
Abstract
Targeted protein degradation using heterobifunctional proteolysis-targeting chimera (PROTAC) compounds, which recruit E3 ligase machinery to a target protein, is increasingly becoming an attractive pharmacologic strategy. PROTAC compounds are often developed from existing inhibitors, and assessing selectivity is critical for understanding on-target and off-target degradation. We present here an in-depth kinetic degradation study of the pan-kinase PROTAC, TL12-186, applied to 16 members of the cyclin-dependent kinase (CDK) family. Each CDK family member was endogenously tagged with the 11-amino-acid HiBiT peptide, allowing for live cell luminescent monitoring of degradation. Using this approach, we found striking differences and patterns in kinetic degradation rates, potencies, and Dmax values across the CDK family members. Analysis of the responses revealed that most of the CDKs showed rapid and near complete degradation, yet all cell cycle–associated CDKs (1, 2, 4, and 6) showed multimodal and partial degradation. Further mechanistic investigation of the key cell cycle protein CDK2 was performed and revealed CDK2 PROTAC-dependent degradation in unsynchronized or G1-arrested cells but minimal loss in S or G2/M arrest. The ability of CDK2 to form the PROTAC-mediated ternary complex with CRBN in only G1-arrested cells matched these trends, despite binding of CDK2 to TL12-186 in all phases. These data indicate that target subpopulation degradation can occur, dictated by the formation of the ternary complex. These studies additionally underscore the importance of profiling degradation compounds in cellular systems where complete pathways are intact and target proteins can be characterized in their relevant complexes.
Introduction
Numerous diseases are linked or attributed to alterations in specific kinase activity, making kinases one of the largest classes of proteins for pharmaceutical development and drug discovery efforts.1–5 Kinase drugs consist primarily of small-molecule inhibitors, which inactivate kinases through targeting the adenosine triphosphate (ATP) binding site or as allosteric inhibitors.1–6 Given the highly related structures and shared domains within the kinome, obtaining highly specific and potent inhibitors is a challenge.3,5 With the emergence of targeted protein degradation as a new therapeutic modality, kinases were an immediately attractive target class, as degradation compounds oftentimes show higher efficacy and improved specificity as compared with their inhibitor counterparts.7–15 Indeed, initial comparison of kinase inhibitors with degraders supported this hypothesis,16–19 and many following studies have shown that kinases in general, including single-pass transmembrane receptor tyrosine kinases, are readily amenable for degradation using heterobifunctional proteolysis-targeted chimera (PROTAC) compounds.16–25
One of the major families within the kinome found to be degraded well with PROTAC compounds was that of the serine-threonine cyclin-dependent kinases (CDKs),19–25 which are key therapeutic targets for cancer and inflammation.1,4,26–31 CDKs regulate cell-signaling pathways, transcription, and transition through cell cycle and cell division.1,14,23,24,29–31 There are 21 family members of the CDK family, although only those involved in cell cycle (CDK1, 2, 4, and 6) and transcriptional regulation (CDK7, 8, 9, 12, and 13) are currently well characterized or understood.1,4,26–31 Even for well-studied CDKs, regulation of activity has shown to be highly complex and is governed by a diverse set of factors, including tightly controlled autoregulatory expression of expression or their binding partners, differential pairing combinations with cyclins or cyclin-like regulatory proteins, interactions with CDK interacting protein/kinase inhibitory proteins (CIP/KIPs), and posttranslational modifications such as phosphorylation or ubiquitination.1,27,30 How all of these various processes or stages might affect degradation efficiency or mechanism is unknown. However, the ability to develop CDK-specific degraders has been demonstrated, indicating that selectivity within the family is achievable.19,21–24 In addition, in an exciting recent development involving CDKs, it was demonstrated that a CDK inhibitor could function as a molecular glue degrader, bringing together the CDK12-cyclin K complex with DDB1, a component of the E3 CUL4 ring ligase (CRL4) complex, which then promoted the ubiquitination and degradation of cyclin K. 32
