
Abstract
A key activity in small-molecule drug discovery is the characterization of compound–target interactions. Surface plasmon resonance (SPR) is a flexible technique for this purpose, with a wide affinity range (micromoles to picomoles), low protein requirements, and the ability to characterize the kinetics of compound binding. However, a key requirement of SPR is the immobilization of the target protein to the surface of the sensor chip. The most commonly used immobilization techniques (covalent immobilization, streptavidin–biotin) are irreversible in nature, which can afford excellent baseline stability but impose limitations throughput for slowly dissociating compounds or unstable targets. Reversible immobilization (e.g., His-tag–Ni-NTA) is possible but typically precludes accurate quantification of slow dissociation kinetics due to baseline drift.
Here we present our investigation of three immobilization strategies (dual-His-tagged target protein, His-tagged streptavidin, and switchavidin) that combine the robustness of irreversible immobilization with the flexibility of reversible immobilization. Each has its own advantages and limitations, and while a universal immobilization procedure remains to be found, these strategies add to the immobilization toolbox that enables previously out-of-scope applications. Such applications are highlighted in two examples that greatly increased throughput for the kinetic characterization of potent kinase inhibitors and kinetic profiling of covalent inhibitors.
Introduction
Surface plasmon resonance (SPR)-based biosensors serve as a key component of the biophysics toolbox during the drug discovery process. 1 In a typical SPR experiment, the drug target is tethered to the chip surface using irreversible immobilization strategies such as N hydroxy succinimide (NHS) and 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)-mediated covalent cross-linking to the chip matrix or pseudo-irreversible capture to covalently preimmobilized streptavidin.2,3 While these strategies can enable stable baselines with minimal drift—a prerequisite for robust kinetic characterization—their irreversible nature precludes the iterative use of the biosensor surface in the case of slowly dissociating compounds or unstable target proteins.
Reversible immobilization typically involves capture approaches, where affinity tags on the target protein (such as a poly-His-tag or antigen peptide motifs) enable tethering via a Ni2+-NTA matrix or via irreversibly immobilized antibodies, respectively.4–6 Most of these techniques have limited use for biosensing purposes, as they are either too weak (e.g., Ni2+-NTA capture) or complicated (e.g., protein DNA-tagging7,8). While these approaches can generate protein surfaces that are efficiently regenerated after use, this reversibility is typically accompanied by increased drift due to dissociation of the tethered target protein. This drift is also larger at the surface densities required for the detection of small-molecule binding. Not only does this drift complicate or even prevent the kinetic characterization of small molecules with small absolute response and slow dissociation rates, but also it results in time-dependent variation of surface coverage, further complicating data analysis.
Different approaches for various degrees of regenerable immobilization have been proposed over the years,7,9–13 but none of them have been widely applied in industry. This is largely due to shortcomings in these approaches and the challenge of ensuring a stable, drift-free baseline, while achieving complete and reproducible regeneration to enable iterative (re-)use of the biosensor surface.
Regenerable protein immobilization would have several benefits. It would provide flexibility to rapidly switch between assays without the need to discard, or even remove, sensor chips from the instrument. Assay development would be accelerated, as various buffer conditions could be tested on freshly immobilized protein in an iterative fashion. Throughput for slowly dissociating compounds would be improved (or testing of irreversible compound enabled), as the surface could be regenerated for the next analyte without waiting for compound dissociation. Finally, inherently unstable targets could be enabled.
For regenerable protein surface strategies to be truly viable and broadly applicable in small-molecule drug discovery, we defined the following set of requirements:
With these requirements in mind, we explored three immobilization strategies: the use of a dual-His-tagged target,14–16 a biotinylated target in combination with His-tagged streptavidin, and a biotinylated target in combination with switchavidin 17 ( Fig. 1 ).
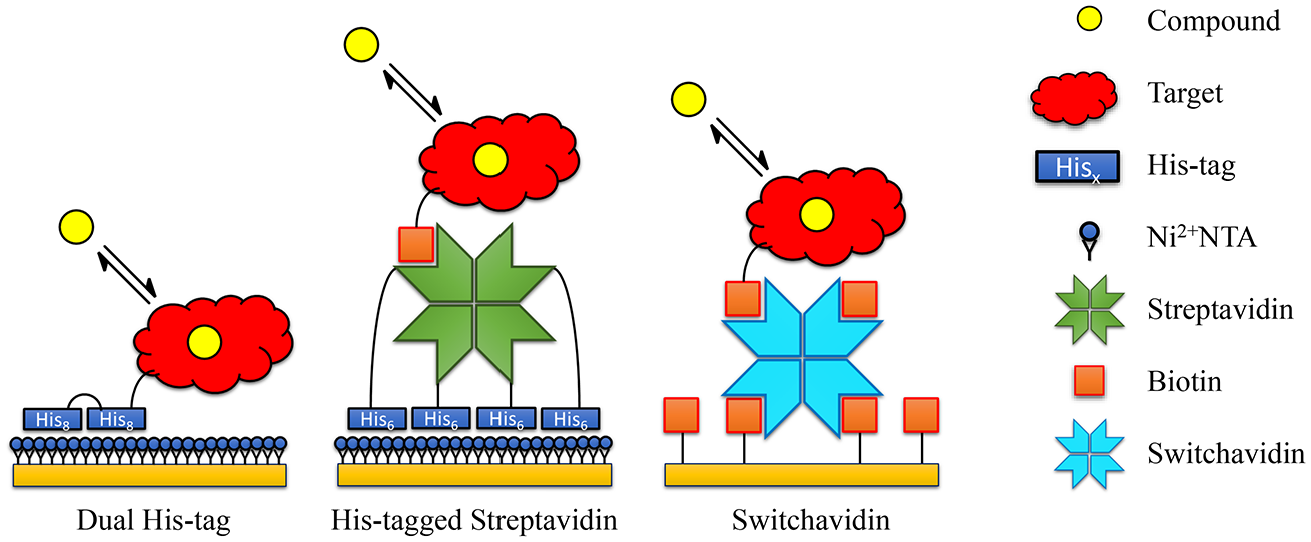
Schematic of the three regenerable immobilization strategies investigated in the current paper: dual-His-tagged target protein captured to Ni-NTA, biotinylated target protein immobilized to His-tagged streptavidin that is captured on a Ni-NTA surface, and a biotinylated target protein immobilized to switchavidin.
