
Abstract
Immunotherapies including PD-L1 blockade have shown remarkable increases in the T cell–directed antitumor response; however, efficacy is seen only in a minority of patients. Recently, pooled CRISPR-Cas9 knockout (CRISPRn) screens in tumor/immune co-culture systems have identified a number of genes that confer resistance to T cell killing in pathways including antigen presentation and cytokine signaling, providing insight into tumor mechanisms that cause resistance to immunotherapies. The development of an arrayed CRISPRn screen in a tumor/immune co-culture system would allow the identification of novel targets for immuno-oncology, characterization of hits from pooled screens, and multiple assay endpoints to be measured per gene. Here, a small-scale arrayed CRISPRn screen was successfully developed to investigate the effects on a co-culture of T cells and Cas9-expressing PC9 lung adenocarcinoma cells modified to express anti-CD3 antibody on the cell surface (PC9-OKT3 T cell system). A focused CRISPRn library was designed to target genes involved in known resistance mechanisms (including antigen presentation, cytokine signaling, and apoptosis) as well as genes involved in immune synapse interactions. The viability of PC9 cells was assessed in two-dimensional adherent co-cultures via longitudinal imaging analysis. Knockout of epidermal growth factor receptor (EGFR) and PLK1 in tumor cells cultured alone or with T cells resulted in increased tumor cell death, as expected, whereas knockout of the test gene ICAM1 showed subtle donor-specific resistance to T cell killing. Taken together, these data provide proof of concept for arrayed CRISPRn screens in tumor/immune co-culture systems and warrant further investigation of in vitro co-culture models.
Introduction
As part of the adaptive immune response, CD8+ T cells play a central role in tumor surveillance. 1 Immunotherapies including PD-L1 checkpoint blockade target the T cell response and have demonstrated remarkable anticancer effects. However, durable clinical responses in most patients are not achieved,2,3 and the mechanisms of resistance remain elusive. In the CD8+ T cell–mediated anticancer response, circulating CD8+ naïve T cells specifically recognize tumor-associated antigens (TAA) displayed on the surface of antigen-presenting cells in the context of major histocompatibility complex (MHC) class I. This antigen-specific interaction is mediated by their unique T cell receptor (TCR) and results in their initial activation through the TCR (signal 1). Costimulatory proteins then provide a second signal (signal 2) via the T cell co-receptor CD28, resulting in full activation and expansion into effector cytotoxic T cells (CTLs) primed for their specific TAA. CTLs efficiently eliminate tumor cells upon recognition of their TAA through the release of cytolytic granules, antitumor cytokines (interferon-γ [IFNγ] and tumor necrosis factor–α [TNFα) and the induction of the Fas/FasL apoptotic pathway. 1 As such, CTL-mediated lysis can be described as an MHC- and antigen-dependent mechanism.
A number of pooled CRISPR screens within in vitro tumor/immune co-culture systems4 –6 and in vivo models4,7,8 have identified tumor genetic mutations that confer resistance to T cell killing. Notably, mutations in genes involved with antigen processing and presentation (including Β2M, TAPBP), the JAK-STAT cytokine signaling pathway (including JAK1, STAT1), and T cell interaction (including CD58, ICAM1) were identified. 6 Indeed, analysis of patient biopsies have shown that mutations in antigen presentation and cytokine signaling genes confer human resistance to immunotherapies.9 –11 Taken together, pooled CRISPR screens have been shown to identify novel tumor immune evasion strategies and resistance to immunotherapies and can accurately replicate the biology seen in human tumors.
Although useful for certain phenotypes such as cell death and resistance, the breadth of biology that can be explored in pooled CRISPR format is limited. Application of CRISPR technology in an arrayed screening format, although technically more challenging, lends itself to analysis of more complex cellular phenotypes in which whole pathways modulating disease can be explored. We therefore set out to develop an arrayed CRISPR screen in a tumor/immune co-culture system to identify mechanisms and pathways involved in tumor evasion from T cell killing. Here, we report the development of a co-culture screen between tumor cells and CD8+ T cells that is amenable to an arrayed CRISPR screening format. The co-culture system was developed with PC9 non–small-cell lung cancer cells expressing a membrane-bound anti-CD3 TCR crosslinker (OKT3) and activated CD8+ T cells, promoting tumor cell lysis through an MHC class I–independent mechanism. We hypothesized that targeting genes involved in T cell interaction, and not antigen presentation, would confer resistance to T cell killing. Using a focused CRISPRn library (including guide RNA [gRNAs] targeting genes previously shown to be critical for tumor evasion from T cell killing), we demonstrate the development of a robust co-culture system fit for purpose for use in arrayed CRISPR screening. Our model identifies 1 of 23 genes whose knockout enhances resistance to T cell killing: ICAM1, which gives a subtle resistance phenotype and decreased interleukin-2 (IL2) secretion in 1 of 2 donors tested. Our approach highlights the promise of tumor immune co-culture systems for identification of resistance but warrants further investigation of co-culture models that can provide larger phenotypic effects.
