
Abstract
Notch signaling is often involved in cancer cell initiation and proliferation. Aberrant Notch activation underlies more than 50% of T-cell acute lymphoblastic leukemia (T-ALL); accordingly, chemicals disrupting Notch signaling are of potential to treat Notch-dependent cancer. Here, we developed a flow cytometry-based high-throughput assay to identify compounds that disrupt the interactions of DNA and RBPJ, the major downstream effector of Notch signaling. From 1492 compounds, we identified 18 compounds that disrupt RBPJ-DNA interactions in a dose-dependent manner. Cell-based assays further revealed that auranofin downregulates Notch-dependent transcription and decreases RBPJ–chromatin interactions in cells. Most strikingly, T-ALL cells that depend on Notch signaling for proliferation are more sensitive to auranofin treatment, supporting the notion that auranofin downregulates Notch signaling by disrupting RBPJ-DNA interaction. These results validate the feasibility of our assay scheme to screen for additional Notch inhibitors and provide a rationale to further test the use of auranofin in treating Notch-dependent cancer.
Keywords
Introduction
The Notch signaling pathway is critical for cell-fate determination and animal development. 1 -3 Notch is a cell-surface receptor. Through cell–cell interaction, a Notch ligand binds to a Notch receptor, which leads to a series of cleavage events and the release of the Notch intracellular domain (NICD). NICD enters the nucleus, interacts with its primary downstream transcription factor RBPJ (also known as CSL, Su(H), CBF1, and LAG1), and initiates Notch-dependent transcription activation together with additional transcription coactivators. A large body of evidence has revealed that aberrant Notch signaling plays a key role in carcinogenesis and tumor progression. 4 -18 For example, aberrant Notch activation accounts for more than 50% of T-cell acute lymphoblastic leukemia (T-ALL). 19,20 Therefore, the Notch signaling pathway has emerged as a therapeutic target of great interest for cancer treatment. 21 -25
There is a need to identify new drugs that target Notch signaling. To date, Notch-pathway targeting drugs have been used in clinical trials and preclinical evaluations; these drugs target either a receptor, ligand, or the γ-secretase complex that performs the final cleavage step necessary for receptor activation. 26 However, the γ-secretase complex is not specific to Notch receptors. 27 An alternative to inhibiting the γ-secretase complex is the use of antiligand or antireceptor monoclonal antibodies. 28 -31 Some antibodies have been developed and are currently being evaluated in clinical trials. Gastrointestinal toxicity is a known on-target side effect associated with the use of Notch inhibitors. Strategies like lowering drug doses or including drug holidays during treatment are used to remedy this toxicity.
Studies over the past two decades have shown that direct targeting of the Notch transcription complex in the nucleus might help overcome the limitations and toxicities associated with the use of γ-secretase inhibitors (GSIs) and monoclonal antibodies. However, as the surfaces of transcription factors generally lack high-affinity binding sites for small molecules, pharmacological targeting of the Notch transcription complex remains challenging. 32,33 Using a combination of molecular docking of small molecules and in vitro complex formation assays, Astudillo et al. identified such a small molecule, IMR-1. 33 IMR-1 selectively inhibits Notch-dependent transcription activation by preventing the binding of Maml1 to the Notch-activating complex, and impairs tumor growth in patient-derived xenograft models. 33 IMR-1’s therapeutic effects are currently being evaluated in clinical trials. More recently, Hurtado et al. identified one small-molecule inhibitor of the RBPJ protein RIN1, which disrupts RBPJ function as both a transcription activator and repressor; 34 it remains to be determined to what extent RIN1 works in cancer therapy.
While there are multiple Notch receptors and ligands, the vast majority of Notch signaling is carried out by the sole DNA sequence-specific transcription factor RBPJ. Modulating RBPJ-DNA interactions is therefore expected to downregulate aberrant Notch activation resulting from Notch receptor or ligand overexpression, gain-of-function mutations, or chromosomal translocations of all isoforms. X-ray crystallographic structure studies indicate that RBPJ binds to DNA via its beta-trefoil domain (BTD). RBPJ specifically recognizes its cognate DNA site through a combination of major- and minor-groove contacts that achieve tight binding coupled with DNA sequence specificity. 35 Furthermore, RBPJ is found to bind to mitotic chromatin, and RBPJ has been suggested to transmit Notch signaling through cell division. 36,37
Here we developed a robust, flow cytometry-based high-throughput assay to monitor RBPJ-DNA interactions. Our strategy is to bind a GST-RBPJ fusion protein to polystyrene glutathione microspheres, which can be used with flow cytometry. Fluorescently labeled DNA containing an RBPJ-binding motif is then added. DNA binding to RBPJ is subsequently analyzed by flow cytometry using the HyperCyt platform. 38,39 Our screens of 1492 compounds, followed by cell-based assays, have identified the compound auranofin that disrupts RBPJ-DNA interaction, downregulates Notch-dependent transcription activation, and preferentially inhibits proliferation of T-ALL cells with active mutations in the NOTCH genes.
Materials and Methods
GST-RBPJ Protein Purification
The GST-RBPJ protein was overexpressed and purified from insect SF9 cells using affinity chromatography, as described previously. 37 GST-RBPJ was further purified over a Superose 6 column in buffer containing 20 mM HEPES (pH 7.9), 100 mM KCl, 10% glycerol, 0.02% Triton-X, and 0.2 mM EDTA.
Preparation of the Cy5-Labeled DNA Probe Containing the RBPJ-Binding Consensus
Nineteen-base pair (bp) 5′ Cy5-labeled DNA oligo GTTACTGTGGGAAAGAAAG and its complementary oligo were from Integrated DNA Technologies (Coralville, IA). The two oligos were annealed in buffer containing 100 mM NaCl, 50 mM Tris-HCl, and 10 mM MgCl2. The identical DNA oligos without Cy5 were prepared the same and used as the unlabeled DNA probe.