In choosing these three immobilization strategies for scrutinization, it is recognized that more could have been selected, such as DNA- or antibody-based approaches. While it is possible to obtain very tight interactions between antibodies and antigens, a prerequisite for stable baseline, suitable high-affinity antibodies are not readily available for many target proteins. Similarly, antibodies derived against peptide tags (like anti-His-tag antibodies) are in general not potent enough to ensure the minimal drift required for small-molecule kinetic characterization. While both the His-tagged streptavidin and switchavidin approaches are similar to Biotin CAPture, which utilizes DNA-tagged streptavidin to stably capture streptavidin to covalently immobilized DNA, 9 the capture levels with the Biotin CAPture approach are typically not compatible with small-molecule detection.
The dual-His-tag approach consists of two consecutive His-tags fused to the target protein. Different variants have been described previously, such as a long His-tag, two His-tags with a spacer, or one His-tag in each terminus of the target protein.14,15,18 While a traditional single His-tag is insufficient to achieve a stable baseline, two tags will improve the affinity of the target protein to a Ni2+-NTA surface through bivalent avidity, leading to reduced baseline drift. The surface is typically regenerated using standard procedures for Ni2+-NTA (e.g., EDTA).
The second approach, His-tagged streptavidin, operates by a similar principle. Each streptavidin tetramer will carry four His-tags resulting in large avidity effects when binding to a Ni2+-NTA surface, resulting in a very stable baseline. 18 Biotinylated target proteins can subsequently be captured, but in contrast to covalently immobilized streptavidin, regeneration of the Ni2+-NTA surface is possible.
The final approach requires switchavidin, 17 a mutant variant of avidin, and utilizes a slightly different strategy. When subjected to acidic conditions, switchavidin is monomeric, which greatly reduces its affinity to biotin, enabling efficient regeneration. Similar to His-tagged streptavidin, switchavidin mediates immobilization of biotinylated target protein, in this case on biotin sensor surfaces, which enables regeneration of the entire protein complex. The switchavidin procedure has been shown to work well with 2D surfaces and large-molecule interactions (such as antibodies), but the procedure has, to our knowledge, not been described in the context of small-molecule binding characterization.
In this report, we describe a comparison of the three approaches in the context of small-molecule binding and kinetic characterization. We investigate how they compare to traditional immobilization procedures and to what degree they present a truly viable and broadly applicable approach in small-molecule drug discovery ( Table 1 ).
Summary of Advantages and Disadvantages for Various Immobilization Strategies, Including the Ones Examined in This Paper.

Direct covalent immobilization to the sensor surface of the target.
Capture of a mono-His-tagged target to a Ni-NTA surface.
Covalent immobilization of a His-tagged target to an activated Ni-NTA surface.
Capture of a biotinylated target to covalently immobilized streptavidin.
Capture of a dual-8His-tagged target to a Ni-NTA surface.
Capture of His-tagged streptavidin to a Ni2+-NTA surface and subsequent capture of biotinylated target to streptavidin.
Capture of switchavidin-biotinylated target complex to a biotin surface.
Materials and Methods
His-Tagged Streptavidin Expression and Purification
The His-tagged streptavidin was designed to contain N-terminal His6 tag followed with streptavidin residues 25–183. The construct was codon optimized and cloned into pET24a vector (GenScript, Colone, Germany) and transformed to the Escherichia coli BL21 Star (DE3) strain (Invitrogen, Stockholm, Sweden). The culture was grown at 37 °C in Terrific Broth medium until the OD600 nm was 1.0 and then induced with 0.2 mM isopropyl β-
Switchavidin was purchased from BioMediTech (Tampere, Finland) and produced as described by Taskinen et al. 17
Dual-His-Tagged ERK2 Expression and Purification
The dual-8His-tagged ERK2, with a construct name 2XHis8-ZZ-TEV-ERK2, was designed to contain two N-terminal His8 tags separated by a multi-amino acid spacer: a ZZ tag (domain of protein A), a TEV protease cleavage sequence, and human ERK2. The construct was cloned into a pET vector (GeneArt, Loughborough, UK) and transformed to E. coli BL21 GOLD (DE3) (Agilent Technologies, Stockport, UK). The culture was grown at 37 °C in autoinduction medium until the OD600 nm was 0.6 and then the temperature was reduced to 18 °C and the culture was incubated overnight to allow protein expression. The culture was harvested, and the protein was purified by IMAC, followed by size-exclusion chromatography with a Superdex-200 column (GE Healthcare) in buffer (20 mM HEPES, pH 7.9, 150 mM NaCl).
