
Abstract
The serum- and glucocorticoid-regulated kinase (SGK) family consists of three isoforms (SGK1, SGK2, and SGK3) that have been implicated in the regulation of tumor growth, metastasis, autophagy, and epithelial ion transport. SGK1 and SGK3 play essential roles in protein kinase B (AKT or PKB)-independent phosphoinositide 3-kinases (PI3K)-mediated tumorigenesis, as evidenced by the significantly elevated expression levels of SGK1 and SGK3 in many cancers, including prostate cancer, colorectal carcinoma, estrogen-dependent breast cancer, and glioblastoma. Therefore, SGK is a potential target for anticancer therapy. A small kinase-focused library comprising 160 compounds was screened against SGK1 using a fluorescence polarization–based kinase assay that yielded a Z’-factor of 0.82. Among the 39 compounds obtained as initial hits in a primary screen, 12 compounds contained the thiazolidine-2,4-dione scaffold. The inhibitory mechanisms of the most potent hit, KMU010402, were further investigated using kinetic analyses, followed by determination of the inhibition constants for SGK1, SGK2, and SGK3. Molecular modeling was used to propose a potential binding mode of KMU010402 to SGK1.
Keywords
Introduction
The phosphoinositide 3-kinases (PI3Ks)/protein kinase B (AKT or PKB)/mammalian target of rapamycin (mTOR) pathway is one of the most activated pathways in human cancer.1,2 Although inhibition of the PI3K pathway has been proven to be highly effective in halting tumor growth, acquired phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) amplification, activating mutations in phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta (PIK3CB), as well as AKT-inhibitor-resistant breast cancer cell lines have been found to severely limit the anticancer efficacy of PI3K/AKT/mTOR inhibitors.3–6 Thus, the therapeutic potential of targeting other kinases within this pathway for the treatment of cancer has recently gained significant attention from both academic research and the pharmaceutical industry. In particular, serum- and glucocorticoid-regulated kinases (SGKs) have recently emerged as important AKT-independent mediators of cell survival and tumorigenesis in the PI3K pathway.
SGKs are involved in the regulation of cellular signaling pathways related to cell proliferation, survival, and stress, as well as the transportation and regulation of ions such as Na+, K+, and Ca2+. 7 These are members of the AGC kinase family of serine/threonine (Ser/Thr) kinases, which consist of three highly homologous isoforms: SGK1, SGK2, and SGK3. SGKs, which share 80% sequence identity in the kinase domain, are regulated via PI3K and phosphoinositide-dependent kinase-1 (PDK1)-mediated phosphorylation. 8 All three SGKs consist of an N-terminal variable domain, a Ser/Thr kinase domain, and a C-terminal regulatory domain.8,9 SGK1 and SGK2, unlike AKT and PDK1, lack an N-terminal pleckstrin homology domain that interacts with phosphatidylinositol (3,4,5)-trisphosphate, while SGK3 possesses a Phox domain, which is involved in binding 3′-phosphorylated inositide phospholipids. 10
The SGK family shares a regulatory pathway with AKT, since both kinases exhibit some overlapping substrate specificity and directly phosphorylate p27 at T157, which leads to impaired nuclear import and cytoplasmic retention.11,12 Recent studies, however, have shown that human breast tumors, as well as many PIK3CA-mutant cancer cell lines, are PI3K/PDK1-dependent but independent of AKT activation.13,14 It turns out that there are subtle differences in the signaling pathways; mTOR complex-1 (mTORC1) phosphorylates SGK1 at the hydrophobic motif S422, while AKT activation is mTORC2-dependent. 12 Subsequently, PDK1 phosphorylates SGK1 at the activation loop, T256, leading to tumor cell viability. A recent study revealed that SGK3 activation also occurs in a PI3K/PDK1-dependent manner in PIK3CA-mutant cancer cells. 13 Expression levels of SGK1 are significantly high in lung adenocarcinoma cells and metastatic prostate cancer cells.15,16 SGK3 expression positively correlates with human estrogen-dependent breast cancer and androgen-mediated prostate cancer.17,18 Breast tumor cells exhibiting high levels of SGK expression and activation are more resistant to PI3K and AKT inhibitors.6,19 Taken together, these studies demonstrate that SGK1 and SGK3 may be attractive small-molecule targets in tumor types that are resistant to PI3K/AKT inhibitors.
To discover inhibitors of SGK, we developed and optimized a fluorescence polarization (FP)-based immobilized metal ion affinity-partitioning (IMAP) kinase assay. The 5-carboxyfluorescein (5-FAM)-labeled peptide derived from glycogen synthase kinase-3 (GSK3) functions as a substrate for SGK, resulting in the phosphorylation of serine residue in the presence of adenosine triphosphate (ATP). Binding of nanoparticles derivatized with trivalent metal ions to the phosphorylated substrate increases the polarized signal owing to the slow tumbling of the bound complex. We screened a small kinase-focused library against the SGK1 isoform in a 384-well plate format and identified hit compounds that were further characterized in mode-of-inhibition studies.