General Assay Plate Assembly
SPHERO glutathione-coated particles (cat. GSHP-40-5; Spherotech, Lake Forest, IL) with immobilized GST-RBPJ were used in the screen. Bead coupling was achieved by incubating ∼6 µg of the GST-RBPJ protein with ∼ 5 million GSH beads in phosphate-buffered saline (PBS) containing 100 µg/mL salmon sperm DNA and 0.02% Triton X-100 at 4 °C overnight with rotation. The beads were washed once with PBS containing 0.02% Triton X-100 and once with the assay buffer (0.012% Triton X-100, 0.12 mM EDTA, 20 mM NaCl, 60 mM KCl, 12 mM HEPES [pH7.9], 10 mM Tris [pH 8.0], 6% glycerol, 7 mM MgCl2, 250 µg/mL bovine serum albumin (BSA), and 0.2 mM DTT) and resuspended in 2.5 mL of assay buffer. Assay plates were Greiner polystyrene 384-well, low-volume plates (cat. 784101; Greiner Bio-One North America Inc., Monroe, NC). The total volume in each assay well was 10 µL. The final concentration of the Cy5-labeled DNA probe was 90 nM, and the unlabeled DNA probe was 45 µM as a positive control. Additions of GST-RBPJ-coupled beads and the Cy5-DNA probe were done using a BioTek liquid dispenser (BioTek, Winooski, VT).
Ten nanoliters of each compound was dispensed into experimental wells using an ECHO 555 (Labcyte Inc., San Jose, CA); an equal amount of DMSO was added to the control wells. Four microliters of assay buffer was added to assay wells and the negative control wells; positive control wells received 4 µL of 112.5 µM unlabeled DNA. Five microliters of coupled beads (2000 beads/µL) were added to all assay wells. Plates were incubated at room temperature for 30 min with rotation, followed by the addition of 1 µL of 900 nM of the Cy5-DNA probe. Plates were incubated at room temperature with rotation for 60 min and the relative binding of the Cy5-DNA probe to coupled beads was analyzed by the HyperCyt platform attached to a BD Accuri flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ).
HyperCyt High-Throughput Flow Cytometry
The HyperCyt system interfaces a flow cytometer and an autosampler, creating a single stream of bubble-separated samples. The samples are delivered to the cytometer and the data accumulate as a single, time-resolved file. The acquired data file is parsed into with specialized software that divides the data stream into 384 separate clusters that are analyzed as separate samples. Library compound information is merged with sample data using spreadsheet templates.
Chemical Libraries
Chemicals used in our screens were the 1280 primarily off-patent drugs of the Prestwick Chemical Library (Prestwick Chemical, Parc d'innnovation, ILLKIRCH, France) and 212 powders of on-patent drugs from MedChemExpress 2015 selection (MedChemExpress LLC, Monmouth Junction, NJ), compiled by Drs. Ursa and Opera.
Cell Culture
The T-ALL cell lines, DND-41 (ACC 525), HPB-ALL (ACC 483), and TALL-1(ACC 521), were purchased from Leibniz Institute DSMZ–German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) and cultured in RPMI 1640 (cat. 10-040-CV; Corning, Corning, NY) medium supplemented with 15% fetal bovine serum (FBS; cat. 97068-085; VWR, Radnor, PA). Jurkat and CUTLL1 cells were obtained from Dr. Matlawska-Wasowska (University of New Mexico [UNM]). CCRF-CEM (ATCC CCL-119) and MOLT-4 (ATCC CRL-1582) were obtained from ATCC and cultured in RPMI 1640 medium supplemented with 15% FBS. HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS. F9 cells (ATCC CRL-1720) were obtained from ATCC (Gaithersburg, MD) and cultured in DMEM supplemented with 10% FBS.
Constructs, Transfection, Luciferase Assays, and Cell Proliferation Assays
The plasmid 4xCSL-luciferase was a gift from Raphael Kopan (Addgene plasmid, cat. 41726; http://n2t.net/addgene:41726;RRID:Addgene_41726), 40 which contains a minimal promoter and four RBPJ high-affinity sites (CGTGGGAA) driving the firefly luciferase gene. NICD cDNA was amplified from the pENTR-NICD1 construct (pENTR-NICD1), a gift from Xin Chen (Addgene plasmid, cat. 4604; http://n2t.net/addgene:46048;RRID:Addgene_46048) 41 and cloned into the UCOE expression vector containing the Flag epitope. The plasmid TK-Rluc containing the TK promoter driving the Renilla luciferase (R-luc) was a kind gift from Dr. S. Ness (UNM). HEK293T cells were transfected with the above constructs as described in the Results section. Forty-eight hours posttransfection cells were treated with varying amounts of chemicals for 24 h; the expression of the reporters was analyzed using a Firefly & Renilla Luciferase Single Tube Assay Kit (Biotium, Fremont, CA). Cell proliferation was assayed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI).
ChIP, Gene Expression, and Real-Time PCR Experiments
HEK293T cells were transfected with NICD and 4xRBPJ-firefly luciferase. Forty-eight hours posttransfection, these cells were treated with 2.5 µM auranofin for 24 h and subjected to chromatin immunoprecipitation (ChIP) and gene expression analyses. Murine F9 cells were treated with 2.5 µM auranofin for 24 h and subjected to ChIP.
ChIP experiments were carried out as previously described. 37,42 Briefly, ∼4 million cells were collected after treatment, fixed, and processed for sonication. The fixed chromatin was sonicated on ice for 12 cycles (30 s on/90 s off) with 40% amplitude using a Branson Sonifier 150 T. Polyclonal anti-RBPJ antibody (FC31) 37 and CTCF antibody (cat. 07-729; EMD Millipore, Burlington, MA) were used at 1:100. Five microliters of protein A-agarose beads (Fisher, cat. 20334) were used in each ChIP of 500 μL. ChIPed DNA was analyzed by real-time PCR using a MyiQ system (Bio-Rad, Hercules, CA) and SYBR green.
Total RNA was extracted from cells using TRIzol reagent (Thermo Fisher, Waltham, MA). cDNA was synthesized by reverse transcription of total RNA (1 μg) using a SensiFAST cDNA synthesis kit (Bioline, Memphis, TN) following the manufacturer’s protocol. Expression levels were assayed by real-time PCR and normalized to beta-actin. Real-time PCR data were analyzed using the ΔΔCt method.
