
Abstract
Targeted protein degradation is an emerging new strategy for the modulation of intracellular protein levels with applications in chemical biology and drug discovery. One approach to enable this strategy is to redirect the ubiquitin–proteasome system to mark and degrade target proteins of interest (POIs) through the use of proteolysis targeting chimeras (PROTACs). Although great progress has been made in enabling PROTACs as a platform, there are still a limited number of E3 ligases that have been employed for PROTAC design. Herein we report a novel phenotypic screening approach for the identification of E3 ligase binders. The key concept underlying this approach is the high-throughput modification of screening compounds with a chloroalkane moiety to generate HaloPROTACs in situ, which were then evaluated for their ability to degrade a GFP-HaloTag fusion protein in a cellular context. As proof of concept, we demonstrated that we could generate and detect functional HaloPROTACs in situ, using a validated Von Hippel–Lindau (VHL) binder that successfully degraded the GFP-HaloTag fusion protein in living cells. We then used this method to prepare and screen a library of approximately 2000 prospective E3 ligase-recruiting molecules.
Introduction
Protein degradation has emerged as an important therapeutic strategy in which small molecules can recruit cellular protein degradation machinery to induce the degradation of selected proteins of interest (POIs). One important subset of degraders that are targeted to specific POIs are proteolysis targeting chimeras (PROTACs), which are bifunctional molecules composed of a ligand to the POI linked to a second ligand that binds an E3 ligase.1–6 Ternary complex formation between the target POI, the PROTAC, and the E3 ligase enables the POI to be covalently modified with polyubiquitin chains, which are then recognized by the proteasome.7–10 Many different POIs have been reported to be successfully degraded by PROTACs, and several PROTAC molecules have now entered clinical trials.11–13 Currently reported PROTACs recruit only a small proportion of the known E3 ligases encoded by the human genome.14–19 However, expanding the available portfolio of functional E3 ligase-recruiting ligands might provide additional versatility for the degradation of challenging targets, as well as potentially offering improvements in physicochemical properties and developability as compared with currently known E3 ligase chemical binders.20–22
One approach to expanding the chemical matter available for the recruitment of E3 ligases is to conduct high-throughput screening campaigns against specific individual E3 ligases using binding affinity methods such as encoded library technology.23,24 This type of screening has the advantage of being high throughput and well established.25–27 However, this approach also means that screened E3 ligases must generally be selected with limited knowledge about their substrate scope and tractability. Additionally, even before a functional PROTAC can be constructed from an E3 ligase screening hit, a medicinal chemistry effort is typically required to improve E3 ligase binding affinity and to prepare and test multiple putative PROTACs with various linkers and linker attachment configurations. In practice, this reductionist single-target screening approach for E3 ligases is costly, inefficient, and slow, and can realistically only be conducted on a small fraction of the total number of potential E3 ligases that might have utility in PROTAC design. 28
To address the inherent inefficiencies posed by serial screening of individual E3 ligases, we have developed and implemented an alternative strategy that combines a cell-based targeted protein degradation phenotypic screen with efficient 384-well high-throughput chemical synthesis, shown schematically in Figure 1 . We based our screening design strategy on the previously described HaloPROTAC method for the evaluation of Von Hippel–Lindau (VHL) E3 ligase binders in a cellular context. HaloPROTAC molecules contain a linker with a terminal chloroalkane group that can covalently bind to the HaloTag protein, 29 and we have previously demonstrated the degradation of a HaloTag-GFP (green fluorescent protein) fusion protein upon cell treatment with a VHL-based HaloPROTAC. 30 We reasoned that the HaloPROTAC assay could be developed into a high-throughput phenotypic screening format, where new ligands would be identified based on their ability to act as functional degraders when tethered to the HaloTag-GFP protein in a cell. This format should be agnostic regarding the underlying mechanism of POI degradation but would effectively multiplex a screening campaign of E3 ligases as well as other intracellular proteins and mechanisms relevant to targeted protein degradation. Any resulting hits will have already demonstrated functional target degradation in cells and would in principle be suitable for incorporation into heterobifunctional degraders.