Materials and Methods
Cell Culture
PC9-OKT3 and PC9-Mock cells were maintained in RPMI-1640 (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS; Gibco, Waltham, MA), 1% Glutamax (Gibco), and 50 µg/mL hygromycin (Invitrogen, Carlsbad, CA). PC9-OKT3-Cas9+ and PC9-Mock-Cas9+ cells were maintained in RPMI-1640 media supplemented with 10% FBS, 1% Glutamax (Gibco), 50 µg/mL hygromycin (Invitrogen), and 1 mg/mL Geneticin (Gibco). Each cell line was passaged with Dulbecco’s phosphate-buffered saline (Sigma) and Accutase (Sigma). CD8+ T cells were maintained in DMEM-Glutamax-I (Gibco), supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin (Sigma).
Generation of PC9-OKT3-Cas9+ and PC9-Mock-Cas9+ Cell Lines
PC9-OKT3 and PC9-Mock cells were generated previously. 12 To introduce inducible Cas9, PC9-OKT3 and PC9-Mock cells were transfected with a doxycycline-inducible Cas9 vector containing the green fluorescent protein (GFP) marker. 13 In each well, 2.5 × 105 cells were seeded in 6-well plates (Corning, Corning, NY) and cultured overnight before Cas9 transfection for 48 h with FuGeneHD (Promega, Madison, WI) in Opti-MEM media (Gibco). Over an 11-day selection period, 1 mg/mL Geneticin was added. Doxycycline (Sigma) 100 ng/mL was added for 24 h to induce Cas9 expression before sorting for GFP+high cells by fluorescence-activated cell sorting (FACS; BD FACSJazz, BD Biosciences, Franklin Lakes, NJ). GFP+high PC9-OKT3-Cas9+ and PC9-Mock-Cas9+ cells were expanded for over 1 wk.
Generation of Activated CD8 T Cells
CD8+ T cells were isolated from peripheral blood mononuclear cell preparations from leukapheresis blood cones (NHS-BT) using the EasySep Human CD8+ T Cell Isolation Kit (Stemcell, Cambridge, MA). CD8+ T cells were then cryopreserved with CryoStor CS5 (Stemcell) prior to activation. Cells were recovered and activated with two rounds of anti-CD3/CD28 stimulation as described in Nelson et al., 12 to generate effector T cells with high cytotoxic capacity. Briefly, 25 µL anti-CD3/CD28 DynaBeads (ThermoFisher, Waltham, MA) per 1 × 106 cells were added in the presence of human IL2 at 20 ng/mL (Stemcell). The T cells were maintained at a density of 1 × 106 cells/mL. After 10 d, the T cells were reactivated with 25 µL anti-CD3/CD28 DynaBeads (ThermoFisher) per 1 × 106 cells in the presence of fresh human IL2 (20 ng/mL) prior to cryopreservation at day 13 with CryoStorCS10 (Stemcell). T cells were recovered from cyropreservation into prewarmed T cell medium and allowed to recover for 2 d before being used in assays.
In Vitro Co-Culture Killing Assay
Effector:target ratios (E:T) were optimized via flow cytometry using Zombie Violet Dye (Biolegend, San Diego, CA) to assess the percentage of live tumor cells after 18 h of co-culture with CD8 T cells, and a 3:1 E:T ratio was chosen. A total of 2500 PC9-OKT3-Cas9+ or PC9-Mock-Cas9+ cells (tumor cells) were seeded in 384-well CellCarrier Ultra plates (PerkinElmer, Waltham, MA) in 40 µL T cell culture medium for 24 h at 37 °C. Using a live cell microscope (IncuCyte, Sartorius, Ann Arbor, MI) at 10× magnification, the phase contrast was scanned every 2 h. Activated CD8+ T cells were recovered and cultured in growth medium containing 20 ng/mL IL2 overnight at 1 × 106/mL. After 24 h of tumor cell culture, 10 µL CD8+ T cells were added to the cultures (at a ratio of 3:1 CD8+ T cells to tumor cells), or 10% DMSO was added as a positive killing control. Co-cultures were imaged on the live-cell microscope for 4 d. The confluence (%) was calculated by analysis on the IncuCyte using the phase contrast images. Ten percent DMSO was added as a positive killing control.