43
All primers used for quantifying RNA levels and ChIP DNA are listed in
Chemicals Used for Cell-Based Assays
(R)-Lansoprazole (HY-13662B), benserazide (HY-B0404A), tigecycline (HY-B0117), Methacycline (HY-B0449), bedaquiline (HY-14881), ceftibuten (HY-B0698), disulfiram (HY-B0240), R-(–)-apomorphine (A4393-1G), ebselen (HY-13750), and fenoldopam (HY-B0735) were purchased from MedChemExpress. Auranofin (cat. 15316) was purchased from Cayman Chemical Company (Ann Arbor, MI).
H2O2 Measurement
H2O2 was quantified using the ROS-Glo H2O2 assay (Promega, cat. G8820) according to the manufacturer’s instructions. Briefly, 4 × 104 cells were transferred to white F96 MicroWell plates (Nunc, cat. 136101) and incubated for 2 h in the presence of 20 µL of H2O2 substrate solution. One hundred microliters of ROS-Glo detection solution was subsequently added to the plate. After 20 min of shaking at room temperature, luminescence signal was measured using a Synergy Neo2 Hybrid Multi-Mode Reader (BioTek) with autogain.
Results
Flow Cytometry-Based Assay to Monitor RBPJ-DNA Interactions
We developed a flow cytometry-based method to monitor RBPJ-DNA interactions. RBPJ was expressed as a GST-fusion protein ( Fig. 1A ) and immobilized on glutathione microspheres. A Cy5-labeled 19 bp DNA fragment containing the RBPJ-binding consensus was used as a probe. To determine if the binding of RBPJ to the Cy5-DNA probe was specific, we compared the interactions of RBPJ and the Cy5-DNA probe in the absence or presence of 50-fold of the unlabeled DNA probe with an RBPJ-binding motif ( Fig. 1B ). If the binding of RBPJ to the Cy5-DNA probe is specific, this interaction can be competed away by an excess amount of unlabeled DNA probe in the binding reaction, resulting in decreased Cy5 signals. The green line represents signals from the RBPJ-bound DNA probe at different concentrations of the Cy5-DNA probe in the absence of unlabeled DNA ( Fig. 1B ). The black line represents the signals from the RBPJ-bound DNA probe at different concentrations of the Cy5-DNA probe in the presence of 50-fold unlabeled DNA. The specific binding of RBPJ to the Cy5-DNA probe is shown as a purple line, which is the difference of the Cy5 signal with versus without unlabeled DNA probe ( Fig. 1B , green minus black).
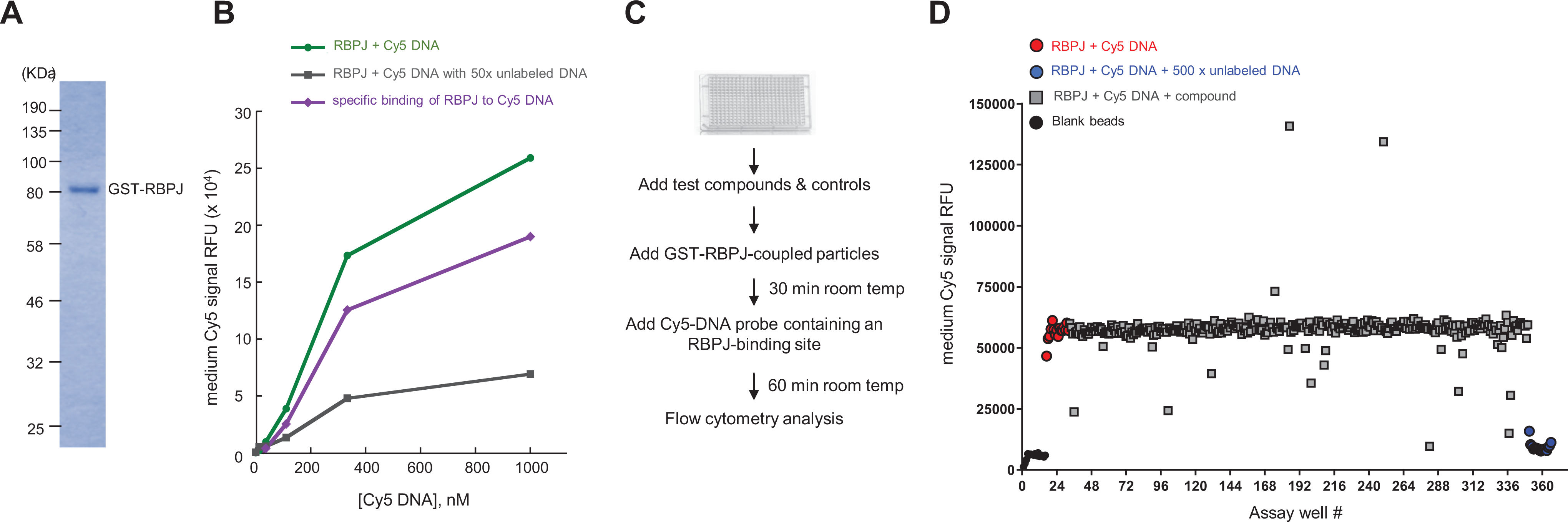
The flow cytometry-based screen for identifying compounds that inhibit RBPJ-DNA interactions. (
To gain insight into whether our assay was selective for the interaction of RBPJ with DNA containing the RBPJ-binding motif or to DNA in general, we compared how DNA with or without the RBPJ-binding motif competed with the Cy5-DNA probe for RBPJ interaction. As shown in
We chose to use 90 nM of the Cy5-DNA probe (which is half of the K
d determined by Friedmann and Kovall
44
) in our screening because at this concentration we detected low nonspecific binding, and that reduces false-negative rates. To determine the robustness of our assay, we calculated the Z′ value by measuring the fluorescence signals of immobilized GST-RBPJ in the presence of a 90 nM Cy5-labeled DNA probe with or without 45 μM of the unlabeled DNA probe as competitor (
Fig. 1C
If any of these compounds truly inhibit RBPJ-DNA interaction, they are expected to alter the Cy5-DNA binding of RBPJ in a dose-dependent manner. Accordingly, we determined how the 37 compounds altered the Cy5-DNA binding of RBPJ using nine concentrations, ranging from 10–9 M to 10–4 M. Nineteen compounds did not show a dose response and were discarded. Eighteen of the 37 compounds decreased the Cy5 signals in a dose-responsive manner with EC50 (concentration of a drug that gives half-maximal response) ranging from 30 nM to 20 μM ( Fig. 2 ).