Materials and Methods
Materials
For the assay, black flat-bottom 384-well microplates were purchased from PerkinElmer (Waltham, MA), 5-FAM-labeled-Crosstide [5-FAM-GRPRTSSFAEG] was purchased from AnaSpec (Fremont, CA), and 5-FAM-phospho-Crosstide [5-FAM-GRPRTS-pS-FAEG] and an IMAP Assay Kit were purchased from Molecular Devices (San Jose, CA). Recombinant human SGK1, SGK2, and SGK3 were obtained from SignalChem (Richmond, Canada). Staurosporine and GSK650394 were purchased from Acros Organics (Morris Plains, NJ) and SelleckChem (Houston, TX), respectively. All other reagents were supplied by Sigma-Aldrich (St. Louis, MO).
Fluorescence Polarization Assay Method
SGK1, 2, and 3 reactions were carried out in a buffer containing 25 mM Tris (pH 7.5), 5 mM MgCl2, 0.5 mM ethylene glycol tetraacetic acid (EGTA), 0.5 mM Na3VO4, 5 mM β-glycerophosphate, 2.5 mM DL-dithiothreitol (DTT), 0.01% Triton X-100, 100 nM 5-FAM-labeled-Crosstide, and 2.0 nM SGK1 (or 2.0 nM SGK2 or 0.50 nM SGK3). The final concentrations of ATP used in the kinase reactions were 67 µM, 105 µM, and 42 µM for SGK1, SGK2, and SGK3, respectively. The reaction mixture was prepared in a total volume of 10.0 µL by adding 5.0 µL of a mixture of the SGK enzyme and ATP and 5.0 µL of 5-FAM-labeled-Crosstide peptide into a black flat-bottom 384-well microplate. IMAP binding reagents (30.0 µL) were added to each reaction mixture to stop the kinase reaction, followed by further incubation for 2 h. The FP was measured at λex = 495 nm and λem = 530 nm on an Infinity F200 plate reader (Tecan, Männedorf, Switzerland). The percentage of inhibition was calculated relative to the signal with 100% inhibition as minimum control and the signal with no inhibition as maximum control. Concentration–response data were fitted to a four-parameter logistic model using GraphPad Prism 6 software (San Diego, CA).
Calibration Curve for Conversion of Millipolarization (mP) Value to Molar Concentration
To convert the FP value in mP to molar concentration of phosphopeptide generated in the kinase reaction, a phosphopeptide calibration curve was prepared according to a previously published procedure.20,21 The total concentration of combined 5-FAM-labeled-Crosstide/5-FAM-phospho-Crosstide was maintained at 100 nM, while the ratios for unphosphorylated and phosphorylated forms of each peptide were varied from 0% to 100% phosphorylation. After 2 h of incubation of each mixture with IMAP binding reagents, the polarization values were measured on an Infinity F200 plate reader.
Determination of ATP Apparent Michaelis Constant (Km, app) Values
To determine the apparent Km (Michaelis constant) values of ATP for SGK1, SGK2, and SGK3, the assays were conducted in a buffer described in the Fluorescence Polarization Assay Method section, and the initial rates were determined by monitoring the phosphorylated peptide in unit time. The apparent Km values were determined from a fit of the data to Equation (1):
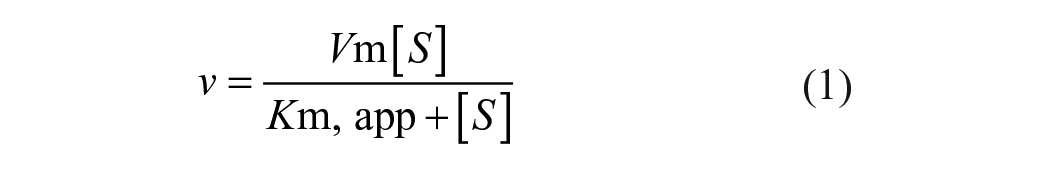
where ν is the initial velocity, Vm is the maximum velocity, Km, app is the apparent Michaelis constant, and [S] is the substrate concentration.
Kinase-Focused Library Screening
Compounds from a kinase-focused library of 160 compounds (KMU010 series) were screened at 10 µM against SGK1 kinase. SGK1 kinase was pre-incubated with compound for 30 min, followed by addition of a mixture of ATP/5-FAM-labeled Crosstide peptide to initiate the kinase reaction. After 1 h of reaction at room temperature, IMAP binding reagents were added to the reaction mixture and further incubated for 2 h. The FP was measured, and the percentage of inhibition was calculated relative to the signal with 100% inhibition as minimum control and the signal with no inhibition as maximum control.
