
Abstract
Bone tissue regeneration and fracture healing remain a significant challenge for physicians, with nonunion failures occurring in an estimated 5%–10% of bone-healing treatments. The autologous bone graft has long been the gold standard of treatment. However, these procedures suffer from persistent donor-site morbidity and extended surgery times, while still having high revision and nonunion failure rates. Cell therapies and tissue engineering strategies utilizing stem cells have been considered as promising alternatives to autologous bone grafts. Here, we explore the concept of using CRISPR-activation (CRISPRa) as a cell-engineering tool to drive osteogenesis without exogenous growth factors. We present a genome-wide CRISPRa screen in adipose-derived stem cells (ASCs) to identify upregulation targets that drive osteogenesis. Top targets from the screen, SPRED2 and ATXN7L3B, demonstrated significant increases in alkaline phosphatase activity and mineralization in monolayer and 3D culture. These results are the first evidence of these genes as osteogenic targets in ASCs.
Introduction
Despite the exceptional regenerative capacity of bone tissue, bone healing and fracture repair remain a significant challenge. Bone fractures are one of the most common injuries leading to hospitalization. 1 While most fractures heal with fragment realignment and/or extended mechanical fixation, over 8% of them have healing complications that require surgical intervention within 2 years. 1 Additionally, several orthopedic procedures, including spinal fusions and primary tumor resections, produce critical-size bone defects that require facilitated repair, making bone the second-most transplanted tissue after blood. 2
Current treatment methods for addressing these healing complications have substantial drawbacks. The longtime “gold-standard” for facilitating bone repair is the autologous bone graft.3–6 Unfortunately, these procedures often require an additional surgery at an otherwise healthy locale, resulting in extended surgical times, unnecessary blood loss, and donor site morbidity that often remains painful for years after the procedure.7,8
The use of graft material from allogeneic sources circumvents some of the major drawbacks of autologous grafts by not requiring the surgeon to harvest tissue from the patient at the time of treatment. Allogeneic bone grafts are readily available from various donors, and they come in many different forms making them highly versatile. However, they are generally not as effective as their autologous counterparts, and are associated with infection, host immune response, and disease transfer.9–11
Alternatives to bone grafting have become more popular clinically in recent years.12,13 Most notable of these, is the application of bone morphogenetic protein-2 (BMP-2) at the treatment site. 14 BMP-2 drives bone formation and healing by recruiting osteoprogenitors and inducing their osteogenic differentiation.15–17 This method can be highly effective for bone growth and healing, but has been connected to serious negative side effects, including ectopic bone formation, excessive inflammation and swelling, and cancer.18,19 Given the risks, limitations, and high revision rates of current bone-healing approaches, alternative methods to promote and improve bone healing are needed. Cell therapies, utilizing multipotent mesenchymal stem cells (MSCs), have been considered for bone healing and regenerative medicine applications due to their multipotency.20–23 Furthermore, MSCs are highly proliferative and exhibit beneficial immunomodulatory characteristics, which makes them excellent candidates for cell therapies and tissue engineering.24–28 Adipose tissue has proven to be an abundant and convenient source of MSCs, due to their ease of isolation from liposuction precedures.29–32 In addition to their ease of acquisition, ASCs can undergo osteogenesis given the appropriate external stimulation. However, these forms of stimuli can be difficult to maintain and control in therapeutic settings leading to limited therapeutic benefit and deleterious side effects.
As an alternative to relying on extracellular factors to affect change in cell phenotype, recent technological advancements have produced methods for engineering the cells themselves. CRISPR-guided gene modulation technologies have been used to engineer cell phenotype.33–35 Such systems allow for highly specific and targeted regulation of gene expression by either upregulating or downregulating target gene expression. Previously, CRISPR-interference was used to suppress the expression of noggin and effectively promote osteogenic differentiation in ASCs. 36 In addition, CRISPR-activation of aggrecan and collagen II drove a synergistic relationship to drive chondrogenic differentiation of ASCs without the use of growth factors.31,37 Therefore, we hypothesize that CRISPR-activation of pro-osteogenic genes could drive osteogenesis in ASCs.