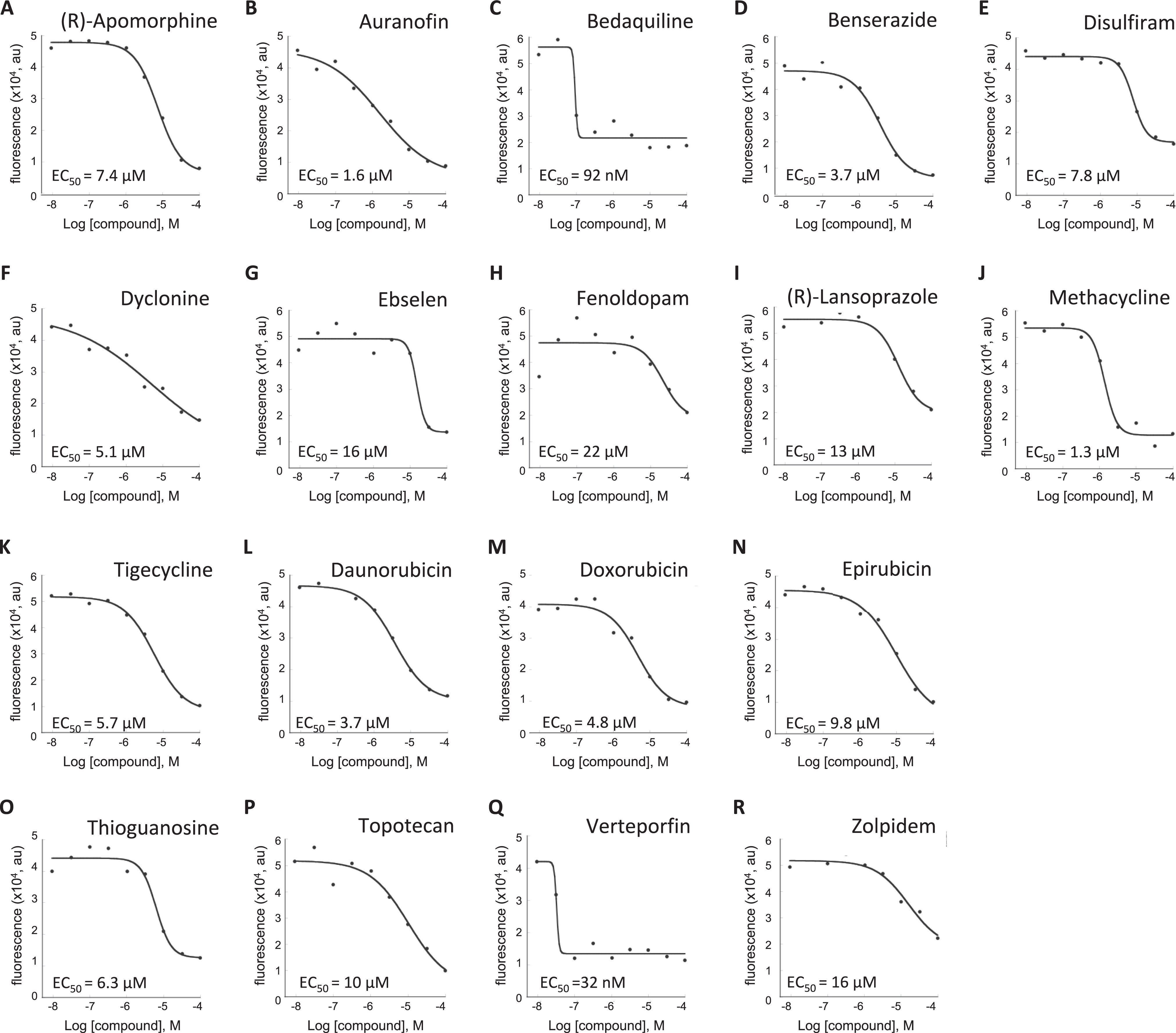
Dose–response analysis of compounds on RBPJ-Cy5 DNA probe interactions. (
Cell-Based Assays to Determine the Impacts of Candidate Compounds on Notch Signaling
The 11 compounds shown in
Figure 2A
If any of these compounds disrupt RBPJ-DNA interactions in cells, they are expected to decrease Notch-induced transcription activation. Therefore, we tested the compounds for their ability to disrupt Notch-dependent gene expression using dual luciferase reporter assays (
Fig. 3A
): firefly luciferase was driven by a minimal promoter containing 4× RBPJ-binding sites; R-luc, used as a normalization control, was driven by a constitutive thymidine kinase (TK) gene promoter. We activated Notch signaling in HEK293T cells by transfecting plasmid DNA containing an active NOTCH1 gene fragment (NICD). In cells without NICD, no firefly luciferase was upregulated. Firefly luciferase activity was normalized by R-luc to control for transfection efficiency or any general impact of the treatments on transcription (
Fig. 3A
). Out of the 11 compounds, auranofin (
Fig. 3B
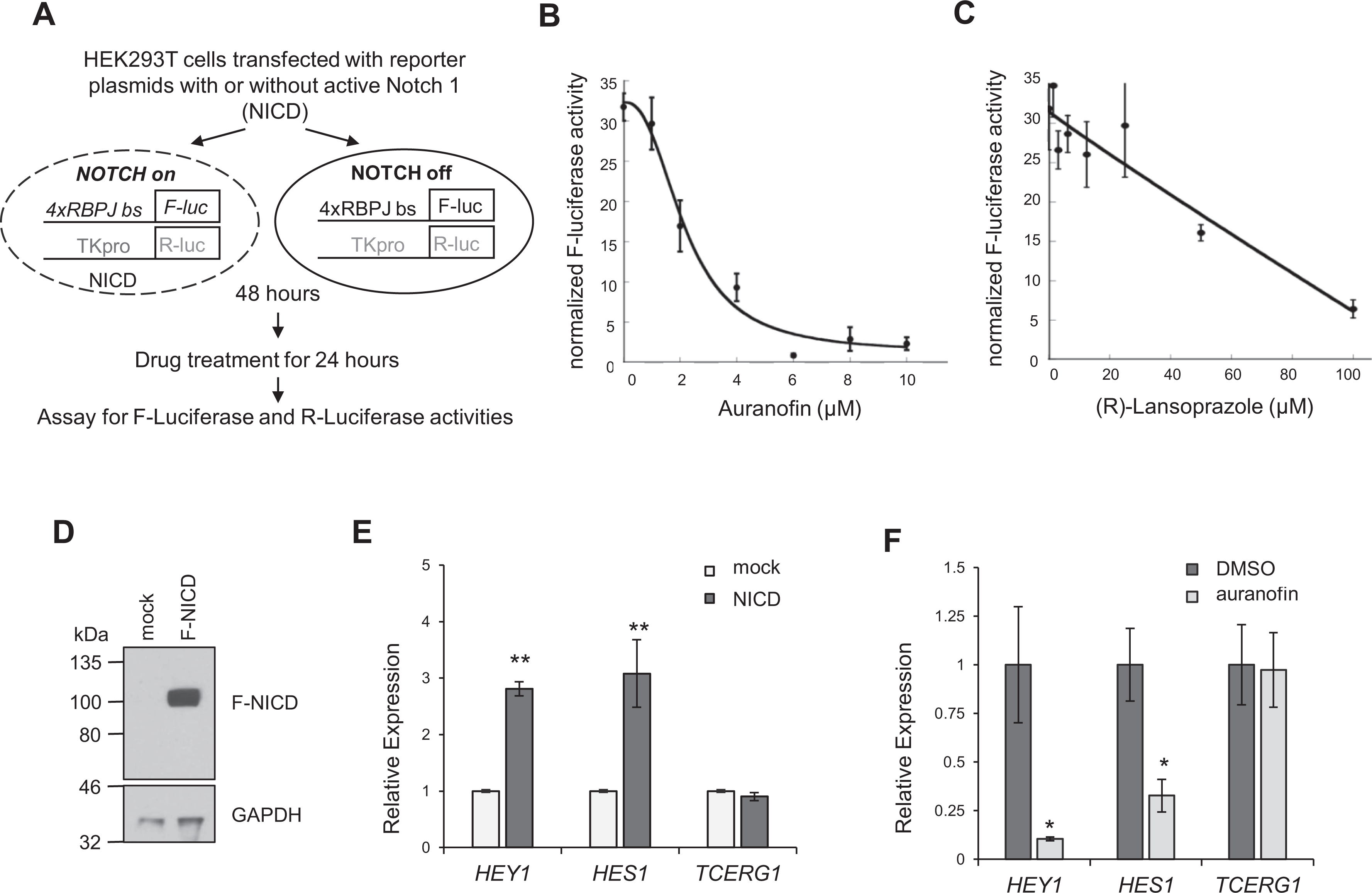
Effects of compounds on Notch-dependent transcription activation
in HEK293T cells. (
We next determined the effect of auranofin on Notch-dependent transcription from endogenous loci of HEK293T cells. We activated Notch-dependent gene expression by transfecting NICD into HEK293T cells; we detected a threefold increase of the two classic Notch target genes, HEY1 and HES1 (
Fig. 3D
To test the hypothesis that auranofin downregulates Notch-dependent transcription activation by disrupting RBPJ-DNA interactions, we used anti-RBPJ ChIP followed by real-time PCR (ChIP-qPCR).
37
As shown in
Figure 4A
