentration of vehicle (DMSO) was 0.2%. After 48 h in culture, cells were preincubated with propidium iodide (PI) for 30 min and imaged live in the GFP and Texas Red channels on an ImageXpress Ultra high-content reader (Molecular Devices) using a 20× objective. Images were analyzed with the multiwavelength cell-scoring application using GFP to define and enumerate cells. The integrated GFP intensity per cell was used to quantify mutant TPI expression, the number of cells was used as a proxy for growth inhibition and/or cell loss, and the percentage of cells that exceeded a threshold for PI based on vehicle-treated controls was used to measure acute toxicity (necrosis).
Flow Cytometry
HEK293 TPIE105D-GFP cells were plated (50,000 cells/well) in the wells of a 96-well plate (100 µl) and treated with 25 µl of a 5× treatment solution of luminespib for 48 h. Cells were detached by adding 25 µl 17.5 mM ethylenediaminetetraacetic acid (EDTA), pH 6.15, for 2 h 14 directly to growth medium (final concentration: 2.5 mM). Plates were subsequently stained with 25 µl PI [5 µg/ml in phosphate-buffered saline (PBS)], dislodged by pipetting, and analyzed by flow cytometry. Samples were acquired on a BD Fortessa SORP cytometer (BD Biosciences, San Jose, CA). Dislodged (resuspended) cells were run on the BD High Throughput Sampler (HTS) with the following settings to ensure even sampling: sample flow rate: 1.5 µl/s; sample volume: 80 µl; mix volume: 40 µl (40% total volume); mix speed: 200 µl/s; number of mixes: 4; and wash volume: 400 µl. An attempt was made to collect at least 25,000 events per sample. Forward scatter (FSC), side scatter (SSC), GFP (488_515_30), and PI (561_582_15) detectors were optimized on untreated viable HEK293T cells. Samples were analyzed for expression of GFP and viability (PI-negative events). Several population patterns were observed, and correlation with FSC/SSC revealed the following states: viable (live), early-apoptotic, and late-apoptotic cells.
Pilot Screen Using the NIH Clinical Collection
HEK293 TPIE105D-GFP cells were plated (10,000 cells/30 µl/well) in collagen-coated 384-well thin-bottom microplates (cat. no. 781956, Greiner Bio-One) and allowed to attach overnight. Assay-ready daughter plates of the NIH Clinical Collection containing 2 µL of a 1 mM DMSO stock were reconstituted in DMEM (60 µL) to yield a 30 µM intermediate treatment concentration. DMEM containing 3% DMSO, 60 µl was added to columns 1 and 24, and 60 µL of 600 nM luminespib was added to columns 2 and 23, to serve as negative and positive controls, respectively. Aliquots of 15 µl were transferred directly to cells in 384-well plates using an Agilent Bravo liquid-handling robot (final concentration in assay, 10 µM). For the 1 µM condition, an aliquot of the 30 µM treatment solution was diluted 1:10 and controls added as above. Aliquots of 15 µL were transferred to cells to yield a 1 µM final concentration. Plates were centrifuged for 30 s at 200 ×g and incubated at 37 °C. After 24 h, plates were imaged live in the GFP channel and returned to the incubator; after 48 h, cells were stained with PI, imaged live in the GFP and PI channels on the ImageXpress Ultra high-content reader (four fields, 20×, capturing approximately 1000 cells), and analyzed as described above. Some dose–response studies were done on a PerkinElmer Phenix Plus, capturing approximately 4000 cells in four fields with a 20× air objective.
Culturing Patient Fibroblasts
Patient fibroblasts were obtained from a male TPI Df patient via skin punch using the Stanford University Institutional Review Board (IRB; Registration 5135, e-protocol 28342, Dr. G. Enns). The cells were de-identified, are known only as FB104, and are homozygous for the common E105D mutation (aka E104D in legacy publications). Fibroblasts were cultured using standard methods (37 °C, 5% CO2) in complete media [DMEM with 10% serum, 100 U penicillin/100 µg streptomycin/ml (Lonza), 2 mM L-glutamine (Gibco), and supplemental non-essential amino acids (Gibco, Carlsbad, CA)].