SPR Experiments with His-Tagged Streptavidin and Dual-His-Tagged ERK2
All SPR experiments were conducted on BIAcore instruments (GE Healthcare) using NTA sensor chips (GE Healthcare). All NTA surfaces were prepared by washing with 0.5 M EDTA prior to loading with 200 µM NiCl2. The normal running buffer was HBS-P+ (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% Tween-20). For buffer tolerance experiments, the capture of dual-8His-ERK2 was typically kept below 4000 RU to prevent baseline drift (see Results and Discussion), and for His-tagged streptavidin, the levels where kept below 5000 RU.
Surfaces were regenerated by injecting 0.5 M EDTA, 5 M GuSCN (Sigma-Aldrich, Stockholm, Sweden, CAS 593-84-0) with 5 mM TCEP (Sigma-Aldrich, CAS 51805-45-9), 1 mg/mL pepsin (Sigma-Aldrich, CAS 9001-75-6) in 1 M glycine, pH 2.5, and 50 mM NaOH with 1 M NaCl using a contact time of 300 s for each solution.
For ERK2 compound interaction studies, 10 mM HEPES, 150 mM NaCl, 0.05% Tween-20, 1 mM TCEP, and 1% DMSO, pH 7.4, were used as a running buffer. Compound (SCH772984) was diluted twofold from 100 nM and injected over the surfaces at 30 µL/min, 120 s contact time, followed by 1200 s dissociation. Resulting sensorgrams were reference and blank subtracted prior to fitting to a 1:1 binding model.
PAD4 Expression and Purification
Peptidylarginine deaminase 4 (PAD4), with the construct name N-6xHis-TEV-Avi-hPADI4(A2-P663), was designed to contain an N-terminal Avi-tag to allow for a controlled enzymatic biotinylation after the purification of the protein. The construct was inserted into a pET24 expression vector and transformed into an E. coli BL21 Star expression strain. The culture was grown at 37 °C in an autoinduction medium buffer until an OD600 nm of 0.6 was reached, upon which the temperature was lowered to 18 °C and the culture was left for incubation for an additional 80 h. After the cells were harvested, the protein was purified with a two-step purification method using Ni2+-affinity chromatography and size-exclusion chromatography by employing a HiLoad Superdex-200 column (GE Healthcare) in a running buffer (50 mM Tris, pH 8.0, 300 mM NaCl, 1 mM TCEP). Site-specific biotinylation was achieved through a 12 h incubation with Biotin ligase (BirA) in the presence of biotin and ATP, which enabled the covalent linking of the free biotin to the lysine of the Avi-tag with a yield of >99%.
SPR Experiments with Biotinylated PAD4
All SPR experiments were conducted on a BIAcore 8K instrument (GE Healthcare). Biotin sensor chips (BD200M; Xantec Bioanalytics GmbH, Düsseldorf, Germany) were washed with 2.5% citric acid and 0.25% sodium dodecyl sulfate (SDS) prior to use. Biotinylated PAD4 (250 nM) was preincubated with 125 nM switchavidin before immobilization on the biotin sensor chip at 25 °C using all eight flow channels, resulting in similar coupling densities of 10,200 RU ± 300 RU in all flow channels. After a stabilization period of 1 h, compounds were injected in twofold dilutions in running buffer (50 mM Tris/HCl, 250 mM NaCl, 1 mM TCEP, 1% DMSO, pH 8.0) using parallel kinetics; that is, all eight concentrations of the dose-response experiment were injected simultaneously over the sensor surface, resulting in one specific compound concentration per flow channel. The highest concentration tested was 50 µM, and the compound was diluted twofold. The flow rate was 30 mL/min with an injection time of 240 s. After a 720 s dissociation period, the surface was challenged with regeneration solution (2.5% citric acid, pH 2, 0.5% SDS) for 60 s to enable a new immobilization cycle. The resulting sensorgrams were reference and blank subtracted prior to fitting to a 1:1 binding model (Biacore evaluation software).
SIK3 Expression and Purification
Salt inducible kinase 3 (SIK3) construct 10His-SIK3-(M1-R327)-Avi was expressed in insect cells. The construct was inserted into a pFastBac expression vector and transfected into Sf21 cells, with a 48 h time of harvest for the expression. The protein was purified and biotinylated with a multiple-step purification scheme containing affinity on Ni2+ resin, removal of the His-tag, size-exclusion chromatography (Superdex-75, GE HEalthcare), ion-exchange chromatography (Resource Q), and site-specific biotinylation. The purification was finalized with a second size-exclusion step (Superdex-75; GE Healthcare) in buffer (40 mM Bis-Tris propane, pH 7.8, 200 mM NaCl, 10% glycerol, 1 mM TCEP).