, RBPJ was significantly enriched at the 4× RBPJ-binding sites present on the reporter construct (
Fig. 3A
). Of great interest, auranofin reduced RBPJ occupancy at this locus by 50%, consistent with results from the dual luciferase assay shown in
Figure 3B
. To rule out the possibility that the decreased RBPJ occupancy at the 4× RBPJ-binding site was because auranofin decreased RBPJ levels, we determined RBPJ protein levels in cells with or without auranofin treatment. As shown in
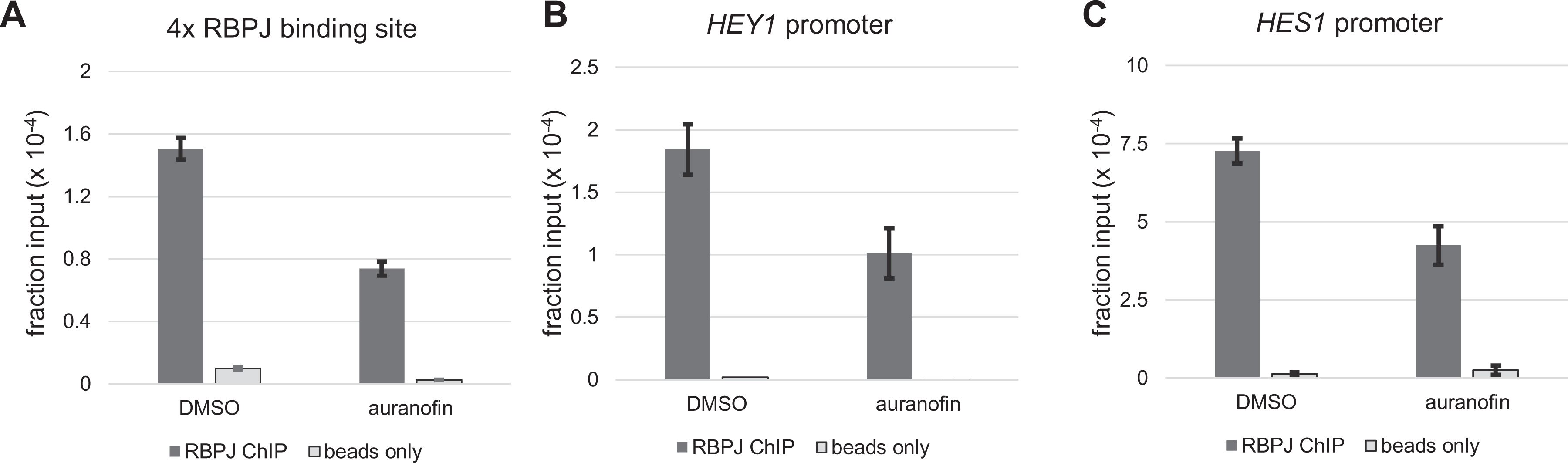
Auranofin decreases RBPJ-DNA interactions in HEK293T cells transfected with NICD. Notch signaling was activated in HEK293T cells by transfecting NICD. Forty-eight hours posttransfection, cells were treated with 2.5-µM auranofin for 24 h and then subjected to anti-RBPJ ChIP-qPCR. (
We next tested how auranofin impacts RBPJ occupancy at endogenous Notch target genes. We found that RBPJ bound to the Notch-dependent HEY1 and HES1 promoters in HEK293T cells expressing NICD (
Fig. 4B
Auranofin Also Disrupts RBPJ-DNA Interactions in Notch-Inactive Cells
To test if auranofin disrupts RBPJ-DNA interactions when Notch signaling is off, we used the murine F9 cells because the RBPJ-binding sites are known and Notch signaling is off in these cells. 36,37 F9 cells were treated with 2.5 µM auranofin for 24 h and subjected to ChIP analysis. RBPJ binds to the promoters of HES1 and ZFP334 in a DNA sequence-specific manner; on the other hand, RBPJ occupies the NAPRT1 and NANOG loci, in part, through the CTCF protein in a sequence-nonspecific manner. 36,37 Actin B is a negative region for RBPJ and CTCF binding. As shown in Figure 5A , auranofin decreased RBPJ occupancy at the promoters of HES1 and ZFP334. Of interest, sites that RBPJ occupies, in part, through the CTCF protein, such as NANOG and NAPRT1, were not effectively disrupted by auranofin ( Fig. 5 ). These results support the notion that auranofin preferentially disrupts the interactions of RBPJ and DNA in a sequence-specific manner regardless of the status of Notch signaling.