Western Blotting
FB104 patient fibroblasts (E105D/E105D) treated with luminespib (200 nM), resveratrol (100 µM), itavastatin (5 µM), or DMSO (0.1%; Sigma Aldrich, St. Louis, MO) were trypsinized (0.05% for 5 min), pelleted, resuspended in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors [phenylmethylsulfonyl fluoride (PMSF; 100 µM), leupeptin (1 µg/µL), and pepstatin A (0.5 µg/µL)], and pulse sonicated. Protein concentrations were determined using a bicinchoninic acid (BCA) assay (Pierce, Waltham, MA). Immunoblotting was performed on whole-cell protein lysates following the addition of an equal volume of 2× sodium dodecyl sulfate–polyacrylamide gel electro-phoresis (SDS-PAGE) sample buffer (4% SDS, 4% β-mercaptoethanol, 130 mM Tris HCl pH 6.8, and 20% glycerol). Proteins were resolved by SDS-PAGE (12%) and transferred onto 0.45 µm polyvinylidene fluoride (PVDF) membrane. The blots were blocked in Odyssey Blocking Buffer (LI-COR, Lincoln, NE) and incubated with anti-TPI (1:5000; rabbit polyclonal FL-249; cat. no. sc-30145, Santa Cruz Biotechnology, Santa Cruz, CA) or anti–beta tubulin (1:1000; mouse polyclonal E7-C; Developmental Studies Hybridoma Bank, Iowa City, IA) diluted in Odyssey Blocking Buffer. Following washes in phosphate-buffered saline with Tween 20 (PBST), the blots were incubated with anti-mouse-IR800 (Fisher Scientific, Waltham, MA) and anti-rabbit-IR680 (Molecular Probes, Eugene, OR) both diluted to 1:20,000 in 0.1% Tween 20 blocking buffer. Blots were washed in PBST and developed using the Odyssey Infrared Imaging System (LI-COR). Quantification of the scanned images was performed digitally using the Image Studio Version 5.2 software (LI-COR). TPI levels were normalized to beta tubulin loading control, and differences in TPI expression were evaluated by a two-tailed Student t test.
Three-Day Variability
Two full 384-well microplates of HEK293 TPIE105D-GFP cells (10,000 cells/well) were treated with vehicle (1% DMSO) or luminespib (200 nM) on 3 consecutive days using a PerkinElmer MDT (Modular Dispense Technology) or Agilent Bravo liquid handler (Agilent, Santa Clara, CA). After 48 h of incubation, cells were imaged live in the GFP channel as described above and analyzed for TPI-GFP expression using the MetaXpress multiwavelength scoring application. Assay performance was assessed between plates and between days by calculating signal-to-background (S/B) ratios, coefficients of variation (CVs), and Z-factors, as described. 15
Results
The “common” TPIE105D mutation is well characterized biochemically and structurally, 16 and it results in a ~50% reduction in steady-state protein levels in patient cells. 17 We developed a HEK TPIE105D-GFP stable knock-in cell line using the Thermo Fisher Flip-In system (Thermo Fisher, Waltham, MA). We previously demonstrated that in compound heterozygotes, the degradation of each mutant protein indeed occurs independently; 18 thus, mutant GFP-tagged TPI degradation will not be significantly affected by wild-type TPI that keeps the cells healthy. HEK TPIE105D-GFP cells provide a convenient optical assay platform of steady-state mutant human TPI protein in the context of a human cell. Figure 1 documents stable and uniform expression of mutant GFP-tagged TPI that is consistent with a ubiquitous nuclear and cytosolic localization, suggesting it could serve as a discovery assay for compounds that increase levels of mutant TPI.