CDK degradation has also been observed in key landmark studies of pan-kinase inhibitors converted to PROTAC compounds.20,25 These data from independent groups revealed pan-kinase PROTACs selectively degraded only a small fraction of their kinases targets, despite being able to bind nearly all with similar affinity as the promiscuous parental inhibitors.20,25 These results showed that target degradation specificity could occur even when using pan-inhibitor precursors and supported a significant paradigm shift from inhibition, wherein binary target:drug interaction affinities, although necessary, were neither predictive nor correlative with degradation success.15,20,25,33–40 The results of this and similar studies substantially influenced PROTAC compound design and established the formation of a ternary complex between target, PROTAC, and E3 ligase as a key metric for successful PROTAC development.15,20,25,33–41 It was further hypothesized that favorable ternary complex formation was crucial for pan-kinase PROTACs to mediate target-specific degradation.20,25 Therefore, degradation specificity can be engineered by the chemical composition, as well as choice of E3 ligase component in PROTAC design, which has been supported by several other studies.33–35,38,39 The formation of the ternary complex, however, is only one of many steps critical for successful degradation, and further compound optimization is often required to enhance structurally favorable and active ternary complexes, which result in robust target protein ubiquitination and ultimately protein degradation.33–35,40
As kinase-targeted degradation efforts increase, assessing the specificity among the related subclass family members will be critical, as will confirmation of the mechanism of action for target-specific degradation and screening for any potential off-target liabilities. Here we present a detailed study of real-time degradation kinetics for a panel of 16 CDK family members in response to the pan-kinase PROTAC, TL12-186, 20 with the goal to better understand the specificity, efficacy, and temporal profile of the degradation response across this broad family class. The CRBN-based PROTAC TL12-186 was chosen for this study as it was shown to degrade numerous kinase targets in both MOLM-14 and MOLT-4 cell backgrounds, most notably nine total CDK family members in each cell line. 20 To enable our studies, we endogenously tagged each CDK protein in HEK293 cells, via CRISPR/Cas9, with the HiBiT peptide,42,43 which complements with LgBiT protein36,42–44 to produce luminescence directly proportional to each endogenous CDK protein level. These studies as well as additional mechanistic characterization of one of the key cell cycle regulators in the family, CDK2,4,26,27,31,45,46 allow for degradation selectivity profiling and characterization of the CDK family in response to this pan-kinase PROTAC. 20
Materials and Methods
Reagents and Cell Culture
TL12-186, nocodazole, 2,3-DCPE, and lenalidomide were purchased from Tocris (Bristol, UK), and TAE-684 and MG132 were purchased from Selleckchem (Houston, TX). HEK293 cells were obtained from the American Type Culture Collection and cultured at 37 °C, 5% CO2, in DMEM (Gibco, Waltham, MA) containing 10% fetal bovine serum (Seradigm, Radnor, PA). Rabbit anti-CDK2 antibody was purchased from Cell Signaling Technologies (Danvers, MA), and rabbit anti-tubulin antibody was purchased from Abcam (Cambridge, UK).
CRISPR/Cas9 Editing and CDK Cell Line Generation
The terminus of each CDK protein for tagging with HiBiT was previously chosen based on performance in a target engagement assay using ectopic expression. One nanomole Alt-R CRISPR-Cas9 crRNA and 1 nmol Alt-R CRISPR-Cas9 tracrRNA were assembled in 50 µL of nuclease-free duplex buffer (Integrated DNA Technologies [IDT], Coralville, IA) by incubation at 95 °C for 5 min and cooling to room temperature to generate gRNA for targeting each CDK. Single-stranded Ultramer DNA Oligos (IDT) were used as the ssODN donor templates. Ribonucleoprotein complexes with recombinant S.p. Cas9 Nuclease V3 (IDT) were assembled by incubating 100 pmol of Cas9 and 120 pmol of gRNA for 10 min at ambient temperature. A total of 2 × 105 HEK293 cells were resuspended in 20 µL of 4D Nucleofector Solution SF, and ribonucleoprotein complexes along with 100 pmol ssODN template were electroporated into cells with the 4D Nucleofector System (Lonza, Basel, Switzerland) using program CM-130. Immediately following electroporation, cells were incubated at ambient temperature for 10 min before transferring to a 12-well plate for culturing. At 72 h after electroporation, edited pools were analyzed for HiBiT insertion using the Nano-Glo HiBiT Lytic Detection System (Promega, Madison, WI) and for measuring luminescence on a GloMax Discover (Promega). Clonal populations of edited cells were obtained by sorting live singlets using a FACSMelody cell sorter (Becton Dickinson, Franklin Lakes, NJ) into 96-well plates. Clones were expanded and screened by luminescence, and genomic DNA was isolated using the Maxwell RSC Cultured Cells DNA kit (Promega). Genomic DNA was amplified using primers designed to anneal ~200 bases either upstream or downstream from the inserted ssODN template sequence, subcloned into a pF5K vector backbone (Promega), and transformed into JM109 cells (Promega), and 24 individual colonies per clonal population were sequenced by Sanger sequencing. Clones were selected based on 100% sequence conformity with no insertions or deletions in the HiBiT-containing open reading frame.