The literature has previously identified various potential upregulation targets that could be pursued for directing osteogenesis via CRISPRa.38–40 However, pursuing each target individually is time-consuming and expensive, with no guarantee of success, especially when the reference studies were conducted in differing cell types or conditions. Recently, CRISPR genome-wide screens have provided a method for identifying gene targets and desired phenotypes based solely on their functional outcome, such as evaluating growth phenotypes and improving cell survival in acidic conditions.41–43 These screens probe each protein-coding gene in the genome individually and can be used to identify effective and potentially novel targets and biology when carried out with an appropriate functional indicator.41,42 There are several candidate genes that could potentially be used as a functional indicator,44–46 however osteocalcin (OCN), which has been shown to mediate biomineralization during osteogenesis, is one of the most widely recognized and accepted markers of late-stage osteogenesis.29,47–49 Therefore, by performing a CRISPR-activation (CRISPRa) genome-wide screen and observing its effect on OCN expression, we can identify gene targets that drive osteogenesis in ASCs.
Here, we utilize an OCN-based osteogenic reporter 50 cell line in a genome-wide CRISPRa screen to identify upregulation targets that cause osteogenic differentiation in ASCs. In monolayer validation cultures, 6 of the top 10 upregulation targets from the screen, including RUNX2, SPRED2, ATXN7L3B, TP53BP1, MTHFR, and CARD16, led to increased alkaline phosphatase (ALP) activity in ASCs, an early sign of osteogenesis. Furthermore, 3D culture on type-I collagen sponges confirmed that the top 2 targets, ATXN7L3B and SPRED2, resulted in increased mineralization, as shown by alizarin red staining and micro computed tomography (micro-CT) scans. Our results display the applicability and power of utilizing CRISPRa genome-wide screens to identify novel gene targets and biology to drive stem cell differentiation. In this study, we systematically engineered ASCs with an enhanced osteogenic capacity while also providing insight on poorly understood mechanisms of osteogenesis in ASCs. Additionally, this study attests to the utility of CRISPRa for controlling driving cell phenotype which has broad applications throughout cell and tissue engineering for the development of therapies treating musculoskeletal disease.
Materials and methods
CRISPRa system optimization and genome-wide screening strategy
Experiments were conducted to identify CRISPRa upregulation targets through a genome-wide screen that would enhance osteogenesis in ASCs. First, we compared three different CRISPRa systems to determine the most potent effector molecule for eliciting robust endogenous gene upregulation in ASCs. Then, to monitor osteogenic differentiation in response to target gene upregulation, a reporter ASC cell line was created by transducing naïve ASCs with two lentiviral vectors: (1) a dCas9-VPR CRISPRa upregulation vector and (2) an osteogenic reporter vector with the dTomato fluorophore under an OCN promoter. The osteogenic reporter cell line was validated and then utilized to perform a genome-wide CRISPRa screen for upregulation targets that would drive osteogenesis as indicated by activity at the OCN promoter. The screen was initiated by transducing the reporter ASCs with a lentiviral library. After a 3-week culture period, osteogenic (dTomato(+)) and non-osteogenic (dTomato (−)) cells were collected separately by fluorescence-activated cell sorting (FACS) and genomic DNA was isolated from each population. Samples were prepared for, and subjected to, next-generation sequencing (NGS) to determine the enrichment of each sgRNA in each cell population. The top gene targets from the screen were identified, cloned into individual expression vectors, and transduced into ASCs for validation of their osteogenic effect. Alkaline phosphatase (ALP) activity, alizarin red staining, and micro-CT were used to evaluate osteogenic differentiation in the validation experiments.
Vector construction
A lentiviral CRISPRa vector containing an EF1α-dCas9-VPR-P2A-PuroR expression cassette (Addgene #99373) and a genome-wide CRISPRa sgRNA vector library containing 5 sgRNAs per protein-coding gene (Addgene #83978) were acquired from Addgene. An OCN reporter lentiviral vector containing an OCN-dTomato CMV-GFP-T2A-Puro expression cassette was developed and validated at the Center for the multimodal manufacturing of structural tissues at Case Western Reserve University. sgRNAs used in the CRISPRa system comparison were designed using the GT-Scan web applet. 51 Using standard restriction enzyme cloning techniques, sgRNAs used for the CRISPRa system comparison as well as those selected for downstream validation were cloned into a lentiviral vector under an hU6 DNA polymerase-II promoter (Addgene, #83919). In addition to the vectors for validation, a nontarget control (NT) sgRNA was designed and cloned into the hU6 DNA polymerase-II promoter vector. This NT vector contains a scrambled sgRNA that does not target the genome, allowing us to control for off-target effects or vector-related artifacts associated with the CRISPRa system directly.