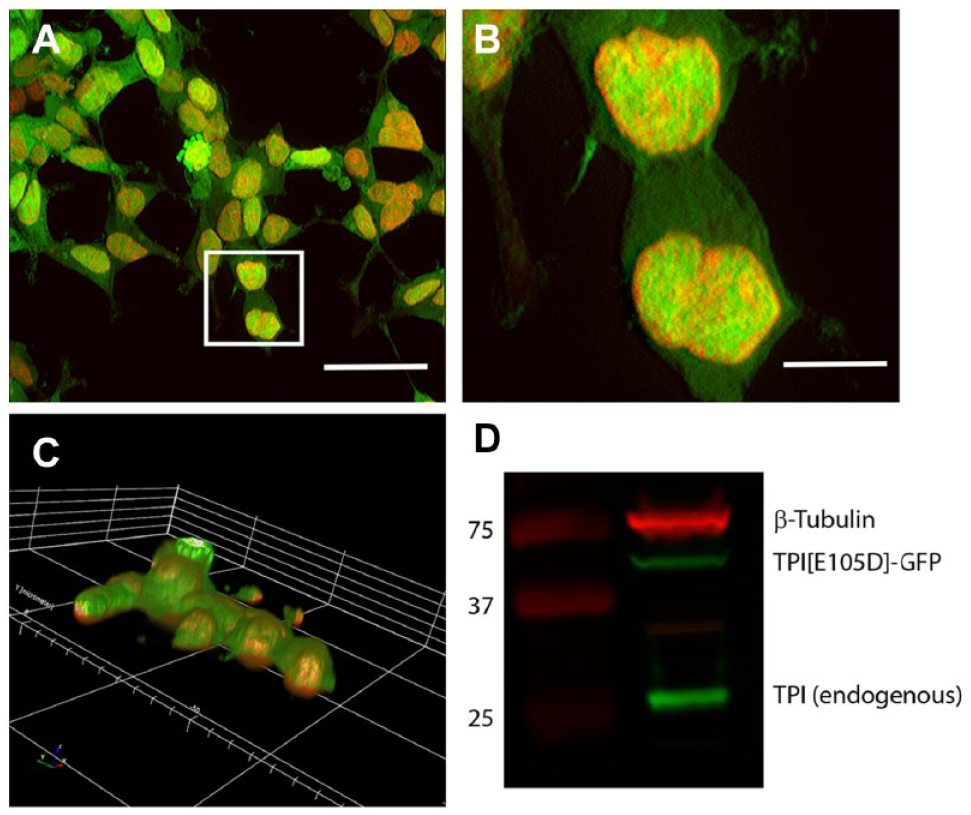
Creation of knock-in cell line. (
Assay Development
We first tested a panel of inhibitors directed at proteins known to target mutant TPI for degradation in Drosophila and examined their effect on HEK TPIE105D-GFP cells via confocal microscopy. Consistent with Drosophila results, bortezomib, cycloheximide, and luminespib all showed a significant increase in TPIE105D-GFP fluorescence, demonstrating that TPIE105D turnover in human cells is similarly regulated as TPIsugarkill (sgk) protein in Drosophila, as well as confirming the utility of the reporter system (
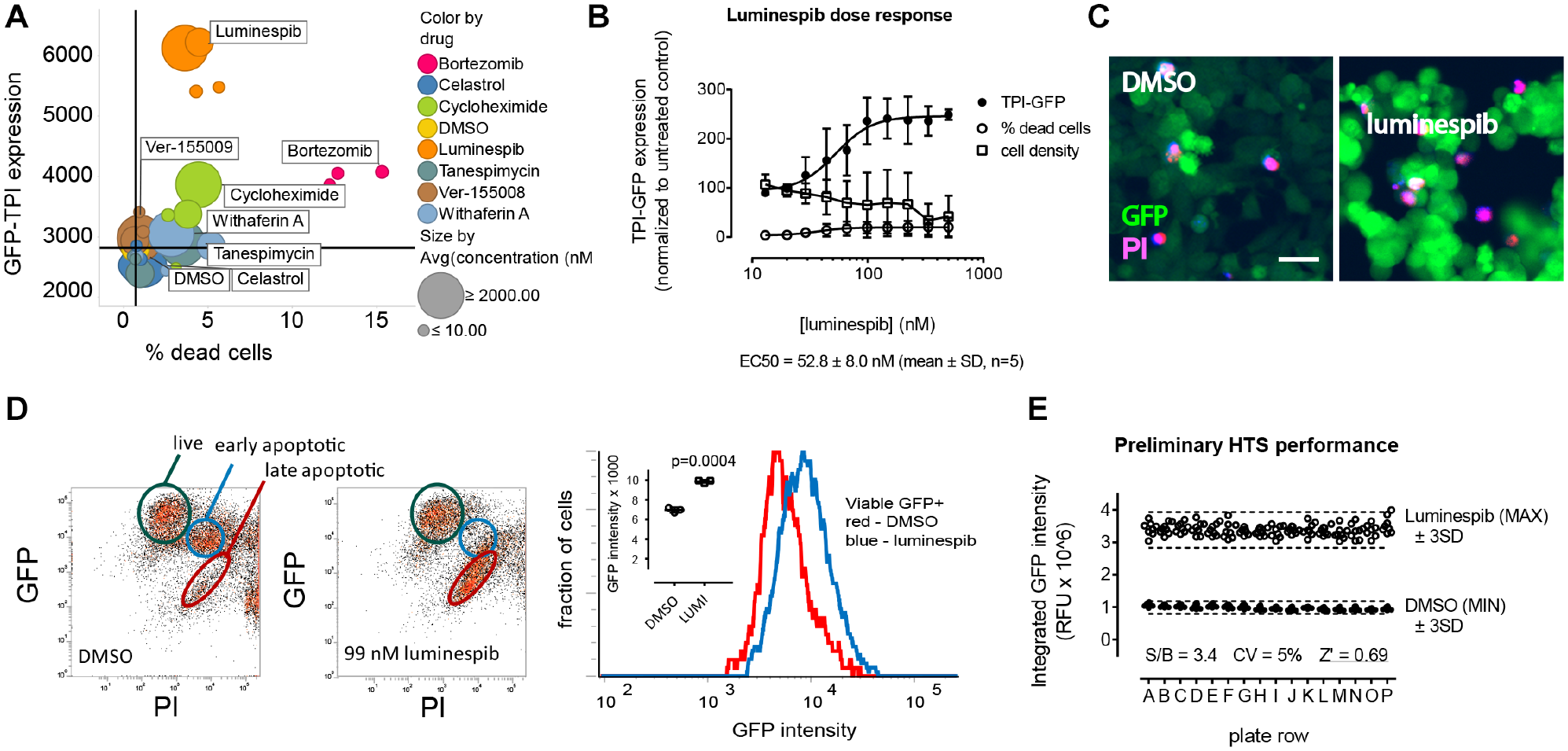
Luminespib increases triose phosphate isomerase–green fluorescent protein (TPI-GFP) expression that is quantifiable by high-content analysis. (
Compound Pilot Screen
We conducted a pilot library screen using the NIH Clinical Collection in the HEK TPIE105D-GFP cell line. The NIH Clinical Collection maintained at the UPDDI contains 446 FDA-approved drugs and small molecules that have a history of use in human clinical trials. The collections were assembled through the Molecular Libraries and Imaging