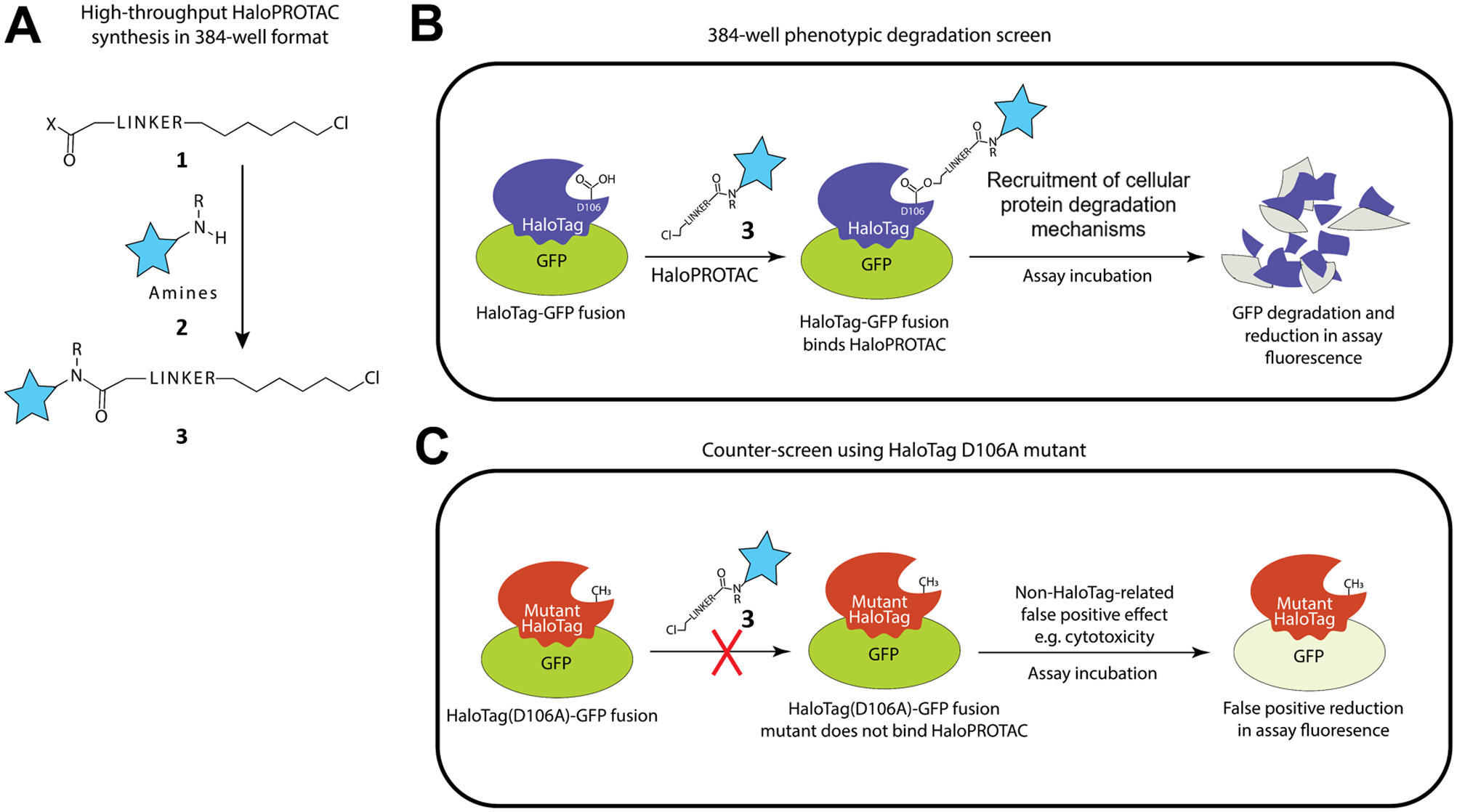
Schematic overview. (
To realize the goal of conducting a high-throughput phenotypic HaloPROTAC screen, the preparation of a suitable HaloPROTAC screening library must be addressed, as we predicted that this step might be rate-limiting. We hypothesized that it might be feasible to prepare a HaloPROTAC library
Since only fully elaborated HaloPROTAC molecules should be capable of degrading the HaloTag-GFP fusion protein, we further hypothesized that it would be possible to screen the HaloPROTAC library in situ, without purification of the individual library compounds. We also speculated that in view of the multiplexed nature of this assay design, which in principle screens all of the E3 ligases and degradation mechanisms present in the cells simultaneously, it might be possible to identify a functional degrader hit from a much smaller compound library than would otherwise be required in an equivalent nonmultiplexed single-target screening campaign. Toward this goal, we describe herein the optimization of a high-throughput amide coupling reaction in low-volume 384-well format, and the efficient synthesis of a HaloPROTAC screening set starting from a curated library of approximately 3000 reactive amines. The resulting HaloPROTAC screening set was used to conduct a phenotypic screening campaign to detect functional HaloTag-GFP degraders in HeLa cells ( Fig. 1B ). A false-positive counterscreen was also implemented using a cell line with a mutated version of the HaloTag protein that does not covalently bind chloroalkanes ( Fig. 1C ). Our work demonstrates a novel application of HaloTag technology for a phenotypic screening approach to detect small-molecule ligands for use in targeted protein degradation.