SPR and Mass Photometry Experiments with Biotinylated SIK3
All SPR experiments were conducted on BIAcore instruments BC3000 and S/T200 (GE Healthcare) using HBS-P+ (10 mM HEPES, 150 mM NaCl, 0.05% Tween-20) as running buffer at 20 °C. An SA sensor chip (SAD200M Xantec Bioanalytics GmbH) was washed with a mixture of 10 mM NaOH and 1 M NaCl before immobilization of biotinylated SIK3 at 100 nM. NTA sensor chip (NID500M; Xantec Bioanalytics GmbH) was washed with a mixture of 10 mM NaOH, 1 M NaCl, and 0.5 M EDTA before the addition of 3 mM NiCl2 and subsequent immobilization of His-tagged SA at 100 nM, followed by biotinylated SIK3 at 100 nM. Biotin sensor chips (BD200M; Xantec Bioanalytics GmbH) were washed with a mixture of 2.5% citric acid and 0.25% SDS prior to use. Biotinylated SIK3 was preincubated with switchavidin in a 2:1 ratio for 5 min before being diluted to 100 nM and immobilized. Compounds were injected in threefold dilutions in running buffer before 60 s regeneration (2.5% citric acid, 0.25% SDS). The resulting sensorgrams were reference and blank subtracted prior to the fitting of a 1:1 binding model (Biacore evaluation software).
Biotinylated SIK3 was preincubated with switchavidin in a 2:1 ratio for 2 min before being diluted to 100 nM and analyzed using a Refeyn One mass photometry instrument (Refeyn Ltd., UK) at a 60 s acquisition time. The resulting histogram was calibrated using a NativeMark protein ladder (ThermoFisher, Stockholm, Sweden) and fitted to multiple Gaussians to extract the peak mass.
Results and Discussion
All Investigated Approaches Result in High and Stable Capture Levels
Dual-8His-Tag and His-Tagged Streptavidin
Dual-8His ERK2 and His-tagged streptavidin could both be captured to high levels before significant drift was observed (
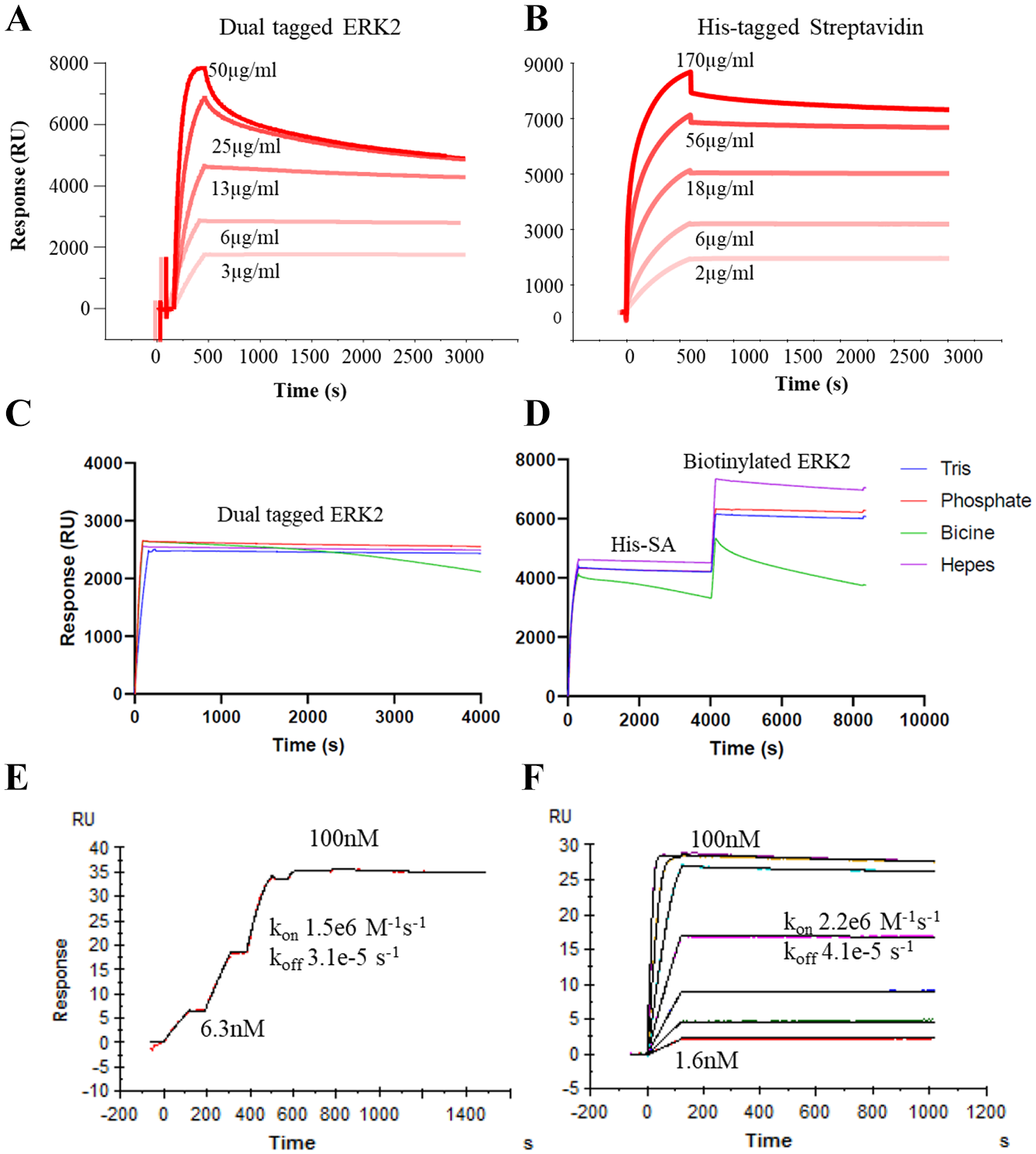
Capture levels using different concentrations of (
Dual-His-tagged proteins and His-tagged streptavidin behaved similarly, in that drift is significantly reduced compared with a single His-tag due to avidity effects.14,15 However, as the capture level increases, the level of drift also increases, as the opportunities for avidity decrease due to neighboring Ni-NTA sites being already occupied. 18 Nonetheless, the capture levels achievable with the dual-His-tagged protein and His-tagged streptavidin are sufficient for typical small-molecule applications.