Kinetic Degradation Experiments
Clonal cell lines edited for HiBiT insertion to CDK family members and parental HEK293 cells were plated in white 96-well tissue culture plates at a density of 2 × 104 cells per well in 100 µL of growth medium with 5% v/v BacMam LgBiT particles (Promega) and incubated overnight at 37 °C, 5% CO2. The following day, the medium was replaced with CO2-independent medium (Gibco) + 10% fetal bovine serum (FBS) containing 1x Vivazine, an extended time-released substrate (Promega), and plates were incubated at 37 °C, 5% CO2, for 1 h before addition of a threefold serial dilution of 1 µM final concentration of TL12-186 or TAE-684 compounds. For cell cycle arrest treatments, CDK2-HiBiT and CDK10-HiBiT cells were transduced with LgBiT BacMam particles and plated at a density of 2 × 104 cells per well in 100 µL of growth medium and allowed to adhere for 6 h before medium replacement with Opti-MEM (Gibco) for serum starvation or addition of 20 µM final concentration of 2,3-DCPE or nocodazole overnight. The following day, medium was replaced with CO2-independent medium (Gibco) +10% FBS containing 1x Vivazine, and plates were incubated at 37 °C, 5% CO2, for 1 h before addition of a threefold serial dilution of 1 µM final concentration of TL12-186. For CDK2-HiBiT control experiments with lenalidomide, a 10 µM final concentration of lenalidomide was added to the plate 10 min before TL12-186 serial dilution. Plates retaining the plate lids were then read every 5 min for a period of 24 h on a GloMax Discover (Promega) set to 37 °C. Fractional relative light units (RLUs) were calculated at each time point, first by subtracting from all conditions the background RLU signal resulting from parental HEK293 control cells, and then dividing all treated conditions by the respective DMSO control for each CDK family member. Dmax values were determined in Excel by calculating the minimum fractional RLU obtained, and percentage degradation was expressed by calculating (1 − fractional RLU) × 100. Dmax50 values were calculated using a log(agonist) versus response model with a constrained Hill slope of 1 in GraphPad Prism.
Western Blot Analysis
Parental HEK293 cells were plated in 10 cm tissue culture dishes and were either untreated or treated with 1 µM TL12-186 for 24 h. Cells were rinsed once with ice-cold phosphate-buffered saline and lysed in 300 µL of Mammalian Lysis Buffer (Promega) supplemented with Protease Inhibitor Cocktail (Promega) for 5 min on ice. Lysates were cleared by centrifugation for 5 min at 14,000 × g. Sodium dodecyl sulfate (SDS) loading buffer was added, and samples were run on 4% to 20% Bis-Tris polyacrylamide gels by SDS–polyacrylamide gel electrophoresis. Proteins were then transferred to a polyvinylidene difluoride membrane using an iBlot 2 dry blotting system (Thermo Fisher Scientific, Waltham, MA) for 6 min. The membrane was blocked for 30 min with blocking buffer (5% bovine serum albumin in tris-buffered saline and Tween 20 [TBST]), and the primary antibody was added at a dilution of 1:500 for CDK2 and 1:1000 for β-tubulin. Primary antibodies were incubated at 4 °C overnight with gentle rocking. After three 5-min washes in TBST, anti-rabbit HRP antibody (Promega) was added in a blocking solution at a dilution of 1:2500, and the membrane was incubated at room temperature for 1 h. The membrane was washed three times for 5 min in TBST before adding enhanced chemiluminescence Western blotting substrate (Promega) for 5 min and imaging chemiluminescence on an ImageQuant LAS-4000 imager (GE Life Sciences, Chicago, IL).
NanoBRET Target Engagement
HEK293 CDK2-HiBiT cells transduced overnight with LgBiT BacMam particles were plated at a density of 2 × 104 cells/well in a white, 96-well assay plate and allowed to adhere for 6 h before medium replacement with Opti-MEM (Gibco) for serum starvation or addition of 20 µM final concentration of 2,3-DCPE or nocodazole overnight. The following day, medium was replaced with Opti-MEM (Gibco). TL12-186 compound dilution was prepared as a concentrated stock solution in DMSO (Sigma-Aldrich, St. Louis, MO) and further diluted in Opti-MEM to prepare working stocks. NanoBRET tracer K-10 was prepared at a working concentration of 20× in tracer dilution buffer and added to cells at a final concentration of 1 µM. Cells were equilibrated for 2 h with tracer and test compound before NanoBRET measurements. To measure NanoBRET in live cells, NanoBRET Target Engagement Substrate (Promega) was added, and filtered luminescence was measured on a GloMax Discover luminometer equipped with a 450 nm BP filter (donor) and 600 nm LP filter (acceptor) using 0.3 s integration time. milli-BRET units were calculated by multiplying the raw NanoBRET values by 1000.