Materials and Methods
General High-Throughput Chemistry Considerations
Experiments conducted under an inert atmosphere were performed inside a MBraun glovebox with a constant N2-purge with oxygen typically <5 ppm. Stock solution reservoirs were polystyrene, 50 mL capacity (Corning Costar, New York, US, cat. 4870); 384-well plates were polypropylene and either Greiner v-bottom (cat. 781280) or Labcyte flat-bottom (cat. P-05525). Plates were covered with a polystyrene lid where indicated (Corning, cat. 3098). Pipetting procedures were completed using an electronic multichannel pipette from stock solution reservoirs (Thermo Fisher, 12 channels, 1–30 μL, cat. 4671030BT with Thermo Fisher ClipTip, cat. 94410103, Cheshire, UK).
HaloPROTAC Library Design and Synthesis
The HaloPROTAC screening library
(
HaloPROTAC library synthesis was conducted directly on 384-well reactor plates containing the selected amines in random order. The plates were prepared to contain 10 μL of 10 mM DMSO stock solution per well and were stored at −20 °C. The selected amine building blocks were delivered on nine plates (including 16 negative control DMSO wells and 16 wells containing the VHL ligand
(
For SS1, a stock solution of the chloroalkane NHS ester 2,5-dioxopyrrolidin-1-yl 18-chloro-3,6,9,12-tetraoxaoctadecanoate
An analysis of the reaction outcomes for each of the nine plates is shown in
Stable Cell Line Preparation
A HeLa cell line expressing enhanced GFP fused at the N-terminus with the previously described HaloTag protein was prepared. 30 Additionally, a second analogous HeLa cell line was prepared containing a D106A mutation in the HaloTag part of the fusion protein to enable counterscreening for false positives. A pBabeD-based retroviral vector system was used for cell line generation (modified version of Cell Biolabs pBabe plasmid, plasmids DU29230 and DU29215, respectively; MRC-PPU Reagents and Services, https://mrcppureagents.dundee.ac.uk). In the event, HEK293-FT cells at ~60% confluence in a 10 cm dish were transfected with 6 µg of the respective pBabeD constructs, 3.8 µg of pCMV-GAG/POL (Clontech, Saint-Germain-en-Laye, France), and 2.2 µg of pCM-VSVG (Clontech) using PEI (2 µg/mL). The medium was exchanged on the next day, and after a further 24 h, virus-containing medium was harvested and cleaned using a 0.45 μm filter. HeLa cells (University of Dundee MRC-PPU stock, obtained from the American Type Culture Collection, Manassas, VA) were then transduced with the virus-containing medium supplemented with 10 μg/mL polybrene for 24 h. Cells were grown for an additional 24 h in normal growth medium and selected for 3 days in 2 µg/mL puromycin-containing medium. Finally, single-cell clones were isolated using fluorescence-activated cell sorting (FACS) based on GFP fluorescence. The cell clones selected for screening and counterscreening both exhibited similar magnitude of fluorescence intensity, although the cells containing the HaloTag D106A mutation were marginally less bright. Stably transduced HeLa cells were subsequently cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM GlutaMAX, 50 U/mL penicillin, 50 µg/mL streptomycin, and 1 µg/mL puromycin, at 37 °C with 5% CO2.
Degradation Screening Assay
Samples of HaloPROTAC test compounds were transferred into black clear-base 384-well tissue culture assay plates (Greiner Bio-One, Stonehouse UK). An Echo acoustic liquid handler (Labcyte, High Wycombe, UK) was used for this procedure, with transfer volumes of 200 nL and 20 nL for 10 µM and 1 µM screening, respectively. For concentration−response curve format assays, typically a 250× test concentration dilution series of each compound was first prepared in 100% DMSO, with up to 11 concentration points differing in half-log unit increments. This series was next further diluted 25× in Fluorobrite DMEM (Thermo Fisher), and 5 µL was then transferred to the assay plate. Cultured cells were harvested at 80%−90% confluency using TrypLE enzyme; resuspended in Fluorobrite DMEM supplemented with 10% heat-inactivated FBS, 2 mM GlutaMAX, 50 U/mL penicillin, and 50 µg/mL streptomycin; and seeded into assay plates containing test compounds at a density of 10,000 cells per well. The final assay volume was 50 µL per well. DMSO concentrations in the assays were 0.4% or less. Assay plates were then incubated at 37 °C with 5% CO2 for 2 days. Hoechst 33342 dye (0.5 µg/mL) was added 30 min before the end of the incubation, and the resulting GFP fluorescence in each well was then measured with a PHERAstar FSX microplate reader (BMG Labtech, Aylesbury, UK). Where stated (
Hit Selection
To select compounds for follow-up from the HaloPROTAC screening library, simple numerical filters were applied to the average (n = 3) 10 µM test concentration target degradation percentage values, and hits were defined as compounds causing greater than 50% degradation of the HaloTag-GFP fusion protein but less than 50% degradation of the HaloTag (D106A)-GFP fusion protein. To make the selection more inclusive, compounds causing greater than 40% degradation of HaloTag-GFP but less than 30% degradation of HaloTag (D106A)-GFP were also included in the hit definition. A small percentage of outlier control wells were excluded before making the percentage degradation calculations.