The Z’-factor, which is a statistical quality indicator for the assay in high-throughput screening, was calculated using Equation (2):
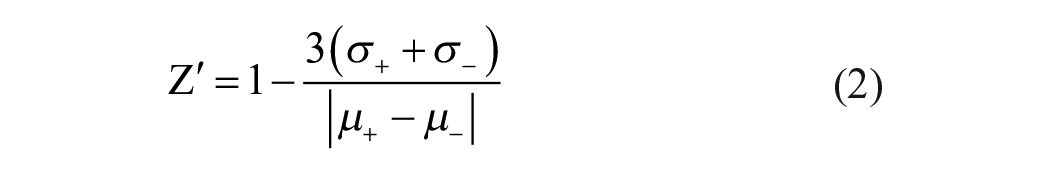
where σ+ is the standard deviation of the positive control, σ− is the standard deviation of the negative control, µ+ is the mean of the positive control, and µ− is the mean of the negative control.
Dose–response inhibition data were plotted as percentage inhibition normalized to DMSO controls (positive controls) and no-enzyme controls (negative controls) with an applied four-parameter logistic [Equation (3)], followed by calculation of the half-maximum inhibitory concentration (IC50) value for each compound.
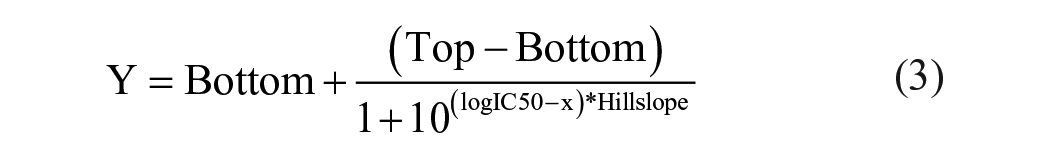
Protocols for the primary screening assay and follow-up IC50 determinations are described in
Mechanism-of-Inhibition Studies
For the mechanism-of-inhibition studies, compounds were assayed at varying concentrations of KMU010402 for SGK1, SGK2, or SGK3, with various ATP concentrations. The initial rates of the time-course data were calculated for each inhibitor concentration at each ATP concentration. The initial rates versus ATP concentrations at each inhibitor concentration were plotted by fitting data to the competitive inhibition model. All data were analyzed using GraphPad Prism 6 software.
The inhibition data were globally fitted to all four kinetic models, including competitive [Equation (4)], noncompetitive [Equation (5)], uncompetitive [Equation (6)], and mixed noncompetitive inhibition models [Equation (7)]:
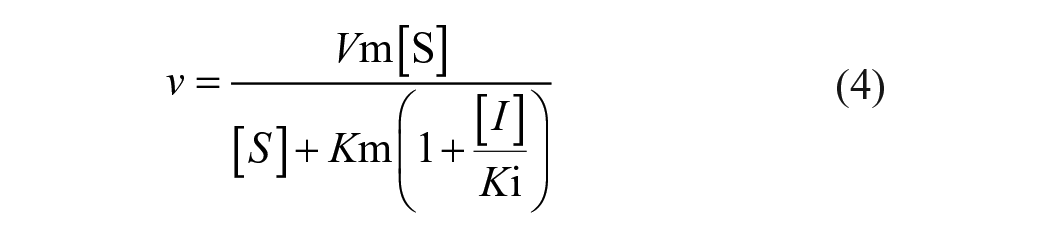
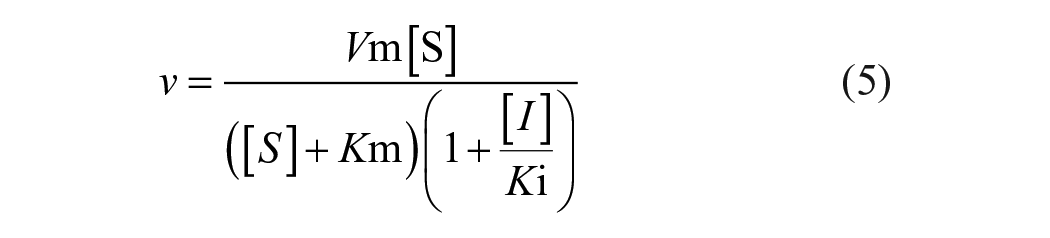
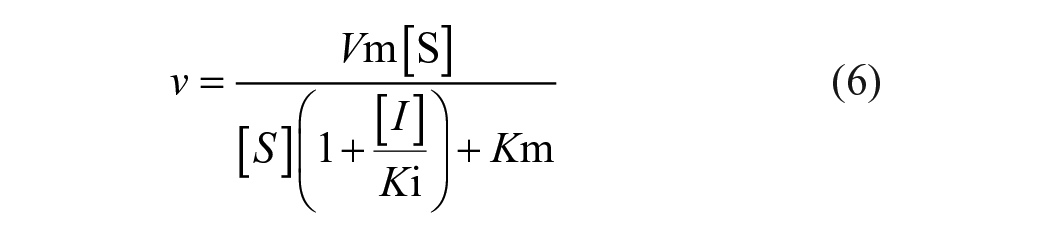
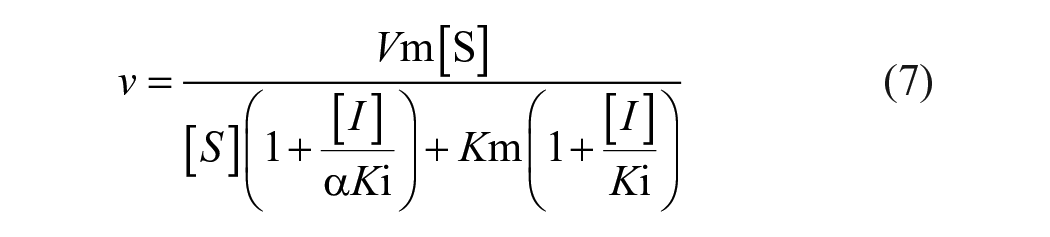
where ν is the initial velocity, Vm is the maximum velocity, Km is the Michaelis constant, Ki is the inhibition constant, [I] is the inhibitor concentration, [S] is the substrate concentration, and α is a thermodynamic cooperativity term.