NanoBRET Ternary Complex and Ubiquitination Assays
HEK293 CDK2-HiBiT cells (8 × 105) transduced overnight with 5% v/v LgBiT BacMam particles were bulk transfected with FuGENE HD (Promega) and 2 µg of HaloTag-CRBN or HaloTag-Ubiquitin (Promega) in 6-well plates. The following day, 2 × 104 transfected cells were replated into white 96-well tissue culture plates in the presence or absence of HaloTag NanoBRET 618 Ligand (Promega), and for ternary complex experiments, they were allowed to adhere for 6 h before medium replacement with Opti-MEM (Gibco) for serum starvation or addition of 20 µM final concentration of 2,3-DCPE or nocodazole and incubated overnight at 37 °C, 5% CO2. The following day, medium was replaced with Opti-MEM (Gibco) containing 1x Vivazine, an extended time-released substrate (Promega), and plates were incubated at 37 °C, 5% CO2, for 1 h before addition of DMSO or 1 µM final concentration TL12-186 compound. Plates were then read every 3 min for the indicated time frame on a CLARIOstar (BMG Labtech, Offenburg, Germany) set to 37 °C. Dual-filtered luminescence was collected with a 460/80 nm bandpass filter and a 610 nm long pass filter using an integration time of 0.5 s. Background-subtracted NanoBRET ratios expressed in milliBRET units were calculated by multiplying NanoBRET ratios by 1000, and fold increase in BRET was calculated by normalizing mBRET ratios to the average mBRET ratios for DMSO controls.
Results
To study endogenous protein degradation kinetics and to profile degradation selectivity among the CDK family of proteins with TL12-186,