Lentivirus production
All cell culture was performed in standard condition (5% CO2, 21% O2, 37°C) with media changes every 2–3 days. Lentivirus was produced in HEK293T cells using a previously described method. 34 Briefly, vectors containing functional expression cassettes were co-transfected into HEK293T cells with the psPAX2 (Addgene #12260) and pMD2.g (Addgene #12259) lentiviral packaging and envelope plasmids using Lipofectamine 2000 (ThermoFisher Scientific; 11668027) according to the manufacturer’s protocol. After 24 h, the cell supernatant was discarded and replaced with fresh medium. Cell supernatant was collected at 48- and 72-h post-transfection, pooled, filtered, and concentrated 100-fold by centrifugation at 20,000 × g for 4 h at 4°C. The supernatant was discarded, and the viral pellet was resuspended in cold PBS prior to being aliquoted and stored at −80°C. Viral titer was determined by transducing ASCs with serial dilutions of the stock virus and determining the number of transduced cells by flow cytometry.
Comparison of CRISPRa systems
Naïve ASCs were transduced with lentivirus containing dCas9-p300Core, dCas9-VP160, or dCas9-VPR as previously described. 34 Briefly, cells were seeded at 10,000 cells/cm2 in growth medium (ADSC Growth Medium BulletKit; PT-4505; Lonza RTP; Morrisville, NC) and allowed to attach after overnight incubation. The following day, the lentivirus was thawed and diluted in growth medium supplemented with 4 μg/ml polybrene to create the transduction medium. The growth medium on the ASCs was replaced with the transduction medium and the cells were transduced for 24 h. Stable cell lines were generated by antibiotic selection and then subsequently transduced with hU6-sgRNA lentiviruses targeting either BMP2 or RUNX2. After 1 week of culture, the cells were lysed, and RNA was processed for qRT-PCR as described below to determine target gene upregulation.
Generation and validation of osteocalcin reporter cell line
dCas9-VPR ASCs were transduced with the OCN-dTomato lentivirus as described in the previous section. A stable reporter cell line was generated by FACS for cells expressing dTomato. Stable OCN reporter cells were then cultured in either growth medium (negative control), or in osteogenic medium (low-glucose DMEM, 10% MSC FBS, 2 mM GlutaMax, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 10 nM dexamethasone, 50 μM 2-phospho-L-ascorbic acid, and 10 mM β-glycerophosphate) supplemented with 100 ng rhBMP2 (ThermoFisher; PHC7141) for 3 weeks. The cells were then analyzed for relative dTomato expression levels by flow cytometry. Thresholds for dTomato (+; top 10% by dTomato fluorescence) and dTomato (−; bottom 50%) cells were established using the negative control.
Genome-wide CRISPRa screen
The workflow for the genome-wide CRISPRa screen is shown in Figure 1. 52.5 million OCN reporter ASCs were plated at 25,000 cells/cm2 in standard growth medium. The following day, cells were transduced with the sgRNA lentivirus library at an MOI of 0.3 with 4 µg/ml polybrene for 24 h as described above. Cells were rinsed with PBS and then cultured for 48 h prior to FACS for TagBFP-positive cells to select for successfully transduced cells. Sorted cells were plated at 10,000 cells/cm2 and cultured in growth medium for 3 weeks with media changes every 2–3 days. Cells were then trypsinized and subjected to FACS for dTomato expression. The top 10% and bottom 50% of cells by dTomato expression were collected separately. Both populations were lysed, and genomic DNA were isolated and purified using a NucleoSpin Blood kit (Macherey Nagel; 740951.50).

Workflow of the osteogenesis CRISPRa screen.
