
Abstract
Mass spectrometry-based proteomics profiling is a discovery tool that enables researchers to understand the mechanisms of action of drug candidates. When applied to proteolysis targeting chimeras (PROTACs) such approaches provide unbiased perspectives of the binding, degradation selectivity, and mechanism related to efficacy and safety. Specifically, global profiling experiments can identify direct degradation events and assess downstream pathway modulation that may result from degradation or off-target inhibition. Targeted proteomics approaches can be used to quantify the levels of relevant E3 ligases and the protein of interest in cell lines and tissues of interest, which can inform the line of sight and provide insights on possible safety liabilities early in the project. Furthermore, proteomics approaches can be applied to understand protein turnover and resynthesis rates and inform on target tractability, as well as pharmacokinetics/pharmacodynamics understanding. In this perspective, we survey the literature around the impact of mass spectrometry-based proteomics in the development of PROTACs and present our envisioned proteomics cascade for supporting targeted protein degradation projects.
Introduction
Mass spectrometry-based proteomics assays have played a critical role in drug discovery, particularly in the areas of target identification, selectivity profiling, and characterization of biological input materials.1–3 Focused analyses, such as chemical proteomics and global proteomics profiling, permit the identification and quantification of thousands of proteins in a single sample. These techniques have proven to be essential elements in characterizing the mechanism of action of candidate drugs as they relate to efficacy and safety. Proteomics studies have played an indispensable role in the development of the protein degradation field, starting with the identification of the E3 ligase complex containing cereblon, DDB1, and Cul4A as the target of thalidomide. 4 Global proteomics profiling was next applied to characterize the degradation selectivities of the first cereblon-based proteolysis targeting chimeras (PROTACs), compounds that arguably opened the door for therapeutic applications of protein degradation.5,6 More recently, a global proteomics study helped identify SALL4 and other neosubstrates of degradation by cereblon responsible for the teratogenic effects of IMiD drugs. 7 In this perspective, we outline our visions and outlooks for how we expect advances in mass spectrometry-based proteomics techniques to impact the field of protein degradation in three main areas: degradation selectivity with downstream pathway effects, quantitation of E3 and targets of interest, and protein turnover.
Impact of Proteomics for PROTAC—Where We Stand Today
Selectivity is one of the most important parameters to understand for PROTACs, as off-targets of binding and degradation can often inform on previously unknown safety flags. The relationship between off-targets of binding and degradation, however, can be complicated. This is particularly true with degraders derived from promiscuous kinase inhibitors. Recent publications8,9 around this topic showed that binding selectivity and degradation selectivity do not track with one another; a promiscuous kinase inhibitor can be designed into a selective degrader, and weak binding interactions can result in a potent degradation, depending on interactions between the E3 complex and the target kinase. Here, a combination of chemical proteomics and global proteomics will deliver the key information around binding and degradation selectivity, respectively. 10 What aspects of the phenotype is driven by binding and what is driven by degradation can be further teased apart by estimating the concentrations of the E3 ligase and target protein and measuring the actual amount of target engagement by the PROTAC to both binding partners. 11 Profiling the global proteome across a time course of compound exposure can distinguish the pathways affected (and ultimately link to phenotype) as a result of binding compared with degradation, as exemplified in the differences in gene transcription between treatment with a CDK9 inhibitor versus degrader. 12 In these situations, pathway effects are read at the transcriptome level and can be complemented with global proteomics at the same time points. 13
The amount of a target of interest in the cell and how quickly it is naturally replenished will affect both how quickly the protein can be degraded and the maximum level of degradation that is achieveable. Understanding these metrics is important for both safety and efficacy considerations as a highly abundant protein that is rapidly replenished naturally will be more difficult to deplete to significant levels in a given time frame than a protein that is not. This is, of course, also tied to the level of degradation that is required to trigger and sustain any downstream pharmacodynamic effect: achieving 50% degradation with a PROTAC is easier than achieving more than 95% degradation. In addition to the target concentration, the E3 ligase concentration is another important cellular parameter that can essentially make or break a PROTAC-based approach. One elegant example of the impact of E3 ligase abundance is BCL-XL; here, the absence of Von Hippel-Lindau tumor suppressor (VHL) prevents the degradation of BCL-XL in platelets by a VHL-based BCL-XL PROTAC, thereby limiting the toxicity and preventing the development of thrombocytopenia.14–16
Mass spectrometry-based profiling of abundances of protein levels is an important discovery tool for understanding and predicting the efficacy and safety of protein degraders. To determine the abundance of these proteins in tissues of interest, one can use both data-dependent (DDA) and data-independent (DIA) acquisitions.17,18 Relative expression profiles can be determined by global proteomics using multiplexed analysis or with DIA to compare across different cell lines and tissues, and these efforts can be combined with mRNA expressions 13 to gain mechanistic insights into degradation and identify new E3 ligases that are differentially expressed in efficacy and safety models. Furthermore, both DDA and DIA approaches can be applied to also determine absolute quantities of a particular protein of interest,18,19 which is particularly relevant for comparing E3 levels across different species.