Results
Development of a HaloPROTAC–HaloTag-GFP Degradation 384-Well Screen and False-Positive Counterscreen
The HaloPROTAC screen and false-positive counterscreen designs are shown schematically in
To determine the suitability of different approaches to deploy for our screening strategy in 384-well plate format, we investigated a live cell whole-well fluorescence endpoint assay, an imaging-based endpoint assay, and a flow cytometry assay. We found that each of these methods provided approximately equivalent response curves, good reproducibility between technical replicates, and high Z′ values (
High-Throughput Chemistry Optimization and Generation of a HaloPROTAC Compound Screening Library
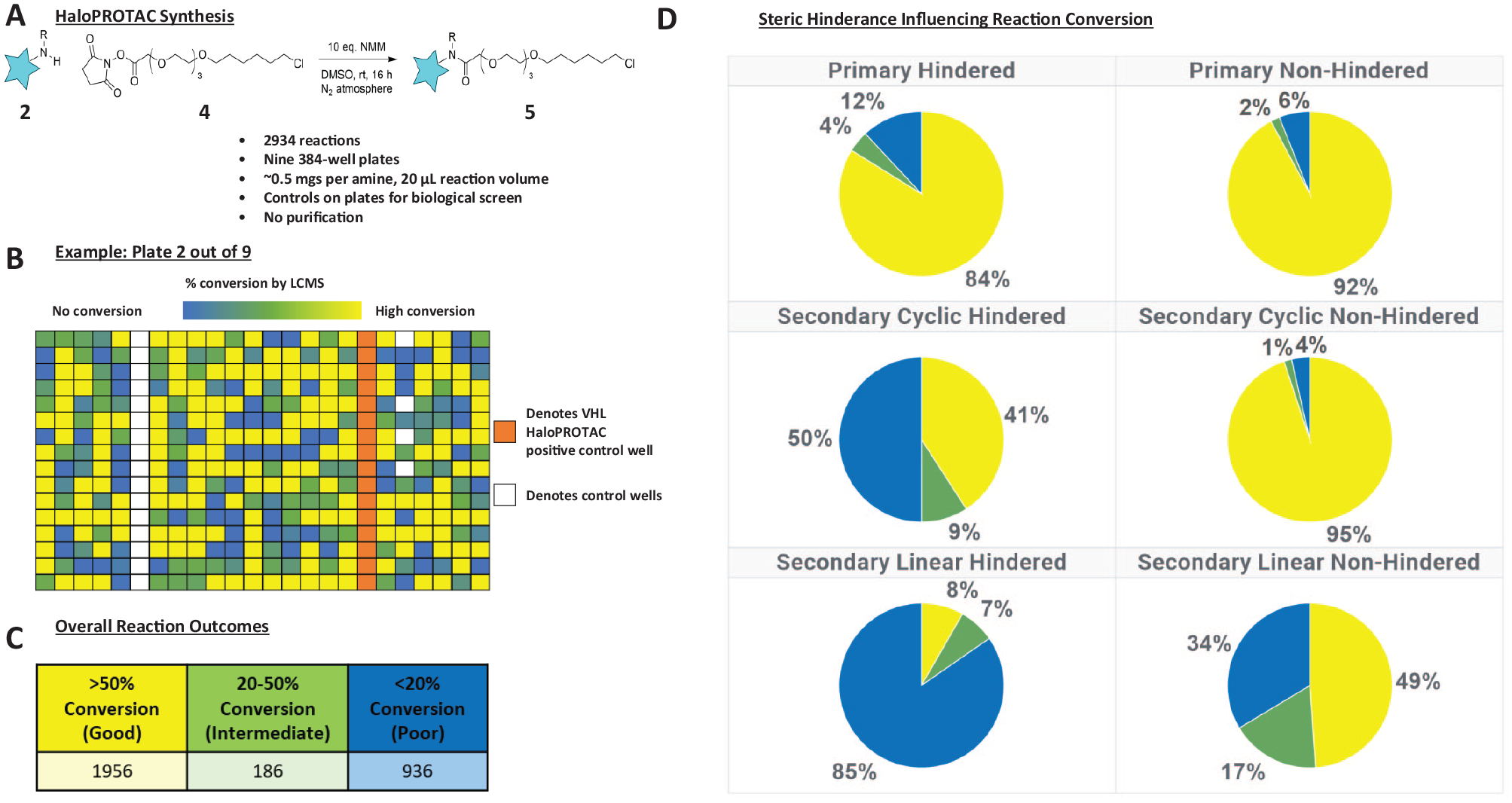
As described above, we designed a simple, one-stage coupling protocol between a collection of diverse amine building blocks
Extensive experimentation demonstrated that robust and reproducible coupling was obtained using DMSO as a solvent with a 1:1 molar ratio of amine
Following plate-based chemistry optimization, we next prepared the screening library of HaloPROTAC test molecules (
Fig. 2B