Capture procedures that rely on avidity may be more sensitive to buffer conditions, as a small weakening in the interaction may result in a significantly weaker overall interaction. Dual-8His-tagged ERK2 and His-tagged streptavidin showed similar baseline stability with a variety of buffers, with the exception of BICINE (
The binding of a compound to a captured target protein using both methods is nearly identical (
While a surface with captured His-tagged streptavidin stabilized quickly (within 5 min), the time for the surface to stabilize after target capture was target dependent. For some targets, it was as rapid as for streptavidin, while for others it could take 30 min or longer. This could be problematic, as any time gained in overcoming slow compound dissociation rates would be lost due to long surface stabilization times. It is worth noting that the biotinylated targets we have used also feature a His-tag, so it is possible that the streptavidin becomes saturated and the target protein then binds to the Ni2+-NTA surface via its His-tag. Here, the dissociation of mono-His-tagged protein from the surface would dictate the stabilization time.
Despite clear advantages, all avidity-based solutions will suffer from a similar limitation, namely, the difficulty of ensuring that all immobilized proteins are bound with maximal avidity. This will limit the immobilization levels possible and, for poorly active or particularly large targets, may limit the utility of such approaches.
Switchavidin
In the original publication, 17 it was shown that for preparation of a 2D surface, sequential capture of switchavidin followed by a biotinylated target was possible. However, while switchavidin alone can be immobilized to a high coverage on a biotin-derivatized 3D matrix, the flexibility of the matrix results in saturation of biotin binding sites, thus rendering the switchavidin unavailable for subsequent target binding ( Fig. 3A ). Despite initiatives to lower the biotin density in the matrix (as low as <20%), all attempts to sequentially immobilize biotinylated targets were highly inefficient.
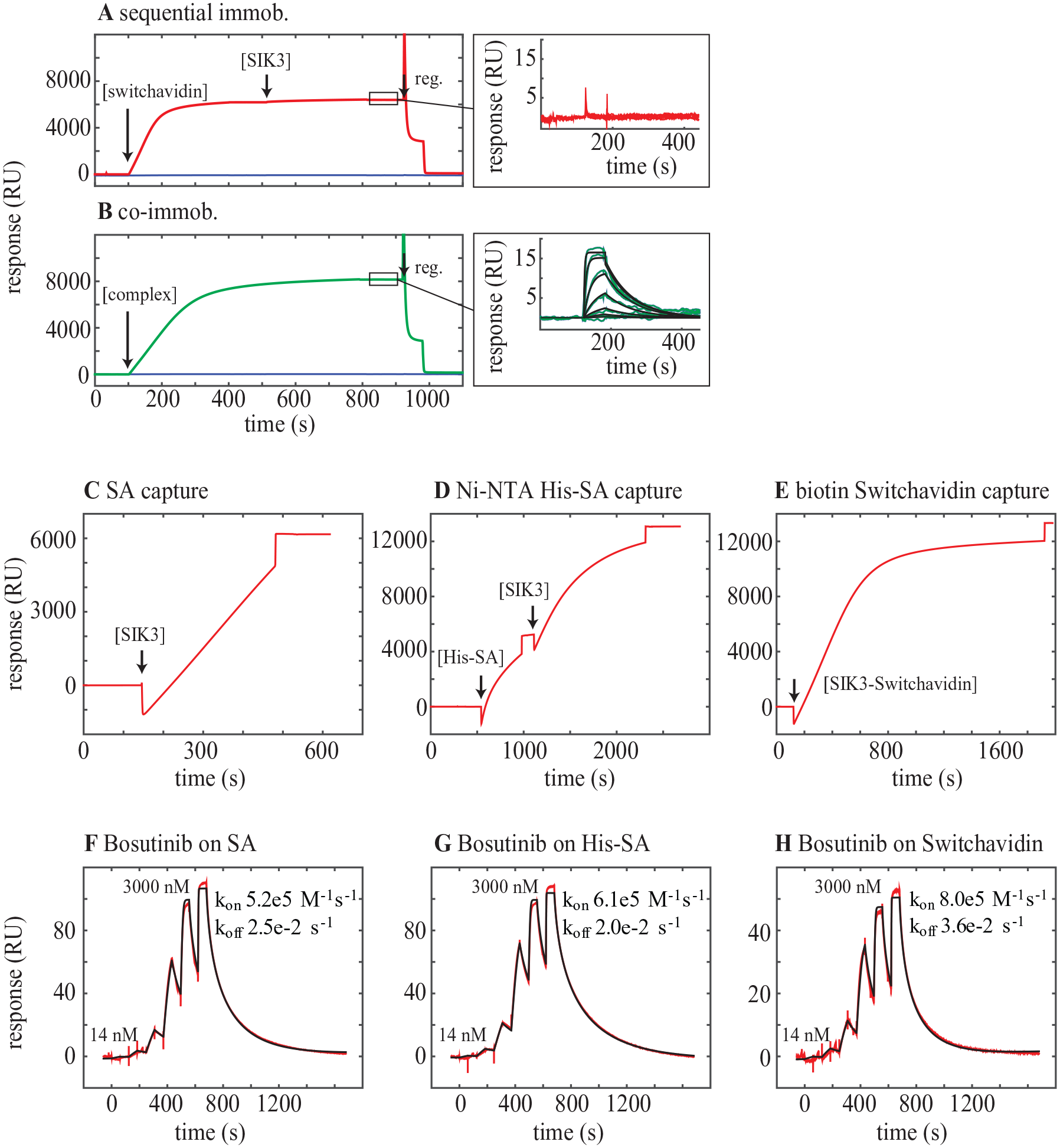
(
To overcome this complication, switchavidin can be premixed with the biotinylated protein of interest and the switchavidin–target complex is captured on the biotin surface ( Fig. 3B ). While the capture of 6000 RU of SIK3 to an SA surface, 6000 RU to a surface precaptured with His-SA, and 12,000 RU of the SIK3-SwA complex ( Fig. 3C–E ) results in almost identical binding profiles for the test compound bosutinib ( Fig. 3F–H ), the maximum response is lower for the SwA surface compared with the other two surfaces. This illustrates one drawback of premixing the proteins in that the binding capacity of the surface cannot be properly determined and immobilization levels cannot be used directly to assess how much of a protein has been immobilized.