20
we engineered HEK293 cells using CRISPR/Cas9 genome editing technology to append the small peptide tag HiBiT42,43 to either the N- or C-terminus of 16 CDK target genes. Four of the CDK proteins, CDK3, 15, 19, and 20, proved difficult for CRISPR insertion and/or HiBiT luminescent detection because of either unavailability of modification at the loci
44
or very low levels of endogenous expression in HEK293 cells. The obtained HiBiT CDK CRISPR clonal cell lines and associated guide and donor sequences are listed in
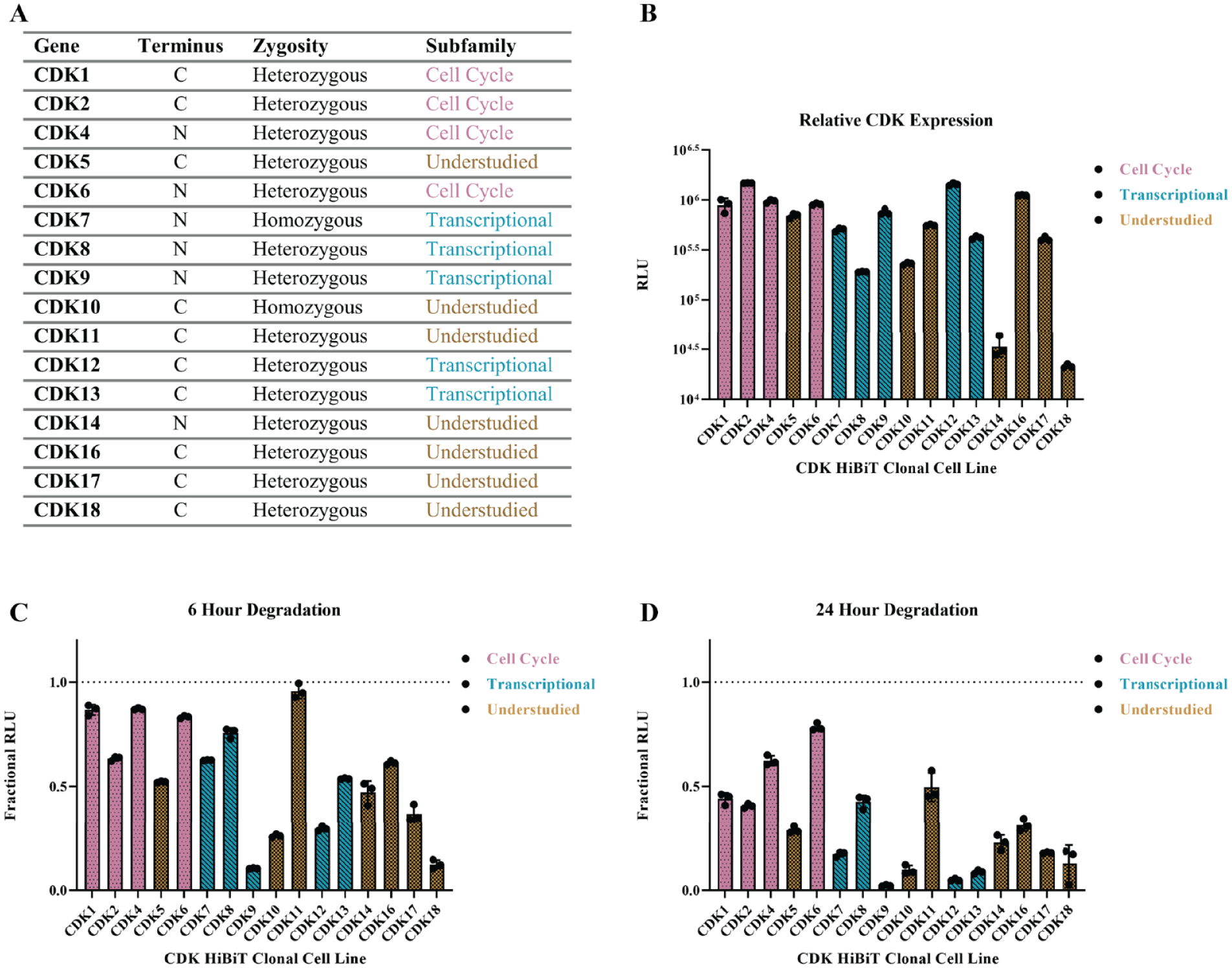
The HiBiT cyclin-dependent kinase (CDK) CRISPR panel in HEK293 cells. (
To more fully resolve the differences in temporal responses observed at 6 and 24 h across the CDK family, kinetic monitoring of a titration of 1 µM TL12-186 treatment was performed and revealed that all 16 CDKs exhibited dose-dependent protein loss (
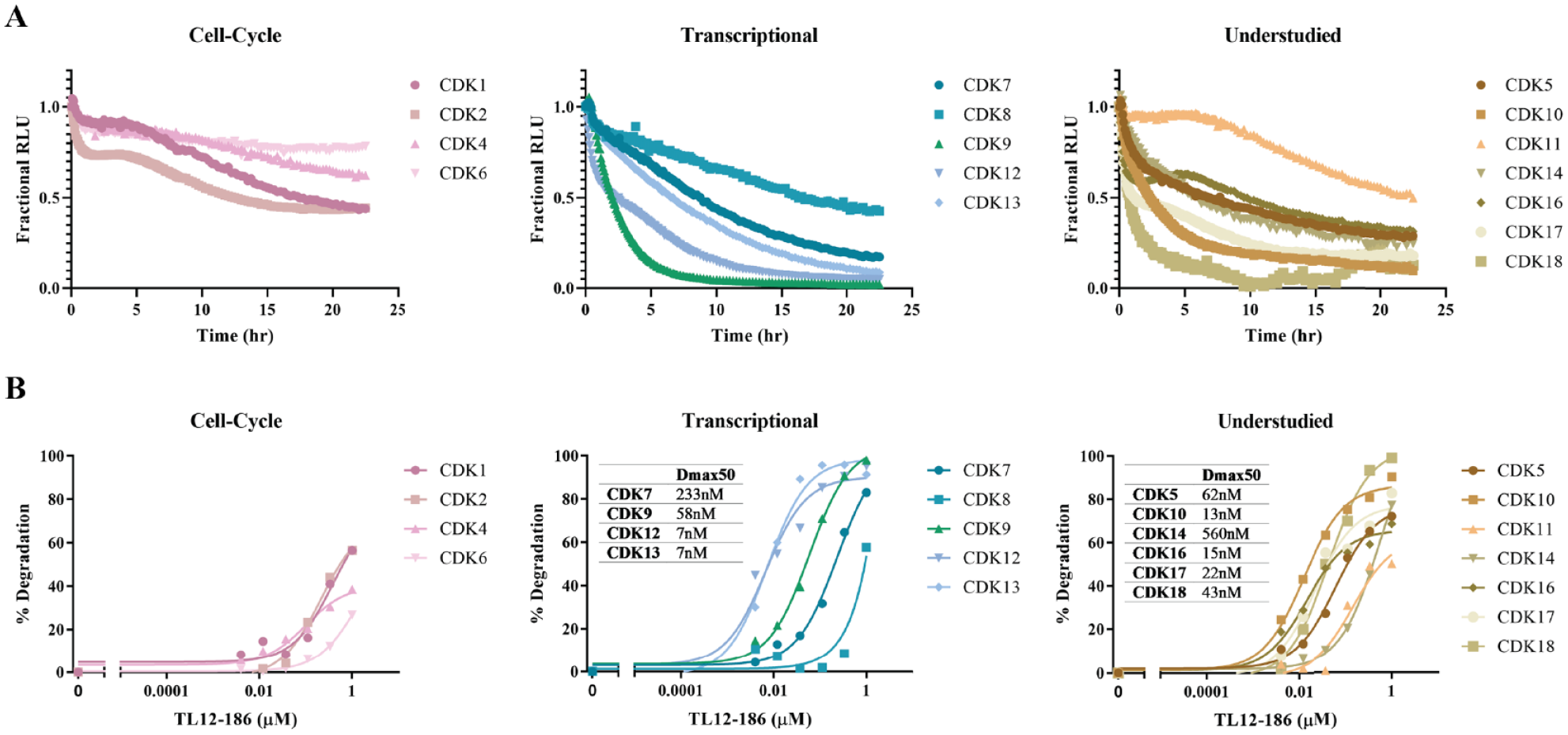
Clustering of cyclin-dependent kinase (CDK) kinetic degradation profiles and potency determinations. (
Because of the atypical degradation profile observed for all CDKs in the cell cycle subfamily, we sought to investigate whether the degradation kinetics could in part result from the differential regulation of these proteins throughout the cell cycle. Given its significant importance as a therapeutic target, CDK2 was chosen for further characterization of degradation mechanism, although we first wanted to confirm the dependency of the response on a direct, PROTAC-driven mechanism. To do so, kinetic degradation control experiments were performed with TL12-186 in the presence of a molar excess of lenalidomide (