Initiative as part of its mission to enable the use of compound screens in biomedical research. To extract as much information from this library as possible, we screened the library in duplicate at two concentrations (10 and 1 µM) and at two time points (24 and 48 h). Cells were plated in collagen-coated 384-well plates and treated the next day with library compounds in duplicate using the Agilent Bravo pipetting robot. DMSO (1%) and luminespib (200 nM) served as, respectively, negative (MIN) and positive (MAX) controls (n = 32 each). Cells were incubated at 37 °C, scanned on the ImageXpress Ultra high-content reader after 24 h, and returned to the incubator for an additional 24 h. Thirty minutes before the 48 h scan, cells were stained with PI and imaged in the GFP and Texas Red channels to assess TPIE105D-GFP levels, loss of cell attachment, and acute toxicity (necrosis). Plates were analyzed with the multiwavelength cell-scoring application in MetaXpress; TPI levels were calculated on a per cell basis. Cell densities and TPIE105D-GFP expression were then normalized to DMSO control.
The performance of the HEK TPIE105D-GFP cellular assay under conditions of compound screening was excellent (
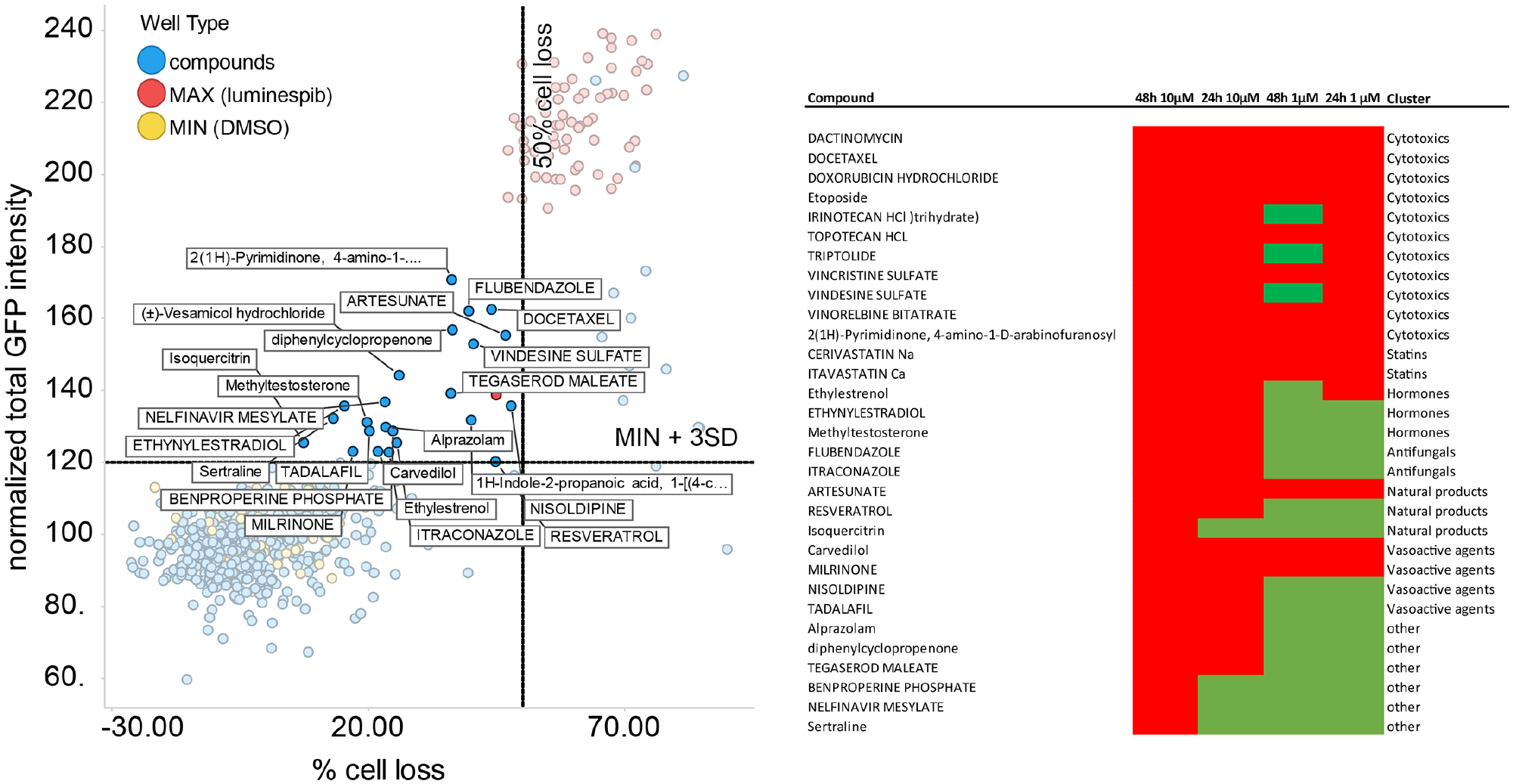
Pilot screen of the National Institutes of Health (NIH) Clinical Collection. The NIH Clinical Collection was screened in human embryonic kidney (HEK) cells expressing the common triose phosphate isomerase deficiency (TPI Df) protein fused with green fluorescent protein (HEK293 TPI-GFPE105D cells) at two concentrations and at two different time points. (
Hit Validation
Using fresh samples repurchased from commercial suppliers, both agents showed concentration-dependent increases in TPIE105D-GFP (
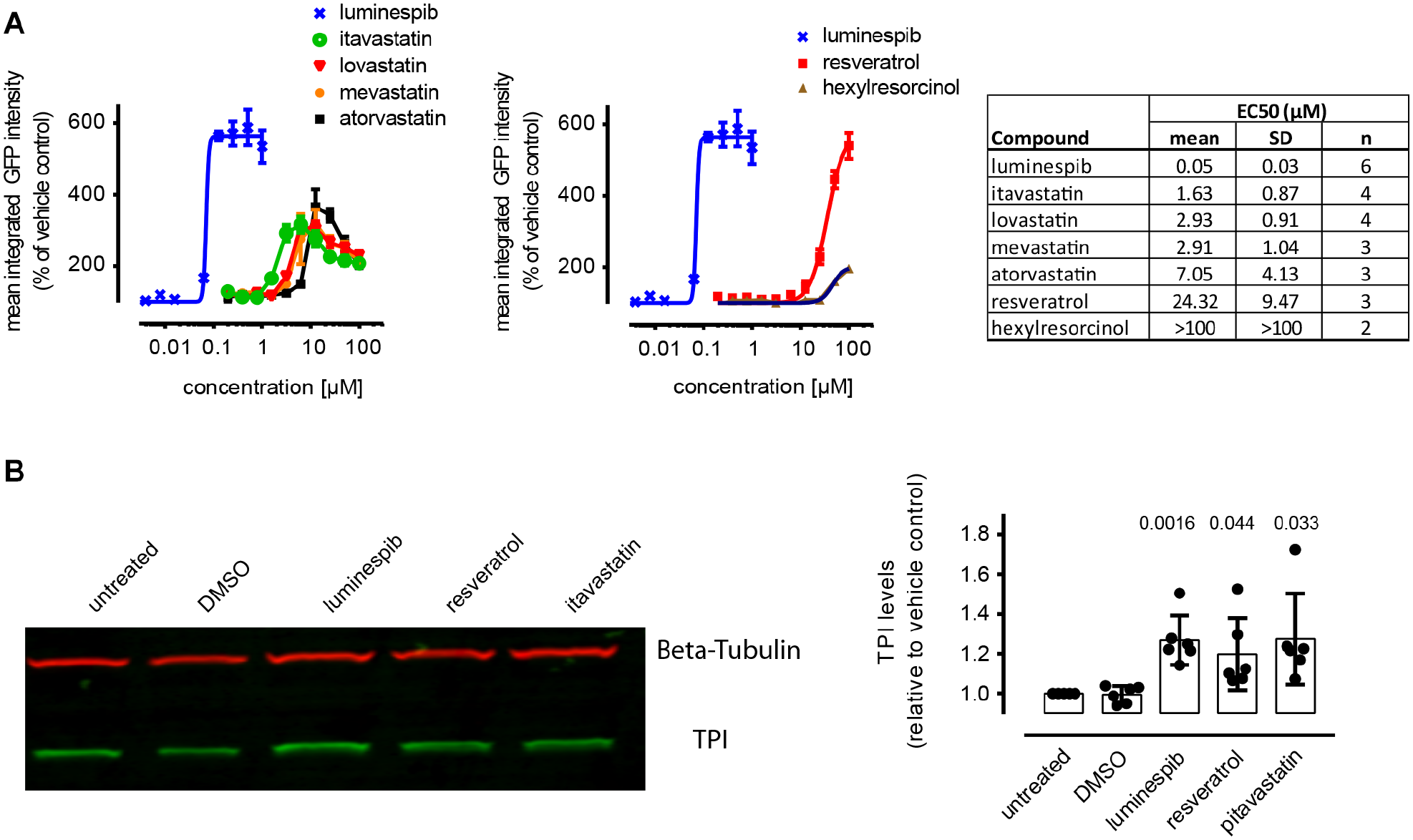
Hit confirmation in triose phosphate isomerase deficiency (TPI Df) patient fibroblasts. (
Based on these results, we performed Western blots using fibroblasts from a TPIE105D homozygous patient and confirmed elevated mutant TPI levels by resveratrol and itavastatin using an orthogonal non-imaging method (
Multiday Variability Assessments
Based on the assay development and validation results above, we performed a formal 3-day variability study (
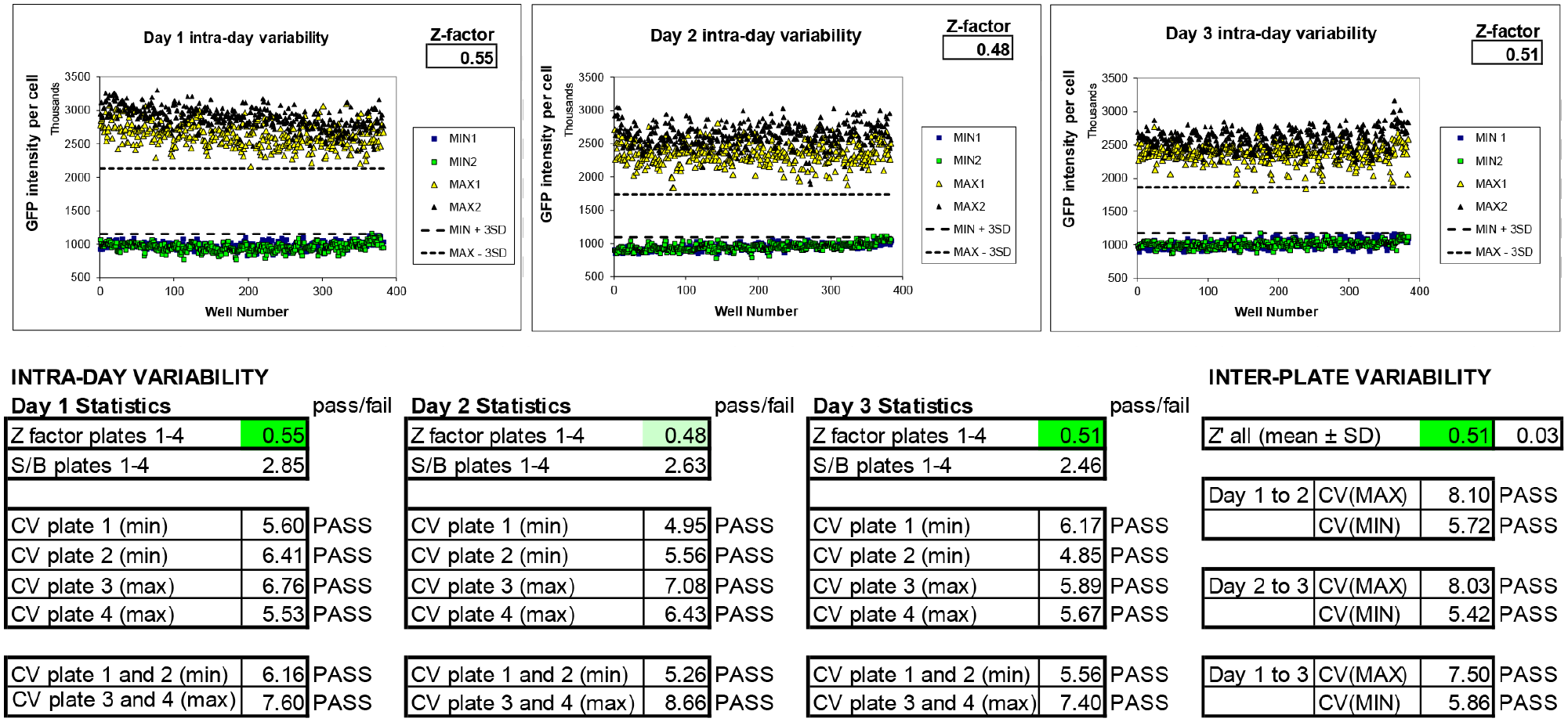