For premixing, the molar ratio between switchavidin and biotinylated protein must be determined empirically, and typically, a ratio of 1:2 between the tetrameric switchavidin and single biotinylated target is optimal. The exact stoichiometry of the switchavidin–target complex was assessed using mass photometry,
19
a novel single molecule-based approach to determine the mass of protein complexes (
On a typical commercial Biotin chip (like BD200M20) it is possible to capture high densities (~10,000 RU) ( Fig. 3 ) of the switchavidin–target complex, sufficient for small-molecule binding characterization, with almost no observed baseline drift. In addition, the stabilization period for immobilization of the switchavidin–target complex is typically very short in contrast to His-tagged streptavidin, where longer stabilization periods are more common. A further advantage of using switchavidin is the possibility of using His-tagged protein as analytes, something that is essentially impossible when using the dual-His-tagged or His-tagged streptavidin immobilization approaches on Ni2+-NTA surfaces.
Surface Regeneration
Ni2+-NTA surfaces are typically regenerated using EDTA. While freshly captured His-SA or dual-His-tagged target proteins are almost fully regenerated using 0.5 M EDTA, we have observed that the regeneration is not always complete and residual protein builds up on the surface over time. In addition, capturing the protein and then not trying to regenerate it for hours makes EDTA regeneration even more inefficient. Hence, to be truly regenerable we needed to establish a more rigorous regeneration procedure. In our hands, the most effective regeneration protocol was a combination of short consecutive pulses of 0.5 M EDTA, a strong chaotropic agent with reducing agent (e.g., 5 M GuSCN, 5 mM TCEP), in situ pepsin digest (1 mg/mL pepsin in 1 M glycine, pH 2.5), and high pH with high ionic strength (50 mM NaOH, 1 M NaCl) ( Fig. 4 ). Using this procedure, inspired by Knoglinger et al., 21 on multiple proteins has consistently resulted in >99% regeneration efficiency with retained chip capacity. Some proteins only require certain steps in this procedure for complete regeneration, and optimized protocols can be tested on a target-by-target basis, but the full protocol ensures broad applicability.
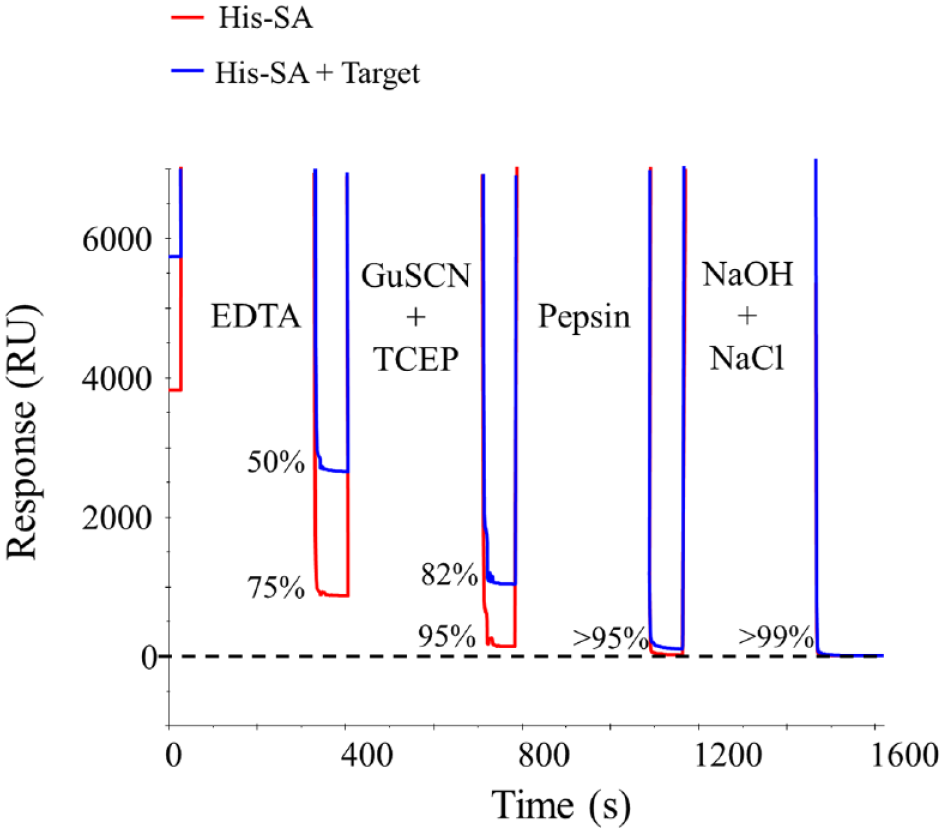
Full regeneration of His-SA (red) and His-SA plus target protein (blue) from a Ni2+-NTA surface requires a combination of 0.5 M EDTA, 5 M GuSCN with 5 mM TCEP, 1 mg/mL pepsin in 1 M glycine pH 2.5, and 50 mM NaOH with 1 M NaCl. The horizontal dashed black line indicates the surface response prior to the capture of any protein.