As CDK2 is highly regulated throughout the cell cycle, has both active and inactive states, and is associated in various complexes with different cyclin protein partners, we sought to determine whether the degradation of CDK2 by TL12-186 could differ within the stages of the cell cycle. To this end, we investigated a series of overnight treatments to synchronize the cells in different phases of the cell cycle that consisted of serum starvation to arrest cells in the G1 phase
48
treatment with 20 µM 2,3-DCPE to arrest cells in the S phase or 20 µM nocodazole to arrest cells at the G2/M phase transition prior to the addition of TL12-186. Strikingly, we observed the greatest degradation of CDK2 following serum starvation (
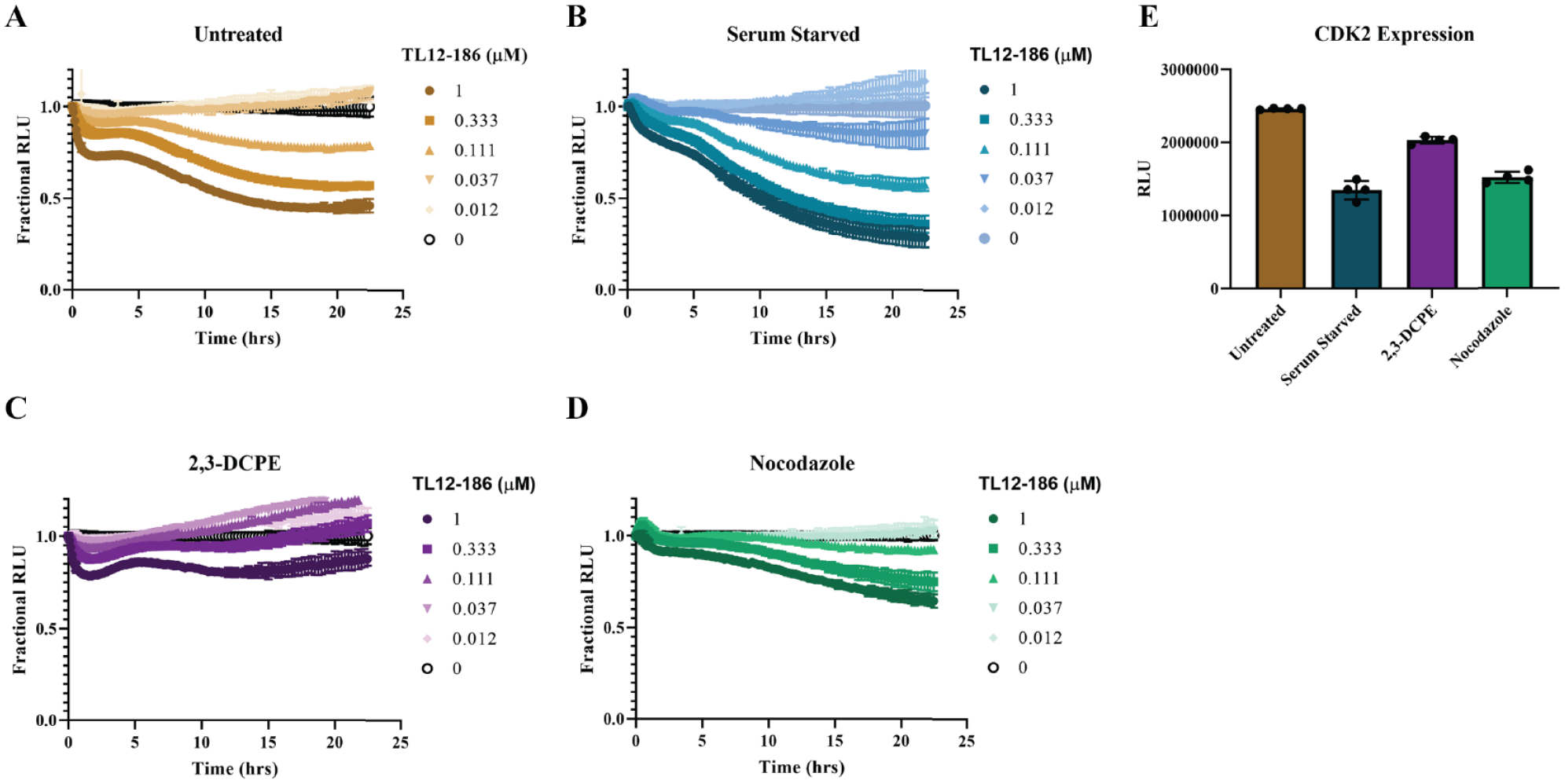
Cell cycle–dependent degradation of cyclin-dependent kinase 2 (CDK2). The 24-h kinetic degradation profiles of CDK2 by TL12-186 following (
To further understand the mechanism and deconvolute the cell cycle CDK2 degradation differences, we investigated the ability of TL12-186 in each phase to engage CDK2 as well as to form a ternary complex with CRBN. Using bioluminescence energy transfer to study the protein–small-molecule interaction by NanoBRET target engagement,
49