Validation of CRISPR Editing by Transferrin Uptake
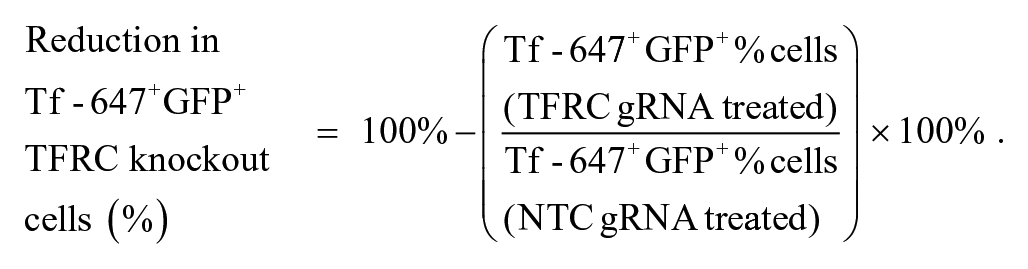
A total of 100 ng/mL doxycycline was added to PC9-OKT3-Cas9-GFP+ and PC9-Mock-Cas9-GFP+ cells at ~60% confluency for 48 h to induce Cas9 expression prior to CRISPR transfection. Four unique transferrin receptor complex (TFRC) or nontargeting control (NTC) Edit-R crRNA constructs (Dharmacon, Lafayette, CO) were pooled and duplexed with Edit-R tracrRNA (Dharmacon) in nuclease-free Tris pH 7.4 buffer (Dharmacon). TFRC ON-TARGETplus SMART pool siRNA (Dharmacon) and siGENOME RISC-free control siRNA (Dharmacon) were also used to compare editing by CRISPR gRNA to knock down by siRNA. gRNA 25 nM or siRNA 50 nM was acoustically dosed into 384-well CellCarrierUltra plates (PerkinElmer). Ten microliters of lipofectamine RNAiMAX (ThermoFisher) was added in serum-free growth medium to the gRNA and left to form transfection complexes at room temperature (RT) for 10 min. Thirty microliters of tumor cells were added in growth medium containing 100 ng/mL doxycycline to the transfection complexes. Tumor cells were incubated at 37 °C for 48- to 96-h transfection before assessment of transferrin uptake by confocal microscopy. Before fixing the cells in 4% paraformaldehyde, 100 µg/mL transferrin–Alexa Flour 647 (ThermoFisher) was added to the cells and incubated at 37 °C for 15 min. Cells were washed and stained with Hoechst Ready for confocal imaging in the channels BP445/45 (Hoechst), BP525/50 (GFP), and BP676/29 (transferrin–Alexa Flour 647). The efficiency of editing in Cas9+ cells was calculated using the following equation:
Arrayed CRISPRn Screen
The CRISPRn guide RNA library was designed based on findings from the previously cited pooled co-culture screens. The library consisted of CD58, ICAM1, ICAM2, CD274, LGALS9 (T cell co-stimulation genes), JAK1, JAK2, STAT1, IFNGR1, IFNGR2, TNFRS1A, PTPN2 (cytokine signaling genes), TAPBP, TAP2, B2M (antigen presentation genes), CASP8, and CARD16 (apoptosis genes). CD14-OKT3, CD14, DPM2, and PIGA genes (involved in the surface delivery of CD14-OKT3) were included as OKT3 surface delivery controls. Epidermal growth factor receptor (EGFR) and PLK1 were included as PC9 growth controls. Edit-R crRNA constructs (Dharmacon) were pooled (four unique crRNA constructs per gene) and duplexed with Edit-R tracrRNA (Dharmacon) in nuclease-free Tris pH 7.4 buffer (Dharmacon) to create guide RNA (gRNA) within an Echo plate (Labcyte, San Jose, CA). A total of 100 ng/mL doxycycline was added to PC9-OKT3-Cas9-GFP+ and PC9-Mock-Cas9-GFP+ cells at ~60% confluency for 48 h to induce Cas9 expression prior to CRISPR transfection. gRNA 25 nM was acoustically dosed into 384-well CellCarrierUltra plates (PerkinElmer). Ten microliters of 0.5% lipofectamine RNAiMAX (ThermoFisher) was added in serum-free growth medium to the gRNA and were left to form transfection complexes at RT for 10 min. Thirty microliters of tumor cells was added in T cell growth medium containing 100 ng/mL doxycycline to the transfection complexes at a density of 2500 cells per well. Tumor cells were incubated at 37 °C for a transfection period of 96 h and then co-cultured with activated CD8+ T cells for 72 h. Phase-contrast images at 10× magnification were taken using the IncuCyte live-cell microscope every 2 h throughout the duration of the assay.
IL2 Cytokine Assay
Supernatants were sampled from co-cultures 24 h after the addition of T cells. Two microliters of supernatant was assayed for IL2 levels using the AlphaLisa IL2 assay (PerkinElmer), and fluorescence was measured using an Envision plate reader (PerkinElmer). Cytokine levels were calculated by interpolation from the standard curve using Graphpad Prism v8.2.1.