Protospacer enrichment and NGS adapter ligation
Purified DNA samples were used as the template for PCR for enriching protospacer sequences. The reaction employed custom PCR primers, which were designed to amplify the inserted protospacer sequence while also attaching adapters for NGS. 42 SPRI beads (SPRIselect; B23318; Beckman-Coulter) were used to isolate the enriched sequences from the PCR products with sequential washes at ratios of 0.65× and 1×.
Next-generation sequencing
Samples were pooled at equal concentrations and analyzed on an Illumina HiSeq 2500 in a single-end 50 bp run with custom sequencing primers targeting the protospacer sequence. 42 Ten percent PhiX was added to the sequencing lane to increase diversity. Reads were mapped, quantified, and analyzed using the ShortRead and base packages of R software, and counts were normalized per 1 million mapped reads for each sample.52,53 For data visualization, the average of the top 3 sgRNAs by absolute value of their log-fold enrichment for each targeted gene were plotted along with their associated Mann-Whitney p-value that was calculated by comparing the targeting sgRNAs to non-targeting sgRNAs within the sgRNA library. Targets were selected for downstream validation by inputting the enrichment data into a previously published gene expression perturbation algorithm called MAGeCK. 54 Briefly, MAGeCK prioritizes sgRNAs and their genetic targets by median normalization of read counts between samples, performing sgRNA mean-variance modeling, ranking the p-values for each sgRNA from that model, and then comparing the distribution of the sgRNA rankings for each gene to a uniformly distributed null model. 54
Generation of validation cell lines
For singleplex validation of each of the top 10 sgRNAs selected by the MAGeCK algorithm, separate cell lines were created for each sgRNA. These cell lines were created by transducing EF1α-dCas9-VPR ASCs with lentivirus created from the individual sgRNA singleplex vectors, as described above.
Alkaline phosphatase assay
ASCs were seeded at 10,000 cells/cm2 in growth medium (n = 6) and cultured for 7 days. Cells were lysed with a 0.2% Triton X-100 solution and ALP activity was determined using a quantitative colorimetric assay using a p-nitrophenol phosphate substrate (BioAssay Systems; DALP-250; Hayward, CA). Quantified ALP activity was normalized to total protein content as measured using a detergent-compatible Bradford protein assay (ThermoFisher Scientific; 23200; Waltham, MA).
3D culture on type-I collagen sponges
Sterile type-I collagen sponges (Integra LifeSciences; ID-1105; Plainsboro, NJ) were cut into 3 mm cubes. The cubes (n = 4) were then immersed in a cell suspension (2.5 million cells/mL in culture medium) before being transferred to individual wells of a v-bottom plate without any additional culture medium. The plate was incubated at 37°C and 5% CO2 for 30-min to allow for cell attachment before the addition of culture medium. After 1 week of culture, the culture medium was changed from growth medium to osteogenic medium. A group of nontarget (NT)-ASCs dosed with rhBMP2 (100 ng/ml) were included as a positive control. Following 4 weeks of culture in osteogenic medium, the samples were fixed in 4% paraformaldehyde in PBS for 24 h and then stored in 70% ethanol.
Histology
Fixed samples were embedded in paraffin, cut into 5 μm sections, and mounted onto slides. Slides were stained with either hematoxylin and eosin (H&E) or alizarin red, as previously described. 55 Semi-quantitative analysis of the alizarin red staining was performed in ImageJ by setting a threshold for positive staining and comparing the stained area to the total section area.