Another key variable in the efficacy of a given PROTAC is the turnover rate of the degraded proteins. Specifically, the protein turnover rate (PTR) will dictate how quickly the target returns after the PROTAC has been cleared in vivo, which has implications for the dosing schedule. As protein resynthesis will be a factor in the recovery of target functionality after degradation, 20 measuring protein half-life early in development can aid project progression. Measuring protein turnover as early as target selection is critical, as turnover can be used to develop a target compound profile and to determine whether a degrader is the best drug modality. Endogenous protein turnover can also be a key parameter in degradation selectivity, as a target with an equivalent degradation rate but slower resynthesis rate will show a larger effective PROTAC response. Later in development, the turnover rate can be combined with protein resynthesis measurements to predict dosing schedules for efficacy experiments.
Protein resynthesis can be measured after PROTAC treatment and washout using traditional methods for quantitating protein levels such as enzyme-linked immunosorbent assay (ELISA). However, the experimentally measured resynthesis rate can vary based on various compound properties, such as maximum degradation level, off-rates, protein binding, and lipophilicity. Another common method for measuring PTRs is to inhibit protein synthesis with inhibitors such as cycloheximide. Given the extensive biological consequences of these inhibitors, and the limited amount of time most cells will tolerate them, turnover rates measured using protein synthesis inhibitors lack accuracy, particularly for longer-lived proteins. 21
The gold standard for measuring protein turnover is pulsed SILAC (stable isotope labeling by amino acids in cell culture) mass spectrometry. 22 This method involves feeding cells 23 or animals 24 (stable isotope labeling by amino acids in mammals [SILAM]) with isotopically labeled amino acids over a time course. New proteins synthesized over the time course will incorporate the labeled amino acids, which can subsequently be distinguished from their unlabeled counterparts by mass spectrometry. These measurements allow for both degradation and synthesis rates to be measured by tracking both the labeled and unlabeled pools of proteins. Protein turnover can be measured for individual proteins using enrichment or nonenrichment methods. Alternatively, turnover can be measured globally for thousands of proteins simultaneously. Recent work by Zecha et al. has combined pulsed SILAC with tandem mass tag (TMT) multiplexing to generate more than 6000 PTRs, each made up of several robust peptide measurements, from a single cell line. 25 This work demonstrates that different proteoforms of proteins can have different turnover rates, which has implications when designing and interpreting targeted PTR experiments. Advances in SILAC quantification have also made turnover measurements in nondividing cells more accurate, particularly for long-lived proteins. 26 This is crucial for PROTACs, as immortalized models might underestimate the relevant physiological resynthesis rate.
A critical aspect of characterizing protein degraders is the ability to distinguish between direct targets of degradation and pathway effects over time. A recent method termed multiplexed proteome dynamics profiling (mPDP) has cleverly combined dynamic SILAC with global proteomics to investigate the mechanisms of protein homeostasis modulators. 27 In this method, cells are grown in labeled SILAC media, and at the time of compound treatment, the media is switched to the opposite SILAC label in duplicate (heavy cells to light media and light cells to heavy media). Multiplexing and mass spectrometry analysis allow changes to mature (preexisting) proteins to be distinguished from nascent proteins (proteins that were synthesized after the compound treatment and label swap). For example, this method was successful is distinguishing a BET inhibitor similar to JQ1 28 from a JQ1-PROTAC by detecting decreased abundance of BR2, BRD3, and BRD4 in the mature and nascent protein pools. Intriguingly, this method also identified FYTTD1, a TREX complex adaptor, as an off-target of JQ1 that leads to downstream arrest in protein synthesis. Due to the upfront requirement for SILAC labeling, mPDP is substantially more time-intensive than a standard global proteomics experiment and is best applied to answer complex mechanistic questions, rather than as a routine compound profiling approach.