Results
Development of the PC9-OKT3 T Cell System
To model CD8+ T cell killing in vitro, the EGFR mutant lung carcinoma cell line, PC-9, was transduced to express an anti-CD3 scFv (OKT3) fused to the transmembrane domain of CD14. PC9-OKT3 cells were co-cultured with activated CD8+ T cells ( Fig. 1a ) at E:T cell ratios between 1:1 and 5:1, and tumor cell death observed in an OKT3-specific manner ( Fig. 1b ) as shown previously. 12
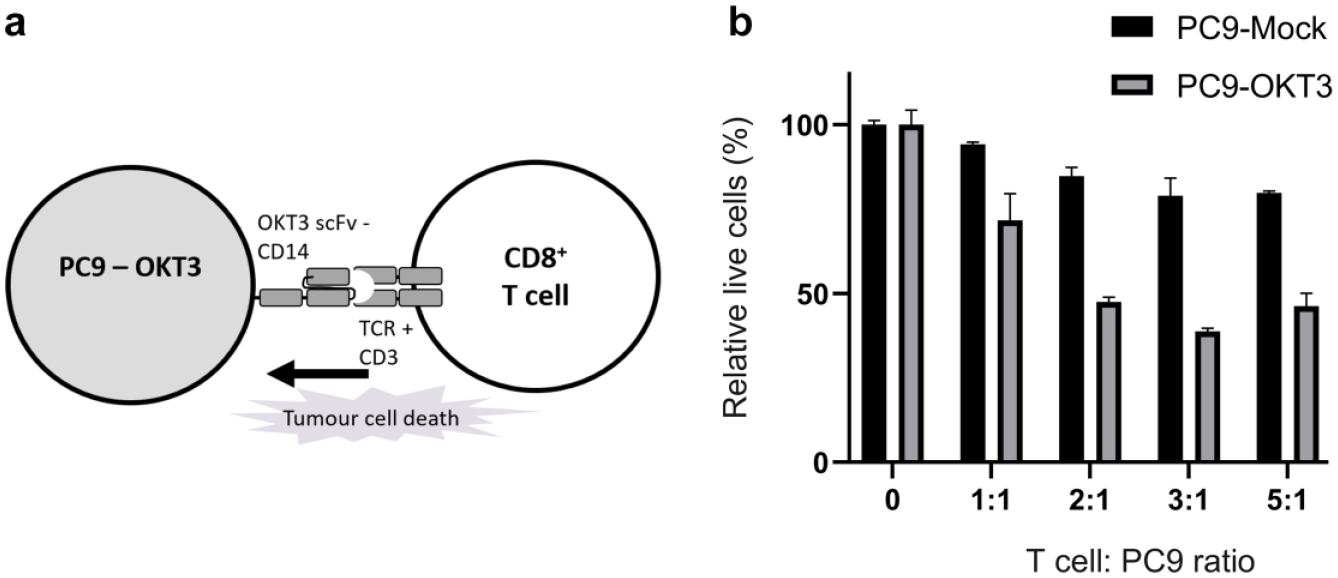
CD8+ T cell–directed tumor cell death is modeled in the in vitro PC9-OKT3 T cell co-culture system. (
Development of Cas9+ PC9-OKT3 Cells for In Vitro Killing Assays
To be able to knockout genes of interest in the PC9-OKT3 cells, we first generated inducible Cas9 PC9-OKT3 and PC9-Mock control cell lines. The plasmid used for introduction of Cas9 also contained GFP, which enabled sorting of pooled PC9-OKT3-Cas9+ and PC9-Mock-Cas9+ cells for GFP+high populations by FACS ( Fig. 2a ). PC9-OKT3-Cas9+ cells showed a significantly higher level of cell death than PC9-Mock-Cas9+ cells after co-culture with CD8+ T cells in the in vitro co-culture assay ( Fig. 2b ).
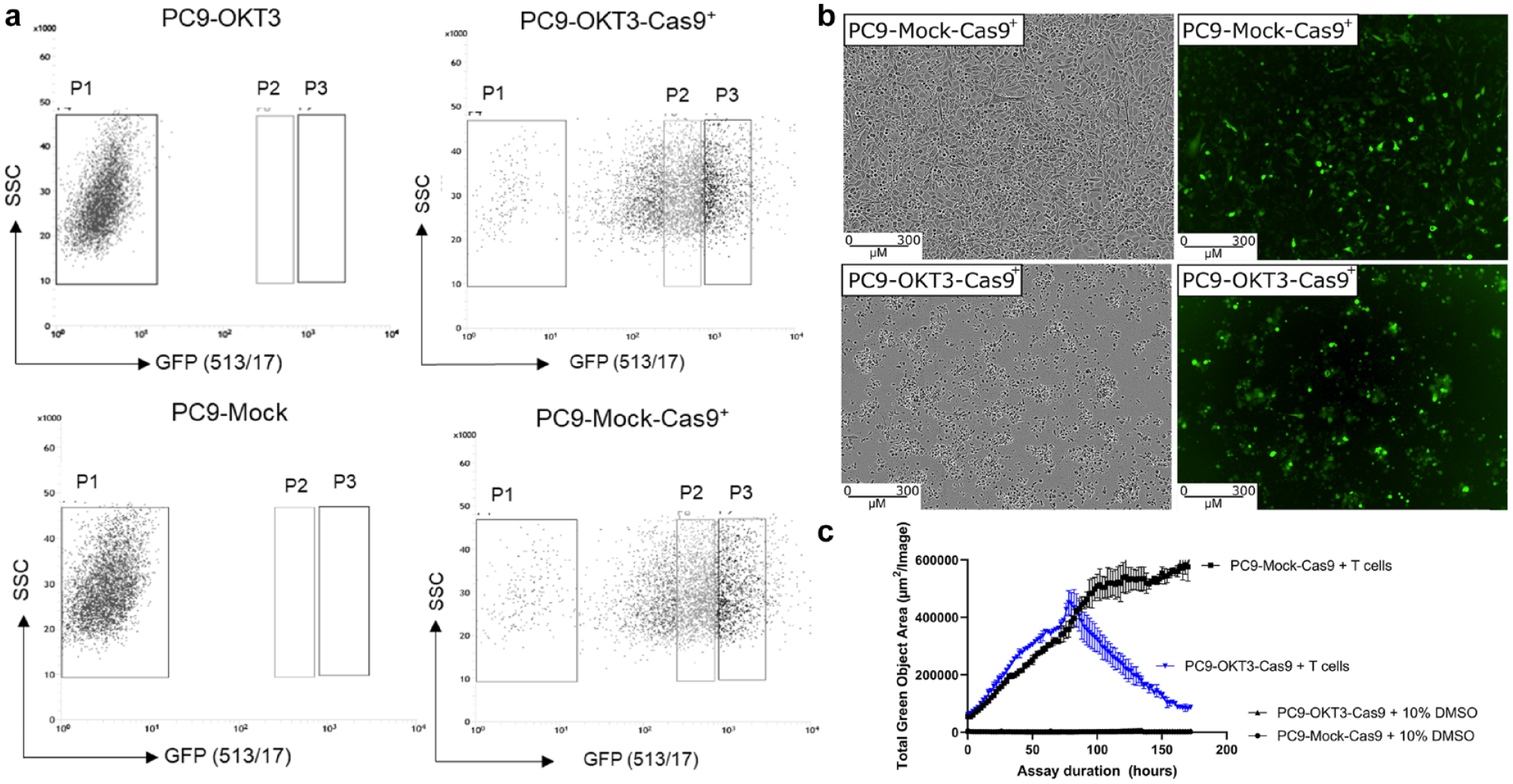
Inducible Cas9+ PC9-OKT3 cells are killed in the in vitro co-culture assays. (