). We selected a total of 2934 amines for use as building blocks for HaloPROTAC library synthesis, and employing our optimized reaction conditions, we observed >50% conversion to product for 64% of amines, 20%−50% conversion for 6% of amines, and <20% conversion for 30% of amines (
Fig. 2C
). Factors influencing reaction conversion appear to be both physicochemical (
Screening of HaloPROTAC Compound Library for HaloTag-GFP Degrading Compounds and False-Positive Counterscreening
The crude HaloPROTAC compounds at 10 µM concentration were tested for their ability to induce a reduction in GFP fluorescence in the HaloTag-GFP cells and in the HaloTag (D106A)-GFP counterscreen cells (n = 3, tested on different days). Screening was also performed at 1 µM concentration (n = 1) using the HaloTag-GFP cells in an attempt to detect more potent degraders. The hit activity histograms are shown in
Figure 4
. The positive control VHL-HaloPROTAC
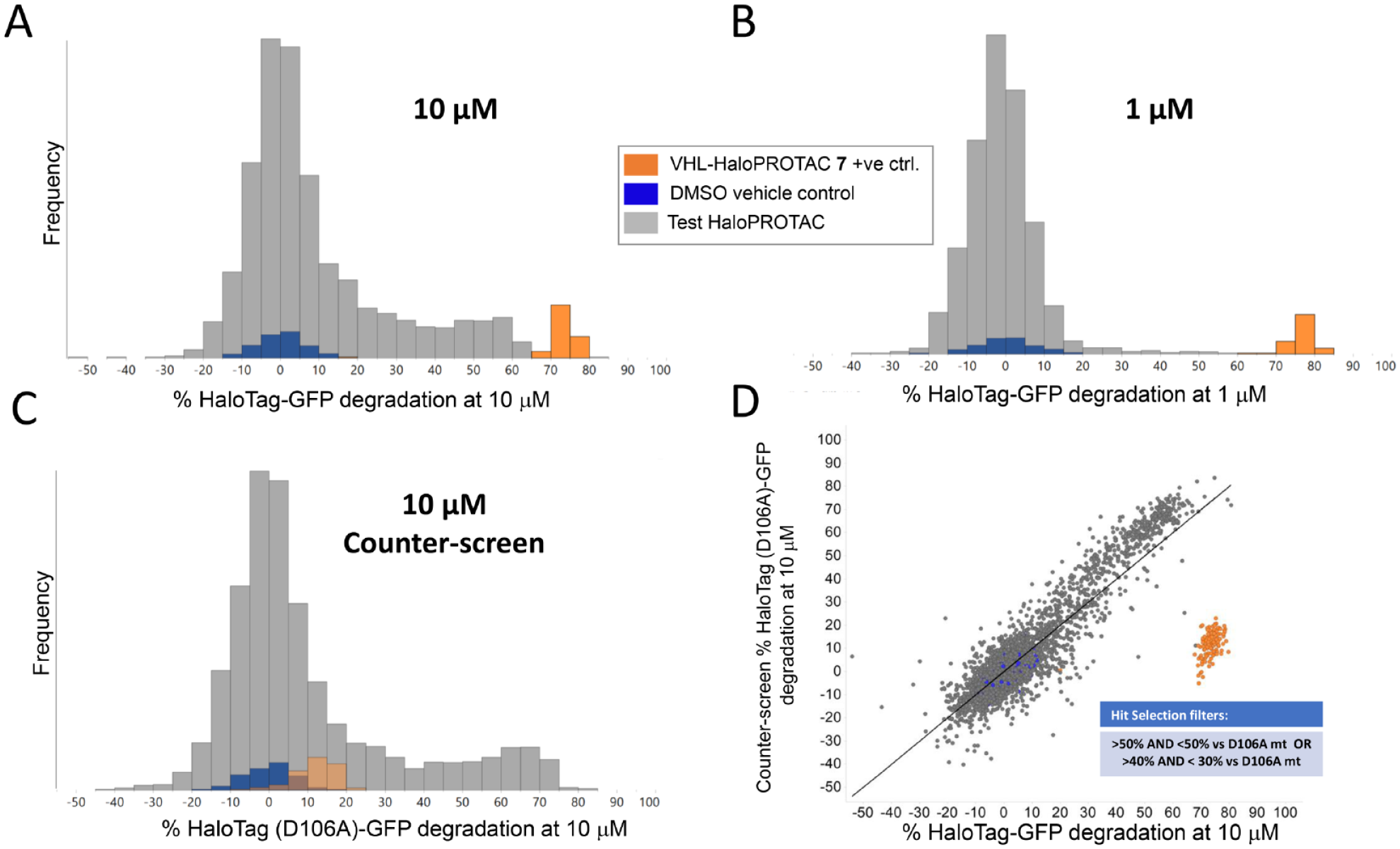
Results of screening the HaloPROTAC set. (
To identify the potential functional degraders from the many apparent false-positive hits, we compared the average effects of 10 µM HaloPROTAC compounds in the screening and counterscreening cells (mean, n = 3) (
Fig. 4D
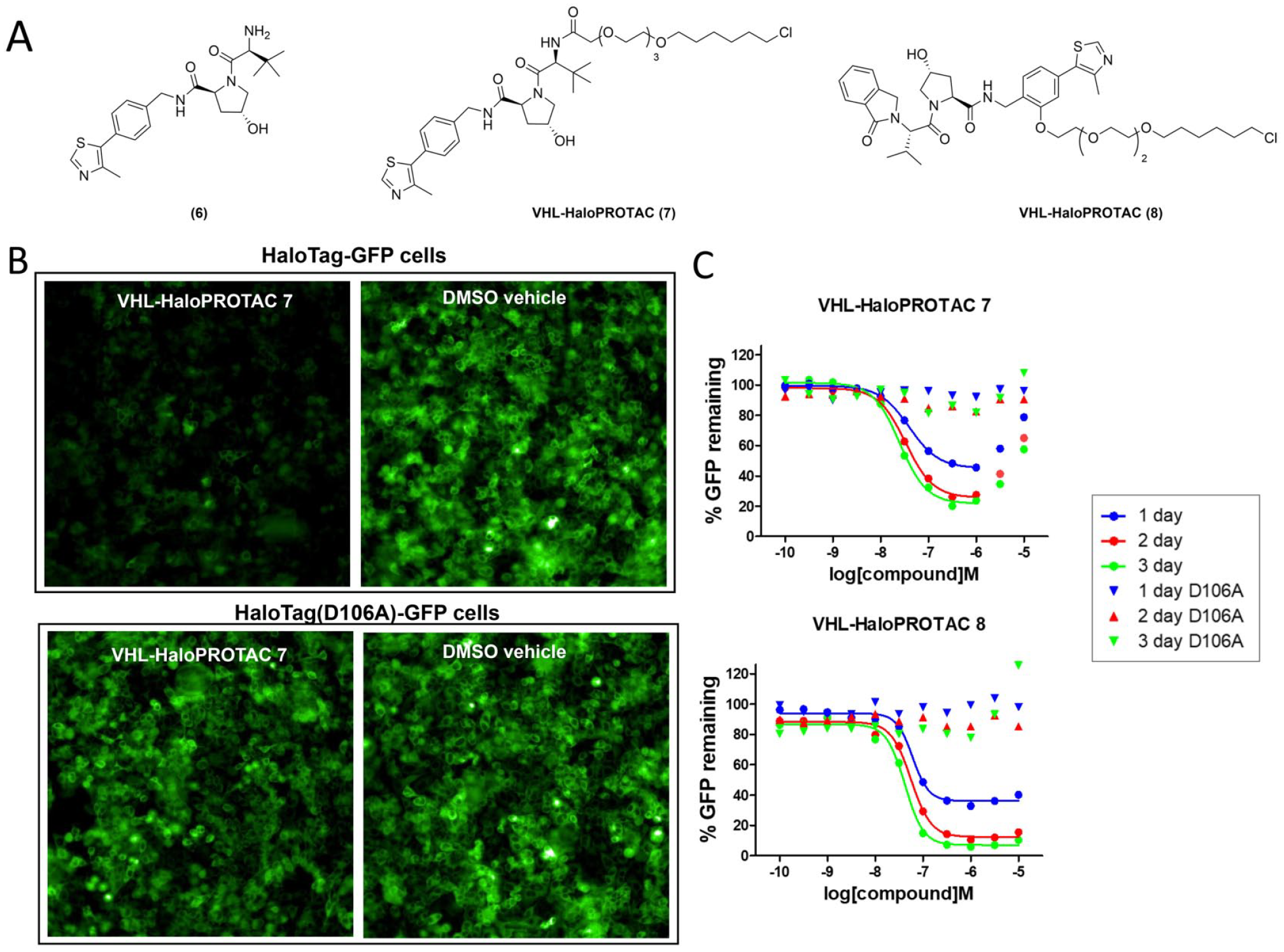