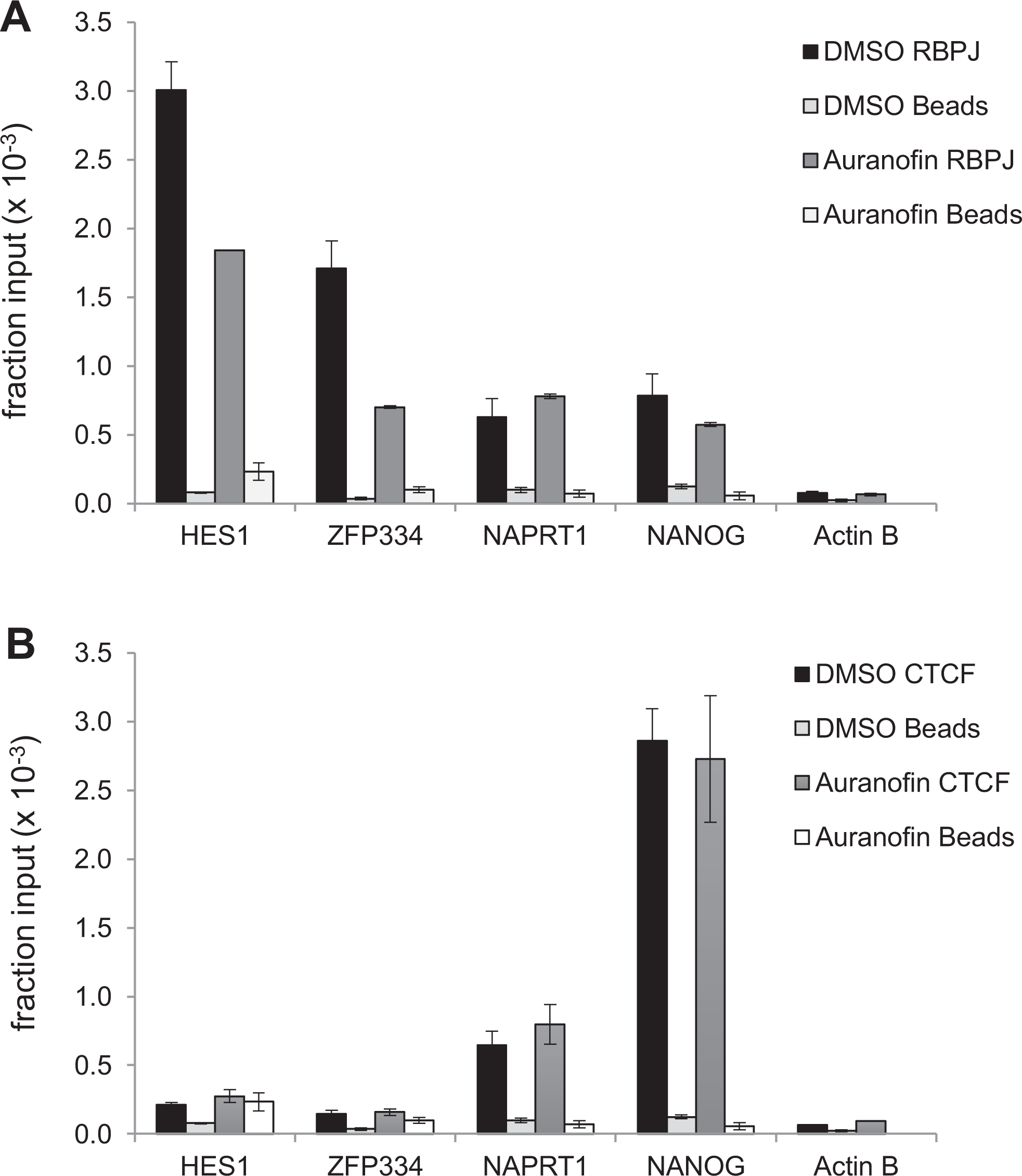
Auranofin decreases RBPJ-DNA interactions in murine F9 cells. F9 cells were treated with 2.5 µM auranofin for 24 h and then subjected to ChIP. (
Auranofin Preferentially Inhibits the Proliferation of T-ALL Cells with Aberrant Notch Activation
If auranofin downregulates Notch-dependent transcription activation, then cells depending on Notch signaling for proliferation should be more sensitive to auranofin than cells independent of Notch signaling. Aberrant Notch activation underlies the uncontrollable proliferation of greater than 50% of T-ALL. 19 Accordingly, we used five T-ALL cells to test our hypothesis. CCRF-CEM and DND-41 contain active mutations in the PEST and HD domains of the NOTCH1 gene that lead to aberrant Notch activation. 19 CUTLL1 has a t(7;9) rearrangement that leads to constitutive Notch activation. 46 These three cell lines are considered Notch-addicted T-ALL cells and require active Notch signaling for their proliferation. JURKAT and MOLT-4 cells serve as Notch-independent controls. 19,20
To determine the sensitivity, we treated these cells with auranofin for 36 h and measured their cellular ATP concentrations, as an indicator of metabolically active cells. As predicted, JURKAT cells were most resistant to auranofin treatment compared with Notch-dependent CCRF-CEM, DND-41, and CUTLL1 cells ( Fig. 6A ). Surprisingly, MOLT-4 cells were more sensitive to auranofin than JURKAT at a level similar to that of CUTLL1 cells, while MOLT-4 cells are classified as NOTCH-independent T-ALL.