Optimization of Reverse Transfection Conditions
gRNA transfection conditions were then optimized in the PC9-OKT3-Cas9+ cell line using gRNA against the TFRC. The uptake of Alexa-647–labeled transferrin was significantly reduced in cells treated with TFRC gRNA compared with the NTC gRNA and was comparable with transfection with siRNA ( Fig. 3a ). Transfection with TFRC gRNA resulted in 95% reduction in transferrin-positive GFP+ PC9-OKT3-Cas9+ cells, and this was used to define the CRISPR editing efficiency ( Fig. 3b ).
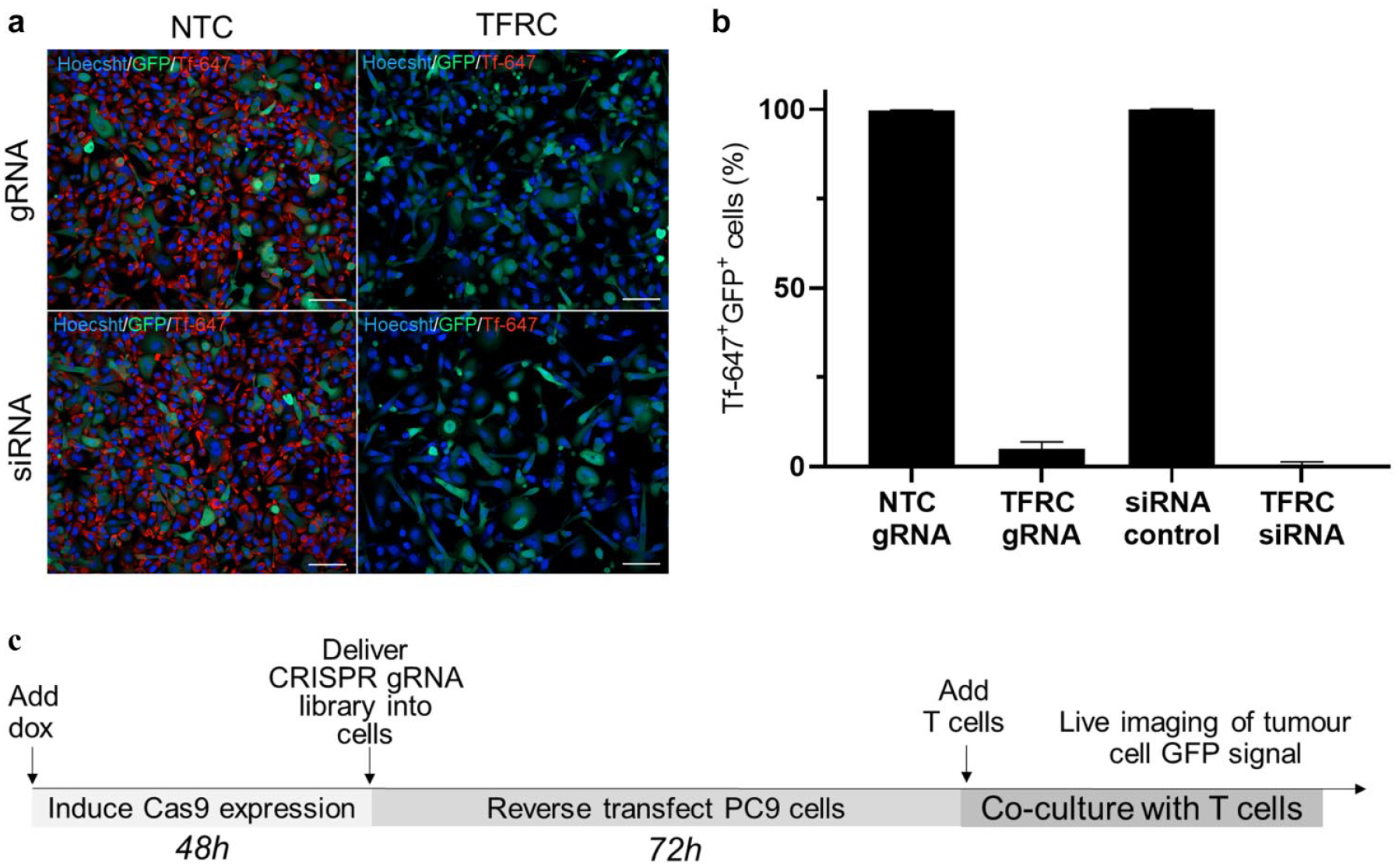
Transferrin receptor complex (TFRC) CRISPR guide RNA transfection was optimized to achieve 95% reduction in transferrin-positive PC9-OKT3-Cas9+ cells. (
Arrayed CRISPRn Screen in the In Vitro Co-Culture Assay
We next sought to determine whether knockout of genes previously identified as important in killing of tumor cells by T cells would modulate killing in the PC9-OKT3 co-culture model. To this end, we designed an arrayed CRISPRn library to target genes involved in T cell interaction, antigen presentation, IFNγ signaling, TNFα signalling, apoptosis, and growth signaling ( Table 1 ). In addition, the library targeted genes involved in the surface delivery of OKT3 and PC9 cell growth as controls (PC9 is an EGFR mutant cell line).