Molecular Docking
A docking method was used by using AutoDock Vina (vina.scripps.edu). 22 The input file needed for AutoDock Vina was prepared using AutoDock Tools. 23 A docking grid, defined as a size of 24 Å × 24 Å × 24 Å, was used for x, y, and z.
Results and Discussion
Fluorescence Polarization Assay Development
FP-based IMAP technology has been successfully applied to the development of several kinase assays.20,24,25 Since the IMAP assay design is based on charge interactions between trivalent metal-containing nanoparticles and negatively charged phosphate groups of the peptide product, it can be applied universally to both tyrosine and Ser/Thr kinase assays. Crosstide is an 11-mer peptide derived from GSK3, in which a serine residue is phosphorylated by several members of the AGC subfamily of protein kinases, including SGK, AKT, and ribosomal S6 kinase (RSK). 26
The first step was to determine the optimal concentration of Crosstide peptide substrate for use in the SGK IMAP kinase assay. Various concentrations of 5-FAM-labeled-Crosstide (substrate) and 5-FAM-phospho-Crosstide (product) were separately captured using IMAP beads. The maximum mP difference (the mP value of 5-FAM phosphorylated product with the beads minus the mP value of 5-FAM substrate with the beads) was achieved at 100 nM (data not shown). Based on this result, a calibration curve was prepared to determine the amount of phosphorylated peptide product produced during a kinase reaction. Mixtures of both 5-FAM-labeled-Crosstide and 5-FAM-phospho-Crosstide at a total concentration of 100 nM were incubated with a fixed amount of IMAP beads ( Fig. 1 ). The calibration curve shows a linear increase in mP value with an increase in the percentage of phosphopeptide (R2 value of 0.9932).