Microcomputed tomography
Formalin-fixed paraffin-embedded (FFPE) samples were analyzed by micro-CT using a Siemens Inveon Preclinical Micro-CT scanner (Siemens AG; Munich, Germany) at a 94 μm resolution and run at 80 kV and 500 μA. Arbitrary intensity units from the scan were converted to Hounsfield units by scanning a water phantom and applying a linear transformation based on the intensity values for water and air. Images were quantified using ImageJ, as previously described. 56
Quantitative reverse transcriptase PCR
Cells were seeded at 5000 cells/cm2 (n = 8) and cultured in growth medium for 2 weeks with media changes every 2–3 days. Cells were then lysed and total RNA was isolated using a Quick-RNA Microprep kit (Zymo Research; R1050; Irvine, CA). cDNA was synthesized from the total RNA using a High-Capacity cDNA Reverse-Transcription kit (Applied Biosystems; 43-688-14; Waltham, MA) and used for quantitative reverse transcriptase PCR (qRT-PCR) with TaqMan Gene Expression Assays (ThermoFisher; 431182) for GAPDH (Hs02786624_g1), Spred2 (Hs00986219_m1), ATXN7L3B (Hs00740521_s1), and Runx2 (Hs00955785_m1) and TaqMan Universal PCR Master Mix (Fisher Scientific; 43-044-37) according to the manufacturer’s protocol. Data were analyzed using the ΔΔCt method. 57
Statistics
Bartletts’s test was used to determine homogeneity of variances for all data sets. One-way analysis of variance (ANOVA) with a Dunnett’s post hoc test comparing treatment groups to the NT group was used to calculate statistical significance for CRISPRa system comparisons and all validation assays. Alpha was set at 0.05 unless otherwise indicated, with all p-values below this value being considered statistically significant. A Mann-Whitney test was performed on the data set from the genome-wide screen, comparing the log-fold change enrichment values for the top 3 sgRNAs (by absolute value) for each gene to the non-targeting controls as previously described. 42
Results
CRISPRa with dCas9-VPR robustly upregulates endogenous targets
The CRISPRa systems were compared by targeting BMP2 and RUNX2 and observing the capacity of each system to upregulate the target genes. Each gene was targeted with two separate sgRNAs (Figure 2(a) and(b)) and one of three CRISPRa systems: dCas9-p300Core, dCas9-VP160, or dCas9-VPR. Both targeting sgRNAs for the VPR and VP160 systems showed statistically significant gene upregulation as compared to their respective non-targeting (NT) control (Figure 2(c) and (d)). The VPR system provided higher upregulation than the VP160 system, with the top-performing sgRNAs achieving 195-fold and 5.55-fold upregulation, compared to 42.6-fold and 3-fold upregulation, for BMP2 and RUNX2, respectively. The p300Core system was largely ineffective on the tested targets. Based on this data, the subsequent screen and validation experiments were performed utilizing the VPR system.