We describe our vision for how to apply proteomics to a PROTAC project in our PROTAC proteomics cascade ( Fig. 1 ), including assays for measuring selectivity, protein abundance, and protein dynamics. For every project, our core activities include comprehensive profiling of degradation selectivity. We perform global proteomics experiments starting from the initial leads all the way to candidate selection. As the leads become more advanced, we increase the depth of our profile to include multiple doses to identify safety-related off-targets at higher concentrations, as well as time course experiments to distinguish pathway effects from direct degradation. Chemical proteomics can be performed to link degradation selectivity with binding selectivity, which can be particularly important for PROTACs from kinase inhibitors. Assays for quantifying protein abundance and measuring protein dynamics can be done on a target-agnostic basis and can actually be used to inform on target selection.
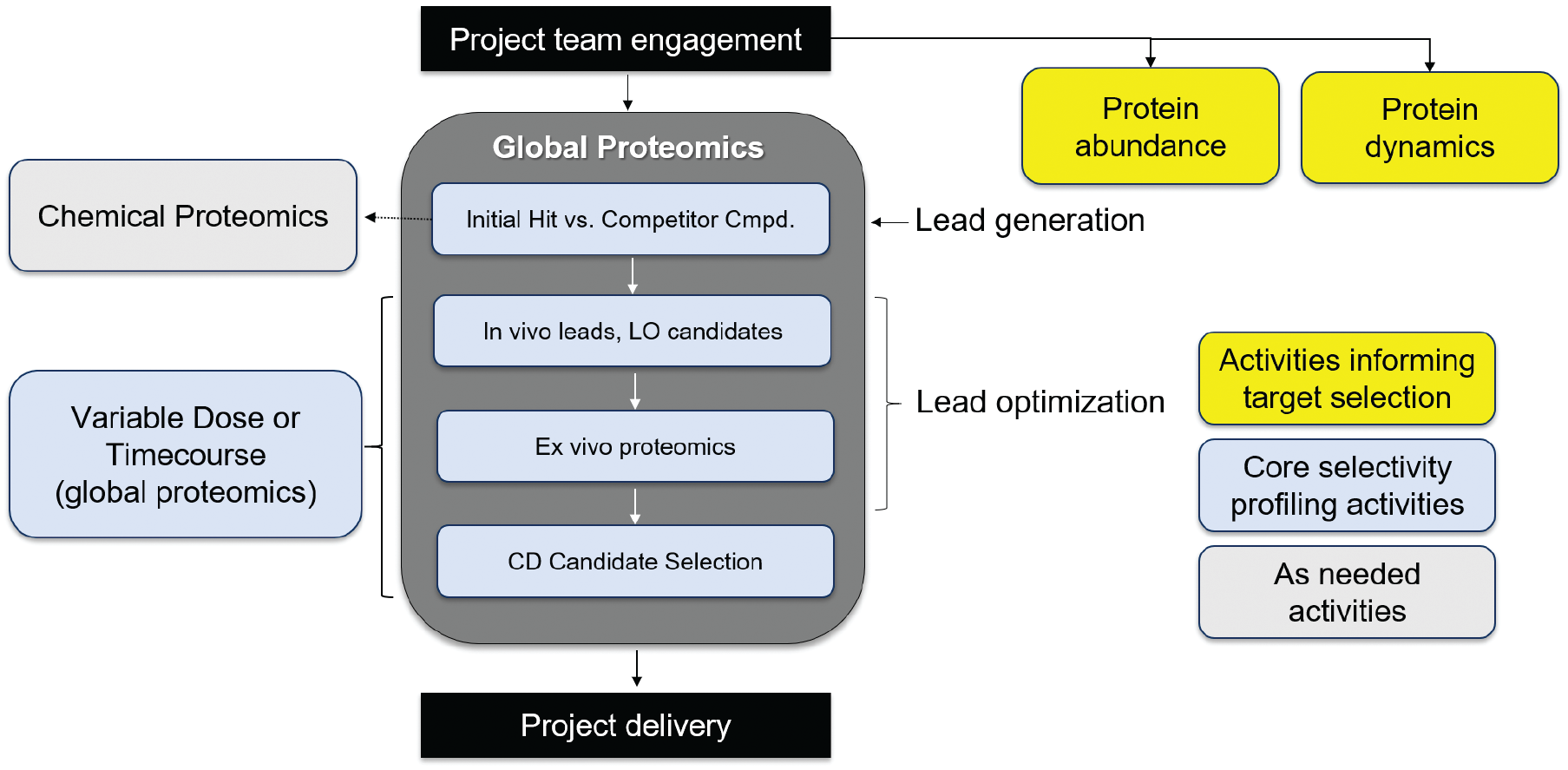
Our vision of a PROTAC proteomics cascade to interrogate selectivity, protein quantitation, and protein dynamics.