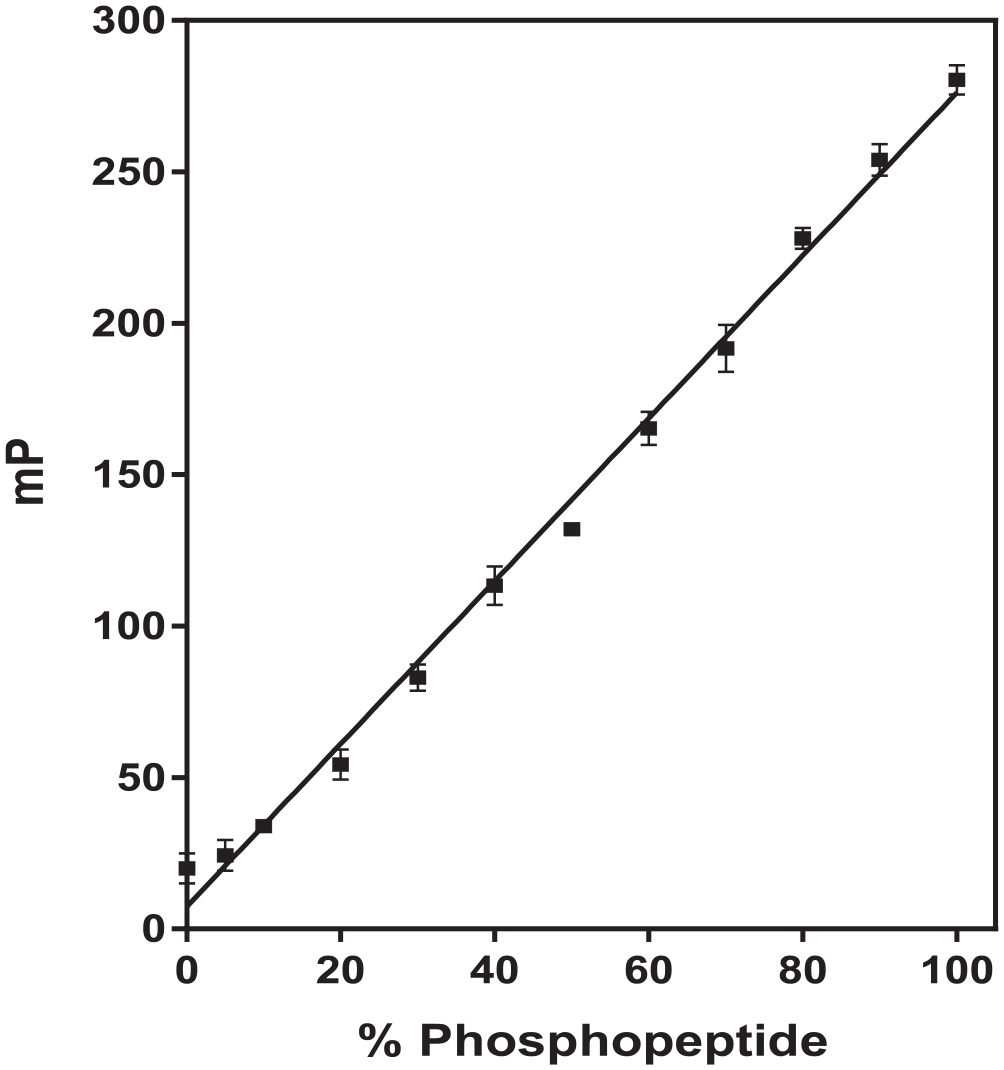
Calibration curve used to calculate the amount of phosphorylated peptide. Mixtures of nonphosphorylated peptide and phosphopeptide [5-carboxyfluorescein (5-FAM)-labeled-Crosstide plus 5-FAM-phospho-Crosstide] were kept constant at 100 nM, while the percentage of phosphopeptide was varied in each sample. Data points and error bars represent the mean and standard deviation of three replicates.
Determination of Apparent Km Values
The kinase activities of all three isoforms of SGK were measured using the 5-FAM-labeled-Crosstide substrate peptide, and the values of the apparent Km were determined to be 67, 105, and 42 µM for SGK1, SGK2, and SGK3, respectively (
Fig. 2
). The IMAP beads bind to not only phosphate groups on the phosphorylated peptide product but also unreacted ATP in the kinase reaction. Therefore, the assay signal begins to cause a reduction in mP when ATP is used at a concentration of greater than 10 µM. Consequently, modified calibration curves were prepared separately using a solution of 5-FAM-phospho-Crosstide and 5-FAM-labeled-Crosstide (the total peptide concentration was kept constant at 100 nM) containing 67 µM ATP for SGK1, 105 µM ATP for SGK2, and 42 µM ATP for SGK3. In addition, the initial reaction rates expressed in mP/min were converted to nM/min. To identify the concentration limit for FP signal linearity of the reaction and reaction time that generates an acceptable signal-to-base greater than 3, the time course of the reaction was measured at enzyme concentrations ranging from 1 to 4 nM for SGK1 and SGK2 and from 0.5 to 2 nM for SGK3, with ATP concentration at the apparent Km value for each SGK and 100 nM 5-FAM-labeled-Crosstide (
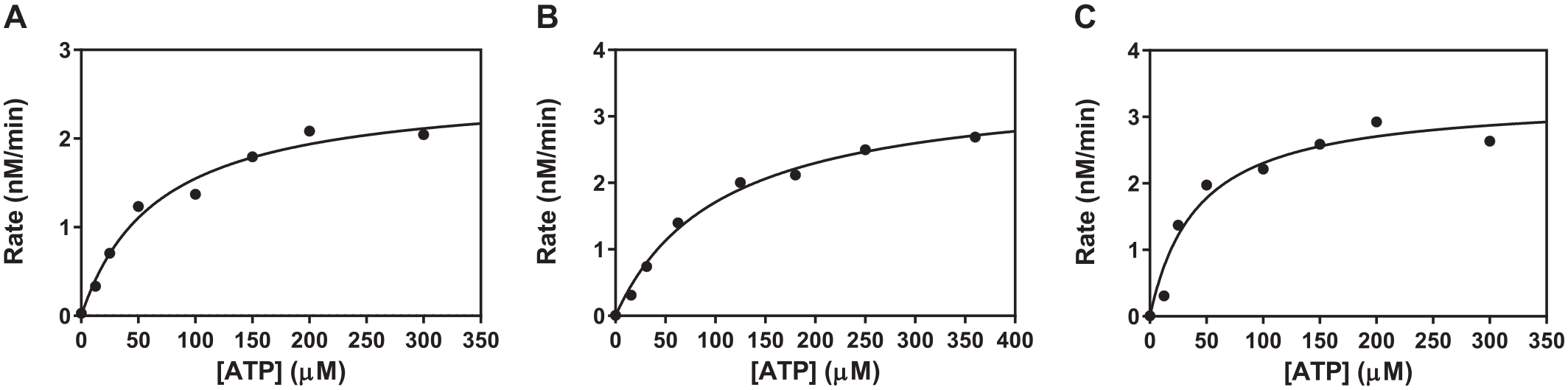
The values of the apparent Michaelis constant (Km, app) for adenosine triphosphate (ATP). (
Z’-Factors and Screening
Because kinase-focused library compounds were dissolved in 100% DMSO at 10 mM concentration, the effects of DMSO on SGK enzyme activity were also investigated. The enzyme activity of SGK1 and SGK2 was not affected up to a concentration of 10% DMSO, while SGK3 activity declined with increasing concentrations of DMSO, with approximately 50% activity remaining at 10% DMSO, because it presumably acts as a protein denaturant (