For switchavidin, the published regeneration procedure comprising 2.5% citric acid and 0.25% SDS
17
worked well with almost all proteins tested (
While these regeneration procedures might suggest an almost unlimited lifetime for a sensor chip, this is not the case. Most chips can be used repeatedly for weeks and even months, but they gradually lose capacity over time. Whether this is due to normal “wear and tear,” accelerated degradation as a result of the regeneration solutions used, or repeated docking/undocking of the sensor chip (undocked sensor chips are typically stored at 4 °C in a humid environment) is still unclear. Consequentially, the capacity of reused sensor chips must be monitored.
Examples of the Benefits with Regenerable Sensor Surfaces
Greatly Increased Throughput for Kinetic Characterization of Very Potent Kinase Inhibitors
Key benefits of the regeneration approach are clearly seen when looking at the profiling of a kinase inhibitor with a long residence time (koff from fit, ~3 × 10−5 s−1) (
Fig. 5
). The cycle time for such a compound based solely on dissociation (traditional approach) would be >24 h/injection. As seen from
Figure 4A
, the cycle time using the switchavidin approach is reduced to only around 40 min, increasing throughput to be >35-fold. Importantly, the kinetic parameters are consistent with data using traditional irreversible immobilization on a chip with covalently immobilized streptavidin (
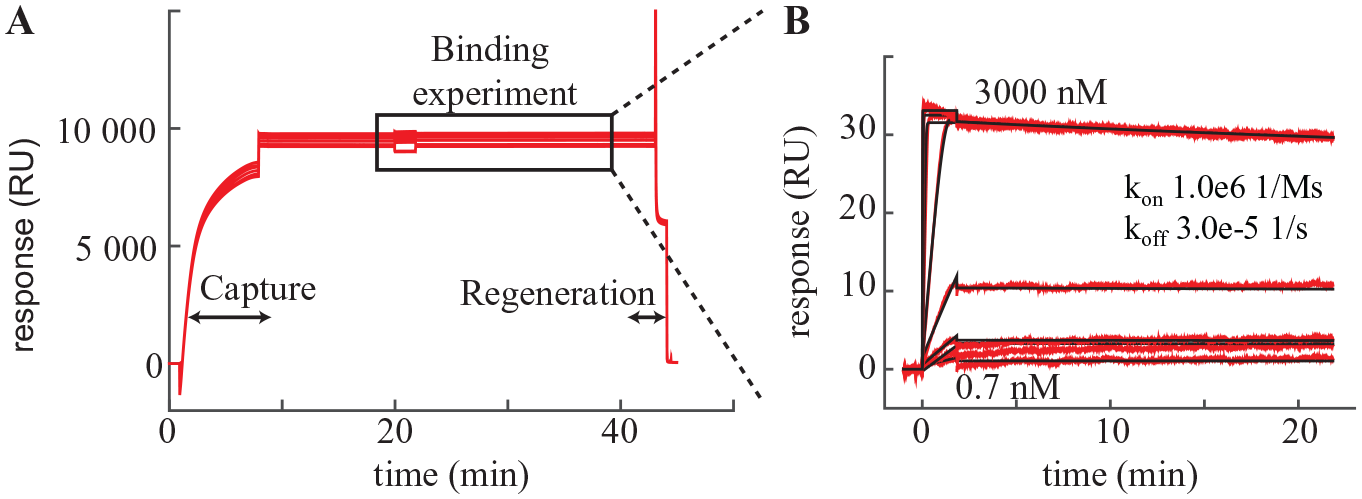
(
Kinetic Determination of the Reversible and Irreversible Steps of Covalent Inhibitors
The possibility of repeated regeneration cycles furnishes the opportunity to study the kinetics of irreversible inhibitors—the most extreme case of long residence time—as shown in
Figure 6
(
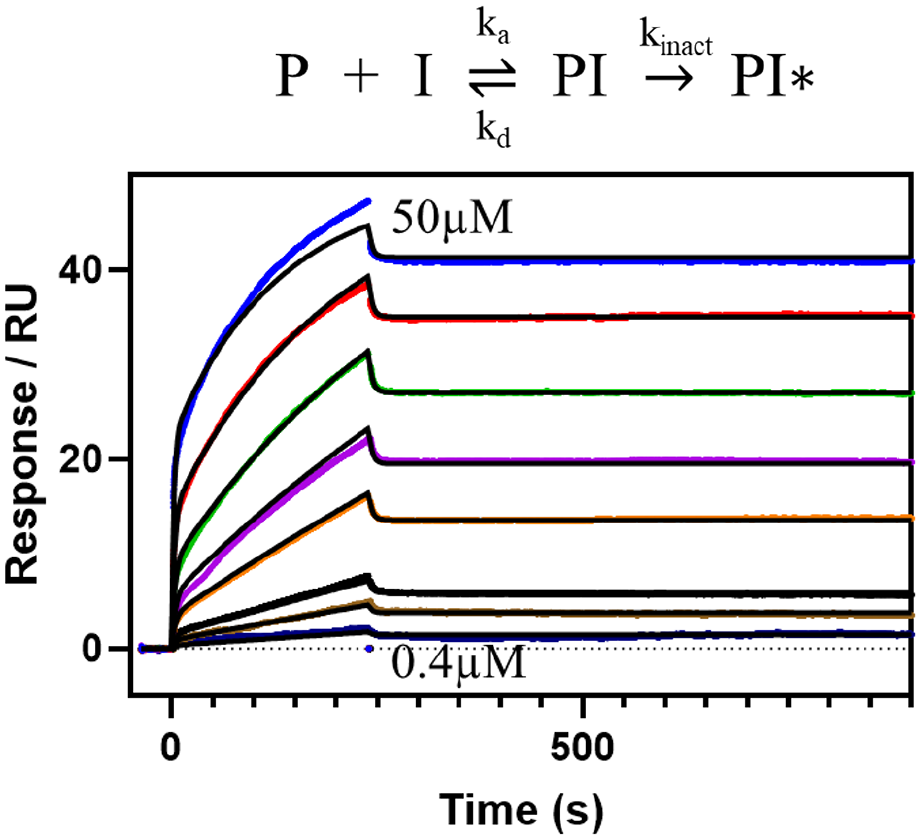
Model and SPR data of the binding of an irreversible PAD4 inhibitor (
No single immobilization strategy will work for all situations ( Table 1 ). They all come with advantages and limitations, but the three approaches investigated here add to the toolbox of available immobilization procedures. The requirements we listed come from a long history of measuring small-molecule interactions to immobilized protein targets and, as such, are probably representative of what the general SPR experimentalists require. That said, it is not possible to cover all situations or requirements, but we hope this will aid in the selection of an immobilization procedure.