) and applied simple thresholds based on the percentage decrease in the GFP fluorescence signal, as described in Materials and Methods. This resulted in nine HaloPROTAC compounds being identified as potential true degrader hits. These nine compounds were then individually re-prepared from stock solutions in the same manner as described above and retested in a concentration−response format using both the screening and counterscreening cell lines. We observed that two of these nine compounds (HaloPROTAC
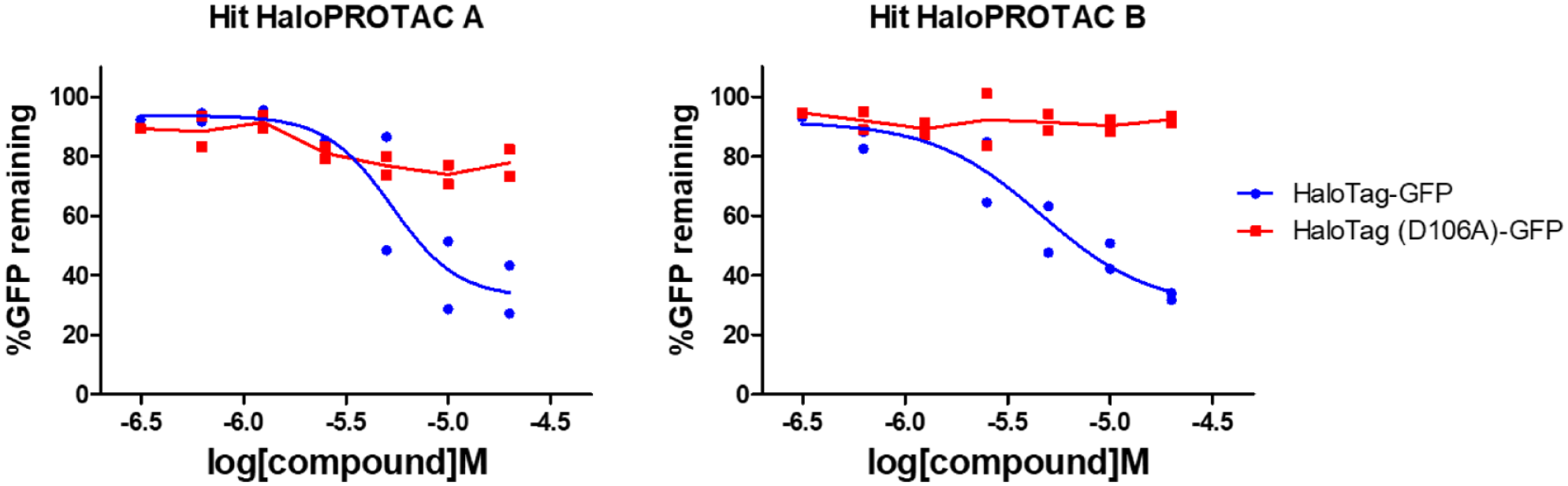
Hit confirmation in concentration−response format showing reproducible differential HaloTag-GFP versus counterscreen HaloTag (D106A)-GFP fluorescence reduction profiles for the two out of nine hits that were confirmed.
Discussion
Targeted protein degradation is a field with significant potential for industrial and academic research applications, and the first examples of PROTACs entering clinical trials have recently been described.5,8,12,33−35 Currently reported PROTAC molecules employ a limited catalog of E3 ligases to induce ubiquitin transfer and subsequent target degradation, and increasing the available E3 ligase repertoire for PROTAC design represents a clear opportunity to expand the scope of PROTAC utility for medicine discovery. 26 However, this goal is challenging to realize due to difficulties typically associated with the expression, purification, and screening of biologically relevant E3 ligase complexes. Consequently, limited examples of successful high- or medium-throughput screening approaches for E3 ligases have been reported to date.36–39
In contrast to a traditional affinity binding or biochemical E3 ligase screening campaign, we envisaged that a phenotypic approach might provide a straightforward solution for the identification of molecules capable of effecting target degradation in cells. 40 Such an approach would have the additional advantage of multiplexed target screening, since the entire cellular repertoire of E3 ligases and other protein degradation machinery would be sampled simultaneously, and we reasoned that a plate-based HaloPROTAC array could provide a good opportunity to implement this approach. 41 HaloPROTACs employ an E3 ligase binder with a linker bearing a primary chloroalkane, and they have been previously demonstrated to covalently bind to HaloTag-POI fusion proteins, resulting in ubiquitin transfer and subsequent degradation of the HaloTag-POI fusion. To the best of our knowledge, the HaloPROTAC concept has not been previously used for screening purposes.