Outlook and Challenges
The methods described above have highlighted the power of “omics” tools to better characterize protein degraders for their selectivity, off-target, and dynamic activities. Nevertheless, these methods can only provide partial information on the full activity of a molecule since only a few cell lines and tissues are usually analyzed due to the high amount of resources required for such analyses. The selection of the models used in proteomics studies is therefore critical in order to maximize the chance of identifying potential off-target liabilities.
This is evident from the example of IMiD neosubstrates that have cell line and tissue expression specificity, such as SALL4, IKZF1, and p63. SALL4 is a transcription factor involved in the formation of tissues and organs during embryonic development, but it is also expressed in hematopoietic stem cells (HSCs), where it has been shown to regulate their differentiation. 7 IKZF1 is a member of the Ikaros protein family and is specifically expressed in immune cells. IKZF1 degradation by pomalidomide and lenalidomide has been linked to the defect in hematopoietic stem cell differentiation into monocytes that can explain the neutropenia effect seen in clinic with these drugs. 29 Finally, p63 is a member of the p53 family and has been linked to the developmental toxicity effect of IMiD in zebrafish. 30 The tissue distribution of these examples of IMiD neosubstrates clearly indicates that a full characterization of PROTAC molecules will be limited to the number of cell lines/tissues analyzed, and a careful selection of the models to be profiled is key to clearly defining the activity and selectivity of a PROTAC molecule.
The bottleneck of the actual “omics” methods is their throughput, which impedes both the analysis of hundreds of PROTAC molecules and their activities in multiple biological samples. The shotgun approach of analyzing highly fractionated, multiplexed samples to get deep proteomics coverage comes with the trade-off of lower throughput; this is especially true in cases where multiple replicates are required for proper statistics. The emergence of higher-throughput mass spectrometry aquisition methods, such as DIA, may change the way we analyze PROTACs, as this provides a means to rapidly process multiple samples. 31 Expedited analysis allows for the possibility of obtaining global profiles in a more iterative fashion to generate structure–activity relationship knowledge. It is worth noting that increased throughput can mean a compromise in data depth; however, the availability of these additional data acquisition methods has given us the flexibility to design our proteomics experiments in a bespoke or fit-for-purpose manner.
As the protein degradation field advances, there is an effort to identify new mechanisms to harness for protein degradation. One aspect of this effort is to design PROTACs to recruit new E3 ligases beyond the more commonly utilized partners VHL and cereblon. Here, key parameters to understand include druggability, expression, subcellular localization, and mechanism of action. Global proteomics, as well as the protein quantitation methods mentioned earlier, will be critical in addressing these questions. In addition, advances in the LOPIT (localization of organelle proteins by isotope tagging) method, such as hyperLOPIT and LOPIT-DC, 32 will allow for higher spatial resolution of E3 ligase expression as well as understanding of degradation selectivity in these subcellular compartments. Here, the subcellular compartments are separated either by density gradient or ultracentrifugation, followed by isobaric tagging (e.g., TMT), to obtain the relative protein quantities in different organelles. In addition to subcellular localization, the mechanism of action of new E3 ligases must be understood, 33 especially around their ability to mediate substrate degradation. To date, the mechanisms of VHL and cereblon have been well characterized, where, depending on the nature of ternary complex formation, different proteins get ubiquitinylated and subsequently degraded. 34 However, we know that ubiquitinylation events do not always result in degradation, 35 and we need to understand if such a phenomenon is associated with novel proposed E3 ligase partners. This would involve mapping out global ubiquitinylation patterns, which can be determined through ubiqutin remnant profiling (also referred to as diGLY proteomics) and compared with actual degradation events to understand whether a particular E3-driven ubiquitination event will lead to protein degradation.36–38
In summary, we have demonstrated how mass spectrometry proteomics methodologies have shaped the targeted protein degradation field and shared our vision for the challenges to be addressed in the coming years. We believe that focused expansion and adaptation of current methodologies and technologies in the directions described above will further engrain mass spectrometry-based proteomics in early-stage preclinical studies in the small-molecule degrader field.
Conclusion
Mass spectrometry-based proteomics and protein degradation are two intertwined fields with an inseparable relationship. In this perspective, we laid out our vision for how proteomics can impact the development of PROTACs from the standpoint of selectivity, protein abundance, and protein dynamics. We envision that as the protein degradation field continues to advance, proteomics studies will play a critical role in identifying new E3 ligases as well as new methods of degradation, such as molecular glues. In the process, our ability to quantify protein abundance and increase protein coverage will generate critical information to inform on potential safety liabilities and candidate efficacy.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.