Three-day variability. Two full 384-well microplates were plated and treated on 3 consecutive days with 1% DMSO (MIN) or 200 nM luminespib using equipment to be used in high-throughput screening. SD, Standard deviation; CV, coefficient of variance; S/B ratio, signal-to-background ratio. Scatterplots illustrate day-to-day performance; tables show calculated intraplate and interplate variability statistics.
Discussion
Decreased stability of mutant TPI, resulting in lower steady-state levels of TPI protein, is the underlying cause for a rare but devastating childhood disease termed TPI Df. TPI Df causes hematological, muscular, and neurological symptoms that manifest themselves as anemia within months of birth; progressively worsening neuromuscular symptoms, typically before age 2; and neurodegenerative disease. TPI Df patients lead a miserable life and usually die within 5 years of diagnosis. Neither symptomatic nor curative treatments exist.
The structural basis for decreased protein stability is not known; however, a recent genetic screen for genes that regulate TPI stability has suggested that lower stability of mutant TPI is governed by a variety of proteins, including known PQC regulators, but also cotranslational modulators and a variety of proteins with unknown functions. 11 Therefore, there is no basis for developing targeted therapies. Thus, we reasoned that a phenotypic assay for TPI expression would be most appropriate to discover novel TPI Df therapeutics because such an assay would capture all mechanisms that lead to stabilization of mutant TPI.
To this end, we developed an operationally straightforward cellular assay that quantifies mutant TPI protein. We created a stable reporter cell line expressing a GFP-tagged mutant TPI fusion protein using a site-directed attB/attP insertion method (Thermo Fisher Flip-In System). The system allows for generation of stable reporter lines expressing mutant proteins while preserving endogenous protein expression, which is required for essential proteins such as TPI to keep cells alive. The stable line was characterized by Western blot and immunofluoresence, and the cells are viable and healthy. The cells allow levels of mutant TPI to be directly measured by quantitative fluorescence microscopy in living or fixed cells.