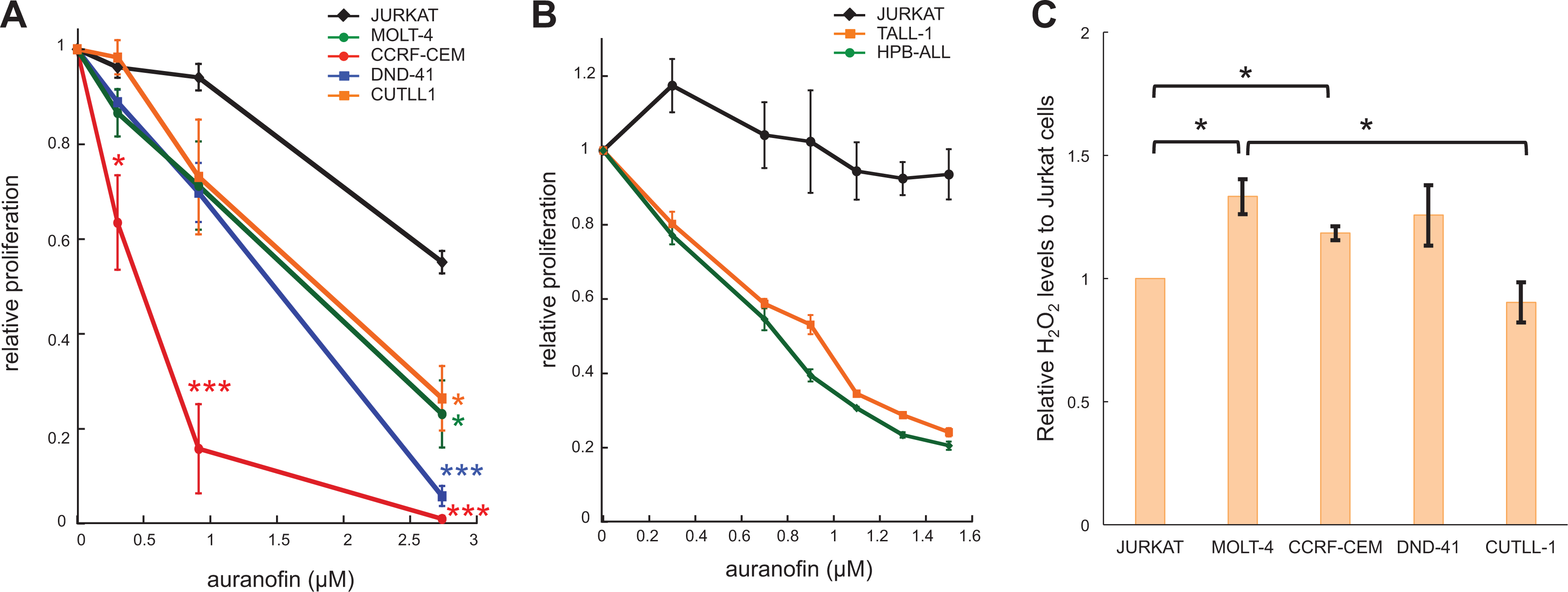
Auranofin impacts cell proliferation. (A) Cells were treated with auranofin for 36 h. The cellular ATP levels were measured to reflect the number of metabolically active cells. Shown is relative proliferation to the DMSO control as mean ± SEM (N = 3). *p < 0.05, ***p < 0.001. (B) T-ALL cell lines were treated with varying amounts of auranofin for 96 h. The cellular ATP levels were measured. Shown is relative proliferation to the DMSO control, mean ± SEM (N = 3). (C) Measurement of cellular hydrogen peroxide levels. Shown is mean ± SEM from three biological replicates. *p < 0.05. No significant difference was found between JURKAT/DND-41, JURKAT/CUTLL1, MOLT-4/CCRF-CEM, and MOLT-4/DND-41.
We also examined two other Notch-dependent lines, TALL-1 and HPB-ALL, with mutations in NOTCH3 and NOTCH1, respectively. 19,20 Given that the cell-division times of these two lines are longer, we treated these cell lines with auranofin for 96 h and then quantified the cellular ATP levels. As shown in Figure 6B , auranofin decreased the proliferation of Notch-dependent TALL-1 and HPB-ALL much more than Notch-independent JURKAT cells, consistent with the hypothesis that auranofin preferentially decreases the proliferation of Notch-dependent T-ALL cells by downregulating Notch signaling.