we interrogated TL12-186 binding to CDK2 in unsynchronized cells and in cells treated under the same conditions to induce arrest in G1, S, or G2/M. With the target engagement assays, we found that in all treatments, TL12-186 binding to CDK2 was similar, as revealed by quantification of IC50 in each cell cycle arrest condition (
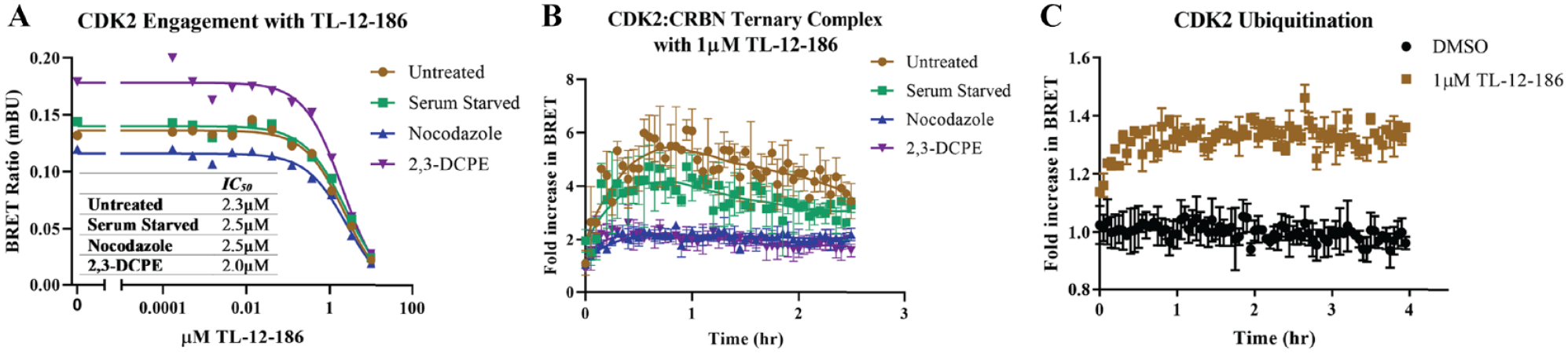
Mechanistic studies of cyclin-dependent kinase 2 (CDK2) degradation in various cell cycle phases. (
Discussion
We present here a comprehensive degradation kinetic study of 16 CDK family member in response to the pan-kinase PROTAC, TL12-186, in HEK293 cells. We observed degradation of all CDK family members tested, including the nine CDKs originally identified as degradable hits by TL12-186 in MOLM-14 and MOLT-4 leukemia cell lines. 20 Of those nine CDKs, we observed similarities among those showing the greatest degradation and potency within our study. In addition to the previously found targets, we observed degradation of additional CDK family members that were not detected in the aforementioned studies 20 using mass spectrometry detection. These discrepancies could be due to a variety of factors, as the studies differ in many aspects: cell background, time point of degradation assessment, concentrations of TL12-186 used, differential means of experimental design and detection, and also different controls used from which to reference protein loss. Given our findings of multimodal degradation of all the cell cycle regulatory proteins, it is also plausible that cell line–specific variations in the regulation of CDK activity and expression could also play a role in these differences.