To enable our screening strategy, we generated a clonal HeLa cell line expressing the HaloTag-GFP fusion protein, and we tested the performance of these cells with the known VHL HaloPROTACs
Chimeric molecules such as PROTACs are not widely represented in typical screening libraries, and the ability to access a library of HaloPROTAC degraders is envisaged to be useful for drug discovery and chemical biology purposes. To this end, we developed a high-throughput chemistry protocol in 384-well plate format to combine a diverse selection of amine building blocks with a single linker bearing a primary chloroalkane HaloTag recognition element. During library construction, we observed conversion rates to product HaloPROTACs of >50% for approximately 64% of the starting amine building block library, which was deemed to be fit for purpose for the evaluation of an initial degradation screen. We also identified steric and physicochemical parameters such as basicity, solubility, size, and flexibility of the amines to be important for controlling the reaction rate and ultimate reaction success, which are envisioned to be useful for future library design and preparation.
Screening of the HaloPROTAC library in the HaloTag-GFP HeLa cells as well as counterscreening in the HaloTag (D106A)-GFP cells revealed that many compounds significantly reduced the fluorescence signal in both cell lines. Because the HaloPROTACs are unable to bind the HaloTag D106A mutant, 42 we speculate that compounds reducing fluorescence in both cell lines are acting primarily through cytotoxicity or other nuisance mechanisms affecting the proliferation and viability of the cells during the 48 h assay incubation, rather than through a true on-target degradation mechanism. This effect demonstrates the well-established need to accurately discern false positives in phenotypic screening campaigns.
We identified nine HaloPROTAC compounds as potential true HaloTag-GFP degrader hits worthy of progression to confirmatory concentration–response testing. Out of these nine compounds, only HaloPROTACs
The identification of HaloPROTAC
Since effective target degradation is dependent on many factors, we speculate that this screening approach can be further improved not only through the addition of more amine components but also by employing additional linker building blocks with different lengths and compositions to increase the odds of effective ternary complex formation leading to degradation. Furthermore, screening HaloPROTAC libraries in different cell lines may also increase the odds of detecting functional degraders, since different cell lines can have differential expression of degradation machinery component proteins. Future iterations of this screening design might also benefit from other established counterscreening approaches, such as co-expression of the HaloTag-GFP with a second fluorescent or bioluminescent control protein.6,44–47
Despite our inability to identify novel functional HaloPROTACs with on-target HaloTag-GFP degradation in this study, we believe that our phenotypic screening approach is fit for purpose for the identification of new chemical matter capable of recruiting E3 ligases or other elements of the cellular protein degradation machinery in an unbiased way and will be a valuable new method for the discovery of target protein degraders.
Supplemental Material
sj-pdf-1-jbx-10.1177_24725552211017517 – Supplemental material for A Phenotypic Approach for the Identification of New Molecules for Targeted Protein Degradation Applications
Supplemental material, sj-pdf-1-jbx-10.1177_24725552211017517 for A Phenotypic Approach for the Identification of New Molecules for Targeted Protein Degradation Applications by Peter Stacey, Hannah Lithgow, Xiao Lewell, Agnieszka Konopacka, Stephen Besley, Georgina Green, Ryan Whatling, Robert Law, Sascha Röth, Gopal Sapkota, Ian E. D. Smith, Glenn A. Burley, John Harling, Andrew B. Benowitz, Markus A. Queisser and Marcel Muelbaier in SLAS Discovery
Footnotes
Acknowledgements
The authors are grateful to Richard Kasprowicz for help with the analysis of fluorescence images and to Blandine McKay and David Battersby for helpful discussions.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: PS, HL, XL, AK, SB, GG, RL, IEDS, JH, ABB, MAQ, and MM are current or former employees and/or shareholders of GlaxoSmithKline, and their research and authorship of this article were completed within the scope of their employment. RW is a former industrial placement student at GlaxoSmithKline. The other authors declare no potential conflicts of interest with respect to the research, authorship, or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by GlaxoSmithKline PLC, GSK/University of Strathclyde Centre for Doctoral Training in Synthetic and Medicinal Chemistry, EPSRC via Prosperity Partnership EP/S035990/1, and UK MRC grant number MC_UU_00018/6.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.