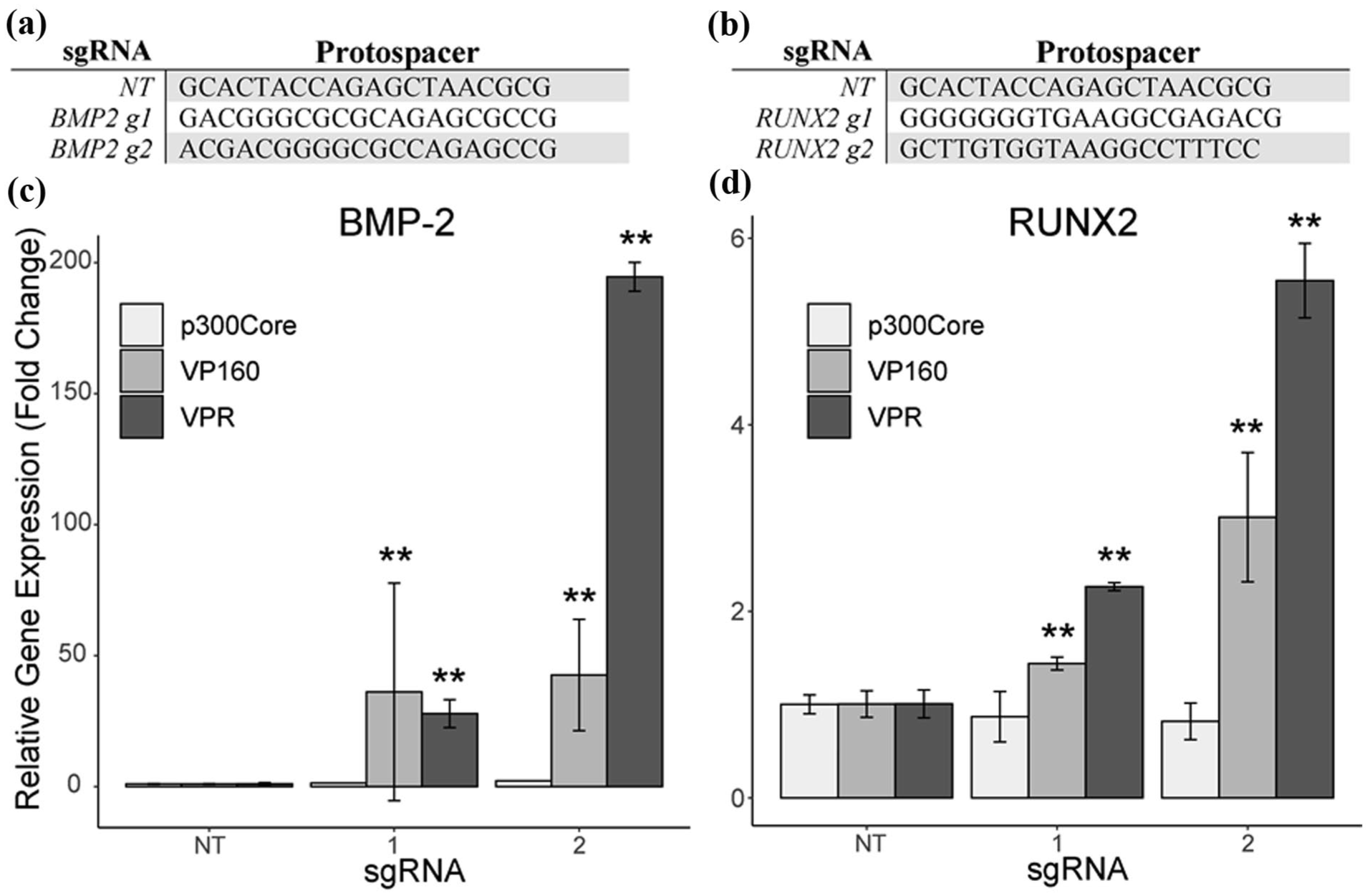
sgRNAs designed using GT-Scan for targeting (a) BMP2 and (b) RUNX2 for comparing different CRISPRa systems. Relative gene expression of (c) BMP-2 and (d) RUNX2 after upregulation with the indicated CRISPRa system.
The osteocalcin reporter identifies osteogenic ASCs
The osteogenic reporter cell line was created by transduction with the dCas9-VPR (Figure 3(a)) and OCN-dTomato (Figure 3(b)) lentiviruses. Successful transduction was validated using flow cytometry and evaluating dTomato expression. The reporter cells undergoing osteogenic differentiation expressed higher levels of dTomato expression, with 76.3% of the reporter cells cultured in osteogenic conditions being above the dTomato (+) threshold, compared to only 10% in the negative control (Figure 3(c) and (d)). Furthermore, only 3% of ASCs grown in osteogenic conditions were below the dTomato (−) threshold, while 50% of ASCs in the negative control were below this value. These results indicate that reporter ASCs undergoing osteogenic differentiation express dTomato as activity at the OCN promoter is increased. Moreover, reporter ASCs grown in non-osteogenic conditions express limited dTomato fluorescence. Therefore, the OCN-based reporter system can be effectively used to identify reporter ASCs that are undergoing osteogenic differentiation.

Expression cassettes for (a) the dCas9-VPR system and (b) the OCN-reporter vector. Flow cytometry results showing dTomato expression in reporter ASCs cultured in (c) growth medium or (d) osteogenic medium with 100 ng/ml BMP-2 during reporter validation. Blue lines indicate upper threshold for dTomato (−) cells and red lines indicate lower threshold for dTomato (+) cells. Thresholds were set based on the negative control.
The CRISPRa screen with the osteogenic reporter identifies potential upregulation targets for driving osteogenesis
Lentiviral transduction efficiency with the genome-wide sgRNA library (Figure 4(a)) was determined to be 28.8% by FACS for TagBFP-positive cells, indicating that the MOI was near the target of 0.3. According to a Poisson distribution, the probability of a transduced cell receiving only one sgRNA was therefore calculated to be 96.6%. The transduction efficiency also implies maintenance of sgRNA representation in the screen, with a calculated 145 transduced cells per unique sgRNA in the library. 17.7 million TagBFP-positive cells were collected and plated for the osteogenesis portion of the screen. Following 3 weeks of culture, FACS showed 12.9% of the cells transduced with the CRISPRa library virus were above the dTomato (+) threshold, and 43.2% were below the dTomato (−) threshold compared to values of 10% and 50%, respectively, in the negative control (Figure 4(b) and(c)).