Furthermore, the performance of assay uniformity and signal variability was assessed in a 384-well format by running the FP assay for each SGK isoform for 3 consecutive days using the optimized conditions (
Lastly, the FP assay was validated by determining the IC50 value of a pan-kinase inhibitor, staurosporine, for 3 consecutive days using the optimized conditions of 100 nM 5-FAM-labeled-Crosstide at 1% DMSO concentration (2.0 nM SGK1 and 67 µM ATP, 2.0 nM SGK2 and 105 µM ATP, and 0.5 nM SGK3 and 42 µM ATP). The average IC50 values of staurosporine were determined to be 0.027, 0.236, and 0.148 µM for SGK1, SGK2, and SGK3, respectively (
Fig. 3
). The IC50 values determined in this study are comparable to reported values.28,29 To further validate the FP assay, the selective SGK inhibitor GSK650394 was chosen as a reference compound, which revealed IC50 values of 0.033, 0.093, and 5.0 µM for SGK1, SGK2, and SGK3, respectively (
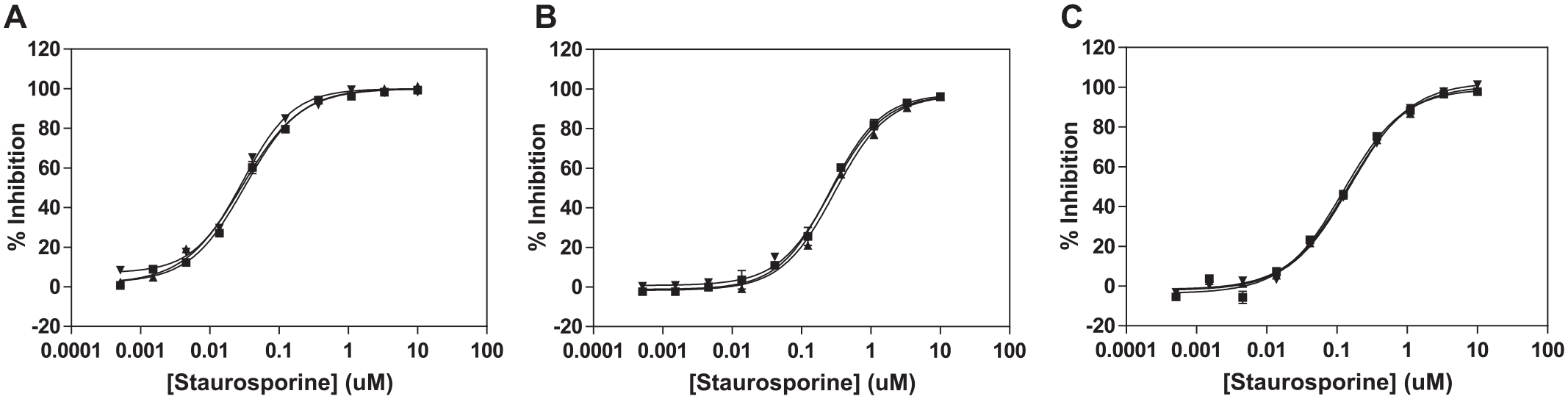
Determination of half-maximum inhibitory concentration (IC50) values of staurosporine for 3 consecutive days. The average IC50 values were determined to be 0.027, 0.236, and 0.148 µM for (
Screening and Identification of Potential SGK Inhibitors
Compounds from a small kinase-focused library of 160 small molecules collected in-house were screened for potential inhibitors of SGK1 at 10 µM concentration of the compound in duplicates in 384-well assay plates. The kinase-focused library set contains high-quality and previously profiled inhibitors from other in-house kinase inhibitor programs. Our primary screening was designed to identify ATP-competitive inhibitors by using a substrate ATP concentration at the apparent ATP Km value, which was determined to be 67 µM for SGK1 in this study. Of the 160 compounds screened, 39 compounds with inhibitory activity greater than 50% were retested using concentration–response curves ( Fig. 4 ). Among our initial hits, 12 compounds contained the thiazolidine-2,4-dione (thiazolidinedione, or TZD) scaffold ( Table 1 ). The most potent SGK1 inhibitor, KMU010402 in the TZD series, was tested for SGK2 and SGK3 inhibitory activity, and its IC50 values were determined to be 5.4 µM for SGK2 and 4.8 µM for SGK3.