These focused studies additionally underscore the advantage of live-cell, high-throughput kinetic screening panels to assess the degradation efficacy and specificity in the context of intact cellular pathways. The scalability of the CRISPR/Cas9 HiBiT-tagging approach 44 allows for broader panel analysis and readily allows for quantification of endogenous protein degradation dynamics 36 as compared with mass spectrometry or antibody-based methods. Before these comprehensive studies, degradation patterns and response clustering among CDK family members were not realized or understood. Coupling of the degradation profiles with NanoBRET functional assays allowed for further understanding of the PROTAC-driven mechanism from other means of protein loss as well as compound binding assessment to discriminate between on- and off-target protein loss and/or degradation.
The variety and complexity of degradation profiles observed in our study, particularly with the multimodal profiles of the cell cycle–associated CDKs, prompted us to investigate the mechanistic aspects of protein degradation of CDK2 in greater detail. To further understand CDK2 degradation in the presence of TL12-186, controls and experiments to understand mechanisms were critical to deconvolute the pathways contributing to protein loss. Monitoring the kinetic degradation of TL12-186 in the presence of excess lenalidomide showed that degradation was driven primarily by the PROTAC, but there also existed a component attributed to the parental kinase inhibitor, TAE-684. 47 Although further experiments are needed to fully understand CDK2 protein loss from the inhibitor, given its broad kinase-inhibition capabilities, this could be due to a variety of pathways, including transcriptional down-regulation. In the analysis of the differential cell cycle degradation of CDK2, it was found that CDK2 is most efficiently degraded in G1 as well as unsynchronized cells, the latter likely due to a high percentage of the global population within this phase, as it is the longest phase of the cell cycle. 48 We attribute the reduction in CDK2 degradation in S and G2/M to the inability to form a ternary complex with CRBN, which in turn suggests a potential structural incompatibility present in these phases, because neither CDK2 expression nor binding to TL12-186 are greatly impacted. As other PROTACs have been developed that can degrade CDK2,23,24 it would be intriguing to see if they also show cell cycle–dependent degradation differences and whether there are any trends with the observations made here. Interestingly, it is in the S and G2/M phases in which CDK2 is found in multiple higher-order complexes, including those with CIPs/KIPs.27,30,45,46 Perhaps it is the association with these inhibitory proteins that prevents the ternary complex formation with CRBN, but they do not preclude the binding to the PROTAC itself, as evidenced by the target engagement studies.
Although only CDK2 degradation mechanisms were further investigated here, we postulate that the remaining cell cycle CDK proteins, CDK1, 4, and 6, might also have cell cycle state-dependent degradation, given the similarity of their atypical degradation profiles to CDK2. If so, this, as well as the findings for CDK2, could have further implications for the design of PROTAC molecules and suggest that PROTACs could potentially be engineered not only to be target specific but even further nuanced in design to be target population specific. This would allow for targeting of proteins in active versus nonactive states, those found only when present in particular complexes or scaffolds, or specific populations at different subcellular localizations. The ability to achieve this type of selectivity by leveraging ternary complex formation in differential target states or complexes presents an exciting new avenue for PROTACs and could result in significant therapeutic benefits, potentially allowing for better discrimination between healthy and affected cells.
Supplemental Material
CDK_Supplemental_Revision – Supplemental material for CDK Family PROTAC Profiling Reveals Distinct Kinetic Responses and Cell Cycle–Dependent Degradation of CDK2
Supplemental material, CDK_Supplemental_Revision for CDK Family PROTAC Profiling Reveals Distinct Kinetic Responses and Cell Cycle–Dependent Degradation of CDK2 by Kristin M. Riching, Marie K. Schwinn, James D. Vasta, Matthew B. Robers, Thomas Machleidt, Marjeta Urh and Danette L. Daniels in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Author Contributions
K.M.R., M.U., and D.L.D. developed the strategy, designed the experiments, and interpreted data. M.K.S. and T.M. designed the guide RNAs and prepared all CRISPR/Cas9 endogenously tagged HiBiT CDK cell lines, including confirmation of expression, sequencing, and clonal selection. CDK family degradation profiles and quantifications of degradation were performed by K.M.R. NanoBRET CDK kinase target engagement assays were developed and optimized by J.V. and M.R. NanoBRET target engagement assays, CRBN ternary complex, and ubiquitination assays were performed by K.M.R. K.M.R. and D.L.D. prepared the figures and wrote the manuscript. All authors read, corrected, and approved the final manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Promega Corporation is the commercial owner by assignment of patents of the HiBiT, NanoLuc, HaloTag, and NanoBRET target engagement technologies and their applications.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All authors are supported and funded by Promega Corporation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.