Arrayed CRISPRn Library.

As indicated in the schematic in
Figure 3c
, we induced Cas9 expression in PC9 cells for 48 h by the addition of doxycycline to the culture media and then delivered the focussed gRNA library by reverse transfection. PC9 cells were co-cultured with CD8+ T cells for up to 72 h with continuous measurement of total GFP signal area to allow a maximal window in which to identify resistance mechanisms. As expected, knockout of essential growth genes (EGFR and PLK1) in PC9-OKT3-Cas9+ cells cultured alone led to a significant increase in tumor death, as shown by a decrease in the total green object area (
Fig. 4
). Knockout of test genes in PC9-OKT3-Cas9+ cells co-cultured with CD8+ T cells did not lead to any resistance or sensitivity to T cell killing in donor 1 (
Fig. 4a
), whereas knockout of the test gene ICAM1 led to higher GFP signal in donor 2 (
Fig. 4b
), indicative of higher tumor cell viability. Knockout of test genes in PC9 OKT3 cells cultured alone did not affect tumor cell viability, with the exception of EGFR and PLK1, which are known to affect PC9 cell growth (
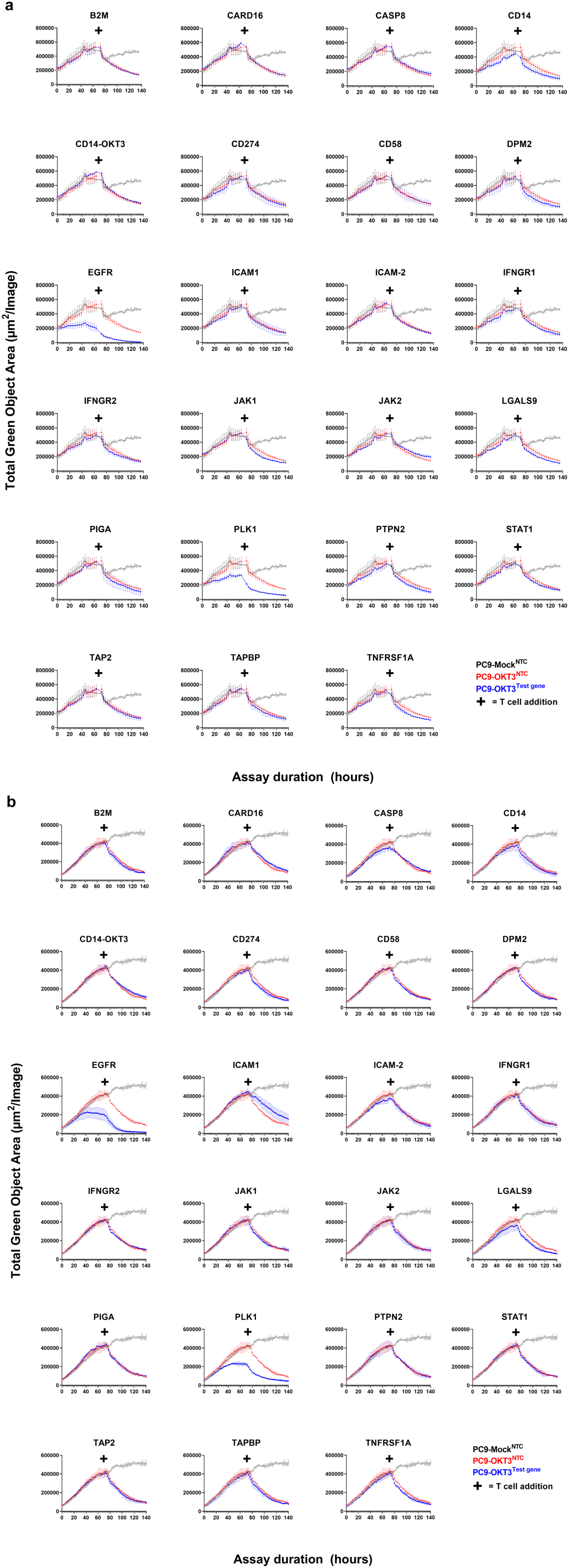
Arrayed CRISPRn screen in the in vitro killing assay. Individual plots for each test guide RNA in the CRISPR screen. PC9-OKT3-Cas9+ cells were transfected with the CRISPRn library and co-cultured with CD8+ T cells in the in vitro co-culture assay. Two CD8 T cell donors were tested in two independent experiments: (
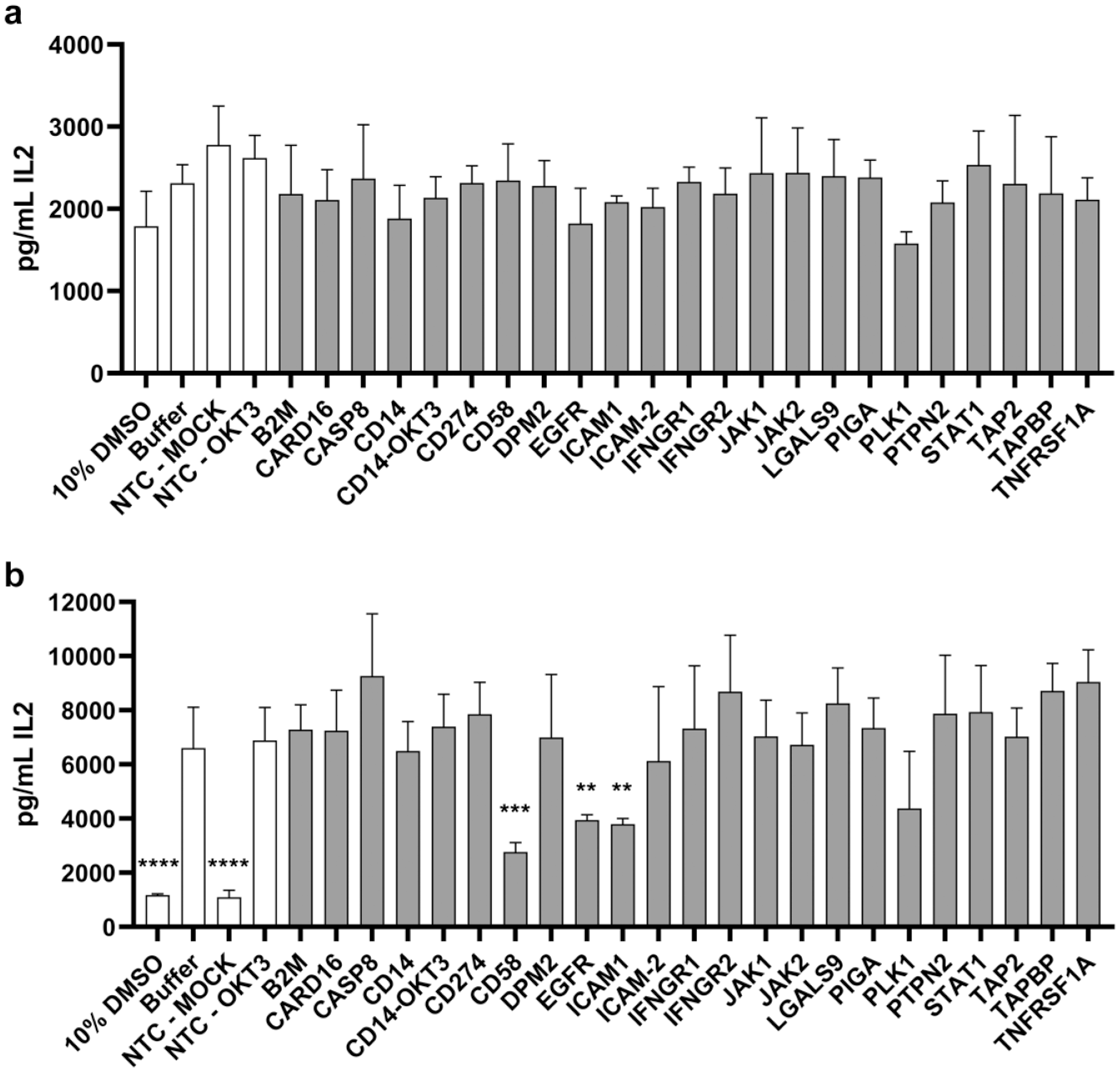
Interleukin-2 levels in co-culture supernatants of the arrayed CRISPR screen. Donor 1 (top) and donor 2 (bottom). Data represent the mean ± SD of n = 4 wells. **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 compared with NTC-OKT3 (unpaired t test).