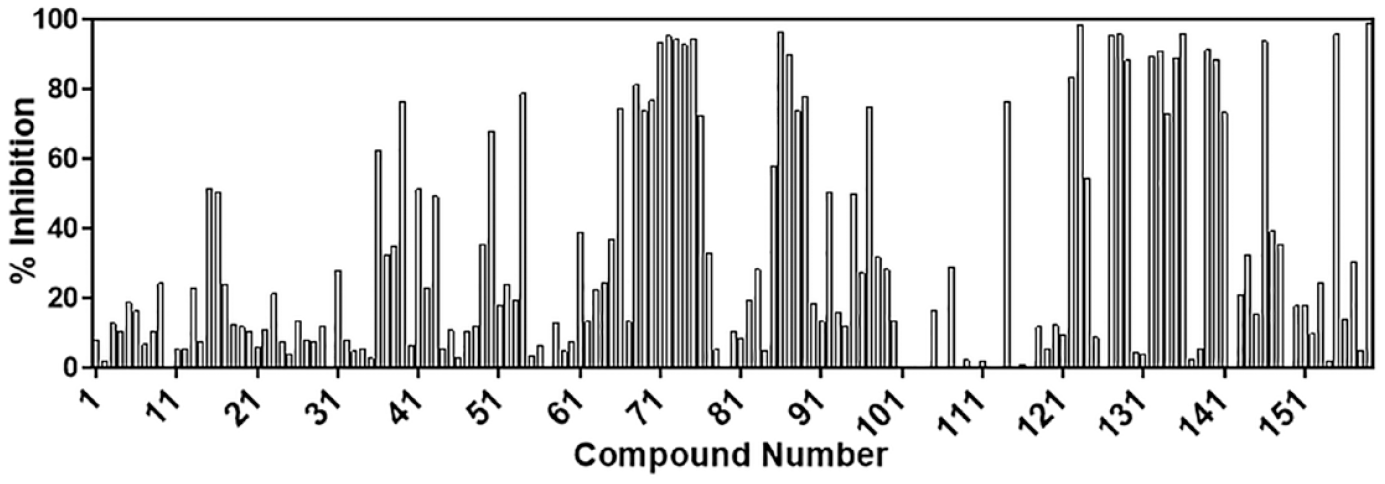
Bar graph of screening results against serum- and glucocorticoid-regulated kinase-1 (SGK1) of a collection of 160 compounds in a small kinase-focused library. Each bar represents the mean of duplicate.
Percent Inhibitions and IC50 Values of Thiazolidinedione Derivative Hits.

IC50, Half-maximum inhibitory concentration.
Mechanism of Inhibition of Thiazolidinedione Scaffold
Next, compound KMU010402 was selected for further mechanism-of-inhibition studies to confirm the TZD scaffold as an ATP-competitive inhibitor for SGKs. Since our FP-based screening assay was mainly designed to identify ATP-competitive inhibitors, it is necessary to determine the mode of action of TZD on ATP as well as the substrate kinetic parameters. To determine whether KMU010402 competes with ATP for binding to the SGK active site, the SGK activity was measured as a function of ATP concentrations at different concentrations of KMU010402. Out of all the models (competitive, noncompetitive, uncompetitive, and mixed noncompetitive models), the inhibition data for KMU010402 were found to best fit a competitive inhibition model. Lineweaver–Burk double-reciprocal analyses with KMU010402 showed the best fit to competitive inhibition, confirming that TZD competes with ATP to bind to the ATP-binding site of each SGK isoform ( Fig. 5 ). The inhibition constants, Ki,app values for SGK1, SGK2, and SGK3 that were determined by Equation (4), are 0.19, 0.21, and 1.2 µM, respectively.