One mechanism of auranofin is to inhibit thioredoxin reductase activity and to increase intracellular levels of reactive oxygen species (ROS). Recently, auranofin was used in preclinical efficacy studies of chronic lymphocytic leukemia cells based on the premise that these cells have higher basal levels of ROS than normal lymphocytes.
47
If inhibiting thioredoxin reductase by auranofin is the primary mechanism by which Notch-dependent T-ALL cells are sensitive to auranofin, we expect higher basal levels of ROS in these cells compared with JURKAT cells under our experimental conditions. To test this possibility, we measured the cellular H2O2 levels, the most stable ROS in cultured cells. As shown in
Figure 6C
, DND-41 and CUTLL1 had similar basal levels of H2O2 to JURKAT; however, DND-41 and CUTLL1 were significantly more sensitive to auranofin than JURKAT (
Fig. 6A
), arguing against the notion that increasing ROS by auranofin preferentially inhibits proliferation of these Notch-addicted T-ALL cells. In addition, CCRF-CEM and MOLT-4 had similar levels of H2O2, but CCRF-CEM, a Notch-dependent T-ALL, is significantly more sensitive to auranofin than MOLT-4, again arguing against increasing ROS by auranofin as the primary mechanism inhibiting the proliferation of these Notch-dependent T-ALL cells (
Fig. 6A
Discussion
Here we described our screen of 1492 drugs for disrupting RBPJ-DNA interaction in vitro. About 1% of the tested compounds inhibit RBPJ-DNA interactions in a dose-dependent manner (
Fig. 2
). We analyzed 11 of these compounds in cell-based assays and discovered that the compound auranofin decreases Notch-dependent transcription activation and disrupts RBPJ-DNA interaction in HEK293T cells (
Figs. 3
Auranofin likely has a general, indirect effect on transcription regulation, as auranofin is known to increase ROS and elevated ROS impacts gene expression. In fact, auranofin decreases transcription from the TK promoter (
Furthermore, the concentrations of auranofin required to inhibit the proliferation of ∼50% of CUTLL1, DND-41, CCRF-CEM, HPB-ALL, and TALL-1 is slightly lower than or close to the EC50 required to disrupt the RBPJ-DNA interaction in vitro ( Fig. 2 ), raising the hypothesis that the effects of auranofin on cell proliferation in general likely result from dual mechanisms: creating ROS and decreasing RBPJ-DNA interactions. Furthermore, they likely have similar weights in our assays ( Fig. 6 ) because (1) we observed MOLT-4 cells that have higher basal H2O2 and are more sensitive to auranofin than JURKAT cells, and (2) among cells with similar basal levels of H2O2, cells addicted to Notch signaling for proliferation are more sensitive to auranofin ( Fig. 6 ).
It remains to be determined how auranofin decreases RBPJ-DNA interactions mechanistically. RBPJ forms both repressive and active transcription complexes depending on the status of Notch signaling.
1
-3
Our data reveal that auranofin disrupts RBPJ-DNA interactions, regardless of the status of Notch signaling (
Figs. 4
Comparison of RIN1 to Auranofin
RIN1 is a small molecule recently found to be an RBPJ inhibitor through a cell-based assay. RIN1 disrupts the interaction of RBPJ (aa2770-3127) and the scaffold protein SHARP. 34 Additional assays suggest that RIN1 also inhibits NOTCH2- and NOTCH3-mediated transcription activation. Surprisingly, RIN1 treatment induces a profile of transcript changes that are distinct from the changes induced by DAPT (a GSI). DAPT treatment decreases the expression of the classic Notch targets, HES1 and HEY1. In contrast, RIN1 treatment increases the expression of these two genes. Furthermore, the Notch-addicted KOPT-K1 T-ALL cells are as sensitive to RIN1 as the Notch-independent JURKAT cells, in strong contrast to DAPT and auranofin. The impact of RIN1 on gene expression is similar to that of siRNA targeting RBPJ. Since RBPJ (aa2770-3127) does not bind DNA, 34 it is unclear whether RIN1 influences RBPJ-DNA interaction.
Comparison of Auranofin to GSI
GSIs prevent the final cleavage step of the Notch receptors necessary for activating Notch signaling. Rao et al. found that NOTCH1 mutation status does not serve as a predictor of GSI sensitivity, a gene expression signature of NOTCH pathway activity does correlate with response, and it may be useful in the selection of patients more likely to respond to GSI.
19,20
In contrast, here we found that all Notch-dependent T-ALL cells tested are more sensitive to auranofin than Notch-independent JURKAT cells. Notably, CCRF-CEM is 10-fold less sensitive to GSI but is as sensitive to auranofin as to the other four Notch-dependent cell lines (
Fig. 5A
In summary, we have developed a screen to identify a novel class of Notch inhibitors. Our study reveals a new mechanism by which auranofin inhibits cell proliferation. Specifically, auranofin disrupts RBPJ-DNA interactions, in both cells and an in vitro reconstituted assay, which leads to downregulation of Notch-dependent transcription activation and impacts T-ALL cell proliferation. Furthermore, our study provides a rationale to use auranofin in animal model and preclinical efficacy studies as a drug to treat Notch-addicted cancer.
Supplemental Material
sup_materials_revised_042920 - A Novel Flow Cytometric Assay to Identify Inhibitors of RBPJ-DNA Interactions
sup_materials_revised_042920 for A Novel Flow Cytometric Assay to Identify Inhibitors of RBPJ-DNA Interactions by Robert J. Lake, Mark K. Haynes, Kostiantyn Dreval, Rabeya Bilkis, Larry A. Sklar and Hua-Ying Fan in SLAS Discovery
Footnotes
Acknowledgments
We thank Dr. Ksenia Matlawska-Wasowska of UNM for kindly supplying us with the JURKAT and CUTTL-1 cell lines. We are also grateful to Drs. Ursa and Opera of UNM, who compiled the 1492 compounds from the Prestwick Chemical and MedChemExpress libraries for screening.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received the following financial support for the research, authorship, and/or publication of this article: This work was supported by a Cancer Center support grant (P30CA118100).
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.