Discussion
Co-culture CRISPR screens have thus far focused on pooled systems, in which T cells are engineered (TCR/CAR-T) to specifically recognize antigens on the tumor cell surface. Pan et al. 5 used the mouse Pmel-1 and OT-1 T cell lines in co-culture with the murine B16F10 melanoma tumor cell line. Patel et al. 6 used TCR-engineered human T cells reactive against the NY-ESO-I antigen in co-culture with NY-ESO-I+ Mel624 melanoma cells. Dong et al. 8 recently used both an in vivo approach measuring the presence of tumor-infiltrating lymphocytes following pooled library transduction and an in vitro pooled approach measuring enrichment of guides in CD107a+ OT-1 T cells after co-culture with peptide-pulsed tumor cells. The PC9-OKT3 system has previously been used successfully to investigate MHC class I–independent T cell killing and modulation by small-molecule inhibitors of the T cell receptor signaling pathway, such as dasatinib. 12 Here, we sought to build on this previous work by investigating whether the PC9-OKT3 T cell system could be used for an arrayed CRISPR screen to identify tumor resistance mechanisms to T cell killing. We used information from the aforementioned pooled CRISPR screens to design a focused CRISPR gRNA library aimed at genes previously identified as important in tumor cell and T cell interactions. We successfully developed the PC9-OKT3-Cas9 T cell co-culture system and showed that measurement of T cell killing is possible in 384-well plate format. We then optimized the workflow for successful CRISPR editing followed by co-culture, as shown by the increased cell death from EGFR and PLK1 knockout. CRISPR editing was optimized to achieve 95% knockout in the Cas9+ cell population. However, when our panel of test gRNAs was tested in this system, we saw no effect on the T cells from one donor and a subtle increase in viability from knockout of ICAM1 in the second. Arrayed CRISPR screens are amenable to multiple readouts, and measurement of IL2 from test wells also showed knockout of ICAM1 and also CD58 to reduce IL2 secretion. Again, this was seen only in our second T cell donor and highlights the need to evaluate both multiple endpoints and donors in co-culture models.
Our PC9-OKT3 T cell system is mediated through an MHC class I–independent mechanism, and consistent with this, the knockout of genes in the antigen presentation pathway did not modulate the system. Similar to our findings, an in vitro pooled CRISPR screen in an MHC class I–independent co-culture system (consisting of a CAR T cell engineered to recognize Her2+ tumour cells) also did not identify hits in the antigen presentation pathways. 4 As the mechanism of action in our system relies on the interaction of OKT3 and the T cell receptor, we targeted genes involved in the surface expression of OKT3 to try to create an artificial positive control; however, we observed no effect on tumor cell death. This may be because the multiple copies of OKT3 inserted via the lentiviral transduction method overcome the action of the Cas9-mediated knockout. In this study, we co-cultured a high-expressing pool of PC9-OKT3 cells together with CD8+ T cells that had been activated with two rounds of stimulation to generate high cytotoxic capacity, as previously shown. 12 Hence, it may be difficult to overcome the level of T cell activation achieved with this model by knockout of endogenous genes.
In addition, we hypothesized that the disruption of genes involved in the T cell interaction including CD58, ICAM1, and ICAM2 would lead to resistance to T cell killing. Of relevance, knockout of CD58 and ICAM1, identified as hits in one of our two donors, was also identified as hits in previous pooled CRISPR screens. 6 Although the MHC class I–independent nature of the PC9-OKT3 T cell system allows investigation of nonclassical resistance mechanisms to T cell–mediated cytotoxicity, this system may not generate a completely physiological immune synapse (e.g., because of the affinity of the CD3 binding domains leading to reduced reliance on ICAM1 to stabilize the interaction) and therefore may not be suited to decipher resistance mechanisms associated with the formation of a competent T cell:target cell synapse. It may be that donor 1 has a lower threshold for initiation of killing, whereas donor 2 has an increased reliance on the co-stimulatory effects of CD58 and ICAM1. Our work shows that prior to larger-scale screening efforts, a large panel of donors should be tested to define donor variability and identify best assay conditions.
We believe that the work performed here sets the foundation for investigating other co-culture models of tumor cells and T cells. Although in vivo CRISPR screening allows investigation of physiologically relevant interactions between tumor tissue and T cells,8,14 it still has the limitation of being restricted to mouse biology. Arrayed CRISPR screens have the potential to play a vital role in the investigation of specific aspects of T cell biology, such as cytokine secretion, activation markers, differentiation status, and “omics” endpoints. For T cells specifically, arrayed screens will be the only way to assess cytolytic activity toward tumor cells following the knockout of individual genes. Finally, arrayed platforms will allow interrogation via other therapeutic modalities such as small molecules and biologics, and as such, the development of arrayed platforms for smaller-scale CRISPR screening is vital to complete the investigator’s toolkit. Our work suggests the necessity of using model systems in which a more physiological T cell tumor cell interaction operates, and future studies will focus on adapting these systems to our arrayed screening platform.
Supplemental Material
Supplementary_Figure_1 – Supplemental material for Developing an Arrayed CRISPR-Cas9 Co-Culture Screen for Immuno-Oncology Target ID
Supplemental material, Supplementary_Figure_1 for Developing an Arrayed CRISPR-Cas9 Co-Culture Screen for Immuno-Oncology Target ID by Sarah Gee, Nadine Nelson, Aurelie Bornot, Nikki Carter, Maria Emanuela Cuomo, Simon J. Dovedi, Paul D. Smith, Davide Gianni and David J. Baker in SLAS Discovery
Footnotes
Acknowledgements
We thank Douglas Ross-Thriepland and Samantha Peel for assistance in designing and optimizing the CRISPR screen and Marcello Maresca for providing Cas9 reagents.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors were employed by and shareholders in AstraZeneca whilst performing the work for this paper.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support for the research, authorship and publication of this article was provided by AstraZeneca.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.