Lineweaver–Burk competitive inhibition curves for serum- and glucocorticoid-regulated kinase (SGK) activity on KMU010402 and adenosine triphosphate (ATP) concentrations. Inhibitor concentrations corresponding to the open circles, closed squares, open triangles, and closed inverse triangles are (
Molecular Modeling
Because KMU010402 is an ATP-competitive kinase inhibitor of all three SGK isoforms, the compound was docked into the SGK1 X-ray crystal structure (3HDM; Protein Data Bank, https://www.rcsb.org) using AutoDock Vina ( Fig. 6 ). The hydroxyl group of KMU010402 forms a hydrogen bond with the backbone carbonyl of Asp177. In addition, the docked model suggests that a nitrogen atom on the heteroarene of KMU010402 may interact with the side chain of Lys127 because they are located within the hydrogen-bonding distance.
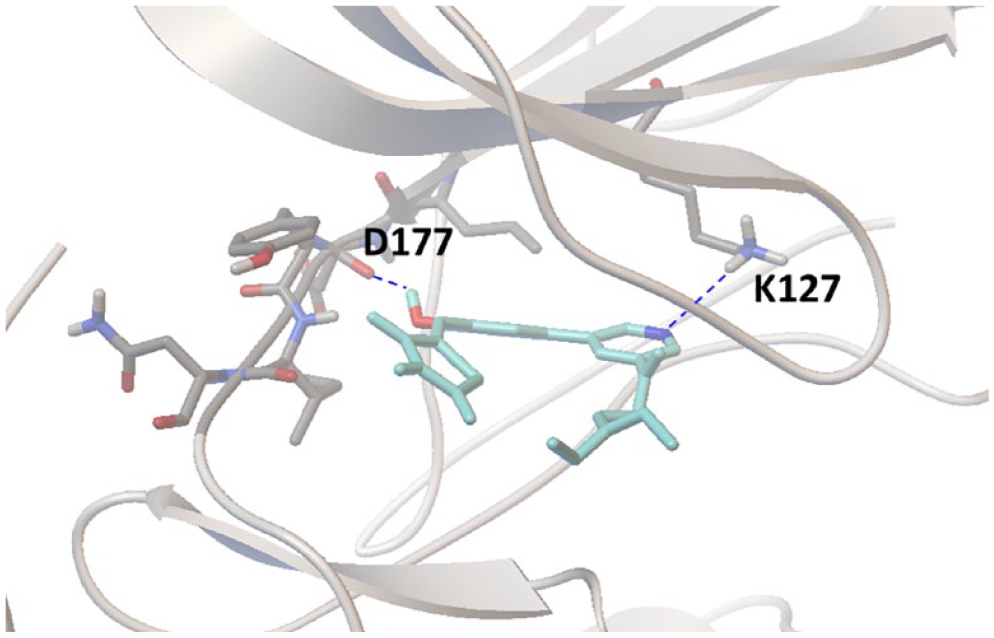
Docking of compound KMU010402 to the adenosine triphosphate (ATP)-binding site of serum- and glucocorticoid-regulated kinase-1 (SGK1) (3HDM; Protein Data Bank, https://www.rcsb.org). Two expected hydrogen bonds between KMU010402 and SGK1 amino acid residues (Lys127 and Asp177) are indicated with dashed lines.
In summary, the SGK family is an essential AKT-independent mediator of the PI3K/PDK signaling pathway that regulates cell proliferation, survival, and stress, as well as transportation of ions. SGK expression is significantly elevated in many solid cancers. Thus, SGK is an attractive target for anticancer therapy. To identify novel SGK inhibitors, we developed and optimized a homogeneous FP-based SGK assay in a 384-well format and screened an in-house kinase-focused library. The identified hit compound, containing the TZD scaffold, was further evaluated for the mechanism of SGK inhibition, resulting in the discovery of a novel pan-isoform ATP-competitive inhibitor of SGKs.
Supplemental Material
sj-pdf-1-jbx-10.1177_24725552211002465 – Supplemental material for Identification and Kinetic Characterization of Serum- and Glucocorticoid-Regulated Kinase Inhibitors Using a Fluorescence Polarization–Based Assay
Supplemental material, sj-pdf-1-jbx-10.1177_24725552211002465 for Identification and Kinetic Characterization of Serum- and Glucocorticoid-Regulated Kinase Inhibitors Using a Fluorescence Polarization–Based Assay by Jeongeun Kim, Donghee Kim, Hyunho Jung, Jinho Lee and Victor Sukbong Hong in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.