It is important to point out that even though these immobilization procedures can increase throughput for slowly dissociating compounds, such measurements will still take time. In order to reliably quantify a dissociation rate, sufficient dissociation data must be collected (at least 5% dissociation, so 1.4 h for koff = 1 × 10−5 s−1), which limits how short these SPR measurements can become. Nonetheless, the main benefit remains: the possibility to regenerate afterward.
The switchavidin and His-streptavidin approaches share the common requirement of biotinylated target protein. This could be achieved via site-directed biotinylation (using, e.g., an Avi-tag 22 ), but can also be achieved chemically at neutral pH and ambient temperatures. 9 While the latter alternative provides less control of position and number of biotinylated residues, it offers the option to modify target proteins to be compatible with regeneration assays after expression/construct design.
While the immobilization strategies investigated in the current paper were chosen to increase the scope of SPR experiments that can be conducted, perhaps the ultimate gain in employing these approaches is cost savings. The SPR sensor chips are relatively expensive, while 1 mg of His-tagged streptavidin or switchavidin, enough for several hundreds of immobilizations, is relatively cheap to produce or purchase.
To summarize, the three investigated regenerable SPR immobilization procedures (dual-His-tag, His-tagged streptavidin, and switchavidin) show great potential in their use for small-molecule binding characterization. While there is no ultimate immobilization strategy, the addition of these to the immobilization toolbox will enable the study of additional targets, compound modes of action (e.g., irreversible), and a higher throughput for slowly dissociating compounds, enabling improved data-driven decision-making.
Supplemental Material
sj-pdf-1-jbx-10.1177_2472555220975358 – Supplemental material for Regenerable Biosensors for Small-Molecule Kinetic Characterization Using SPR
Supplemental material, sj-pdf-1-jbx-10.1177_2472555220975358 for Regenerable Biosensors for Small-Molecule Kinetic Characterization Using SPR by Anders Gunnarsson, Christopher J. Stubbs, Philip B. Rawlins, Eleanor Taylor-Newman, Wei-chao Lee, Stefan Geschwindner, Vesa Hytönen, Geoffrey Holdgate, Rupam Jha and Göran Dahl in SLAS Discovery
Footnotes
Acknowledgements
We thank Dr. Christopher Gray, Dr. Andrea Gohlke, Dr. Jennifer Konczal, and Dr. Justin Bower (Beatson Institute) for providing us with their double-His-tag vector and insightful discussions on the use of double-His-tags for protein immobilization.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors except V.H. are employees of AstraZeneca or were affiliated with AstraZeneca during this study.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.