We implemented the HEK293 cell line assay for HTS. Assay development documented that the line responded to PQC inhibitors with increased levels of mutant TPI. Luminespib, an Hsp90 inhibitor, was chosen as a positive control. Counterassays were developed to eliminate toxicity and morphology artifacts, and preliminary HTS performance parameters in a miniaturized format suggested the assay could be implemented for HTS. A pilot screen of the NIH Clinical Collection identified several classes of agents that increased expression of mutant TPI, two of which were confirmed in fibroblasts from a homozyzgous TPIE105D TPI-Df patient. In particular, resveratrol and itavastatin had properties that might make them candidates for repurposing. Resveratrol as a nutritional supplement would represent an almost immediately applicable treatment for current TPI Df patients. A review of the clinical literature revealed that resveratrol had beneficial effects on skeletal muscle, 24 possibly by attenuating oxidative stress in myoblasts. 25 This could be highly relevant, because muscle wasting is a hallmark of TPI Df and oxidative stress has been observed in the Drosophila model of TPI Df. 26 Statins have been associated with modulation of heat shock proteins, 27 although the clinical picture is heterogeneous. In the right context, however, it is conceivable that statins could mediate protein stability through heat shock protein modulation. Importantly, while the doses of statins that are effective in cardiovascular disease are usually far lower than those observed in the laboratory for other biological activities, itavastatin has been shown to reach peak plasma levels in humans that correlate with concentrations needed to increase TPIE105D-GFP in HEK cells (0.55 µM). 28
How much of an increase in TPI is needed to have a therapeutic benefit or prevent TPI Df altogether? This is not known, and there is not a great deal of evidence that directly addresses this important question. There are limited patient data from asymptomatic controls, but a published case included a heterozygote mother with a reduced TPI of 584 UI/g hemoglobin (Hb; ~25–30% of the adult reference range of 1407–2133) who was completely asymptomatic. 5 In contrast, her child had TPI levels of 289 UI/g Hb (~14–20% of the adult reference range) with severe early-onset TPI Df. 5 These data suggest that fully rescuing TPI to wild-type levels is not necessary to have an extremely significant therapeutic benefit and that modest increases would be of therapeutic value even if they did not fully prevent all disease symptoms. Future studies will be needed to verify whether these compounds work similarly in compound-heterozygous TPI Df patient cells and to test efficacy in vivo.
Based on these results, we performed a rigorous 3-day variability assessment that documented HTS readiness, and we are now positioned to conduct a large-scale screen with the HEK TPIE105D-GFP cell line. Positives that emerge from this effort can be tested in a variety of patient fibroblasts that represent the whole spectrum of disease and in a well-validated in vivo Drosophila model.5,7 These secondary assays will include measurements of mutant TPI half-life by pulse-chase experiments and activity using an enzymatic assay. 17 Final validation will occur in a mouse model of TPI-Df that is currently being developed and, while not having been fully validated yet, shows hallmarks of the human disease (data not shown). Collectively, our data document that the HEK TPIE105D-GFP assay meets HTS performance criteria and can identify low-toxicity, bona fide TPI inducers and stabilizers with different mechanisms of action. The critical path for large-scale HTS will encompass complementary orthogonal assays in TPI-Df patient cells, biochemical characterization of protein stability and activity, and in vivo testing in Drosophila and in a mouse TPI Df animal model that we are currently developing. We are hopeful that this effort will result in the first therapeutics for this devastating childhood disease.
Supplemental Material
sj-pdf-1-jbx-10.1177_24725552211018198 – Supplemental material for A High-Content Screening Assay for Small Molecules That Stabilize Mutant Triose Phosphate Isomerase (TPI) as Treatments for TPI Deficiency
Supplemental material, sj-pdf-1-jbx-10.1177_24725552211018198 for A High-Content Screening Assay for Small Molecules That Stabilize Mutant Triose Phosphate Isomerase (TPI) as Treatments for TPI Deficiency by Andreas Vogt, Samantha L. Eicher, Tracey D. Myers, Stacy L. Hrizo, Laura L. Vollmer, E. Michael Meyer and Michael J. Palladino in SLAS Discovery
Footnotes
Acknowledgements
We thank Drs. Enns and Ruzhnikov for assistance with obtaining the FB104 patient cell line. We also acknowledge the brave patient and generous parents who donated FB104 cells for biomedical research.
Supplemental material is available online with this article.
Abbreviations
GFP, Green fluorescent protein; Hb, hemoglobin; HCS, high-content screening; HTS, high-throughput screening; PI, propidium iodide; PQC, protein quality control; SD, standard deviation; TPI, triose phosphate isomerase
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The University of Pittsburgh has filed a provisional patent application on which Drs. Andreas Vogt and Michael Palladino are listed as inventors. No personal financial benefit has been realized, and there are no pending plans to commercialize that would create a competing financial interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project used the UPMC Hillman Cancer Center Cytometry Core facility that is supported in part by award P30 CA047904 and used shared instrumentation that was acquired with National Institutes of Health grant S10 OD028450. We are grateful to the National Institutes of Health for funding this research project (R21 AG059385, R01 GM103369, and R01HD105311) and the Borofka reseach donation for TPI Df research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.