(a) Expression cassette for the CRISPRa library vector. (b) Naïve reporter ASCs and (c) OCN-reporter ASCs transduced with the CRISPRa lentivirus library after 3 weeks of culture in growth medium. Blue lines indicate upper threshold for dTomato (−) cells and red lines indicate lower threshold for dTomato (+) cells. Thresholds were set based on the negative control. (d) Volcano plot of the CRISPRa genome-wide screen showing the enrichment of each gene target and its corresponding Mann-Whitney p-value. The dashed red line indicates the average enrichment of the NT controls. (e) Distribution of p-values for the gene targets in the screen as determined by MAGeCK with the top 10 gene targets labeled.
Samples were subjected to NGS for quantification of sgRNA enrichment in each population. 88.5 million (847 reads/unique sgRNA) and 108.5 million (1038 reads/unique sgRNA) reads were obtained during NGS for the dTomato (+) group and dTomato (−) group, respectively, indicating a robust sequencing depth for both populations. NGS data analysis revealed thousands of sgRNAs and hundreds of genes as prospective osteogenic targets. Two hundred forty-eight gene targets enriched in the dTomato (+) group, while 1176 were enriched in the dTomato (−) group (p < 0.01; Figure 4(d)). The MAGeCK algorithm calculated that 18,697 sgRNAs were enriched in the dTomato (+) group and 8678 sgRNAs were enriched in the dTomato (−) group (FDR < 0.01). Gene-level analysis of the enriched sgRNAs by the MAGeCK algorithm identified 204 genes enriched in the dTomato (+) group and 231 genes enriched in the dTomato (−) group (p < 0.01; Figure 4(e)). The gene-level analysis ranks best-performing gene targets by considering the enrichment levels of all sgRNAs targeting each particular gene in each population. 54 Further analysis of the top molecular functions, biological processes and phenotypes of the top 204 enriched genes provides additional insight into the functions related to these effected target genes. Utilizing Enrichr, the identified genes can be correlated to the top molecular functions regulated, which include transcriptional regulation, G protein-coupled receptor binding, regulation of ion channel activity, and kinase binding (Figure 5(a)). 58 Further analysis of the transcription factors and protein-protein interactions (PPIs) identifies SMAD 59 and YAP 60 transcription factors which are known to be correlated to osteogenesis and bone formation (Figure 5(b)). 61 Continued analysis of the biological processes and phenotypes affected by these genes shows increases in calcium ion transport and increased bone strength, mineral content, and density, providing additional evidence that the screen proved successful (Figure 5(c)). Given these results, the top 10 ranked targets from the gene-level MAGeCK results were selected for downstream validation of improved osteogenesis in ASCs (Figure 6(a) and(b)).

Analysis of the significant genes from the genome-wide screen displays (a) a UMAP plot with the top Molecular Functions regulated by these significant genes, 58 (b) the top 10 Transcription Factor PPIs that show interactions related to osteogenesis, and (c) the Biological Processes, MGI Mammalian Phenotype, 62 and KOMP2 Phenotypes 63 showing increased processes and phenotypes related to osteogenesis, bone mineralization, and bone formation.

(a) Selected sgRNA sequences for each of the top 10 genes from the CRISPRa screen. (b) The expression cassette that the protospacers were cloned into for validation. (c) ALP activity of dCas9-VPR ASCs after 1 week of culture in growth medium.
Top targets from the genome-wide screen drive osteogenesis in monolayer and 3D cultures
The successful identification of osteogenic upregulation targets was displayed in the validation experiments with 6 of the top 10 targets demonstrating increased osteogenic capacity in monolayer cultures. RUNX2-, SPRED2-, TP53BP1-, CARD16-, MTHFR-, and ATXN7L3B-transduced ASCs all exhibited higher ALP activity than the NT after 1 week of culture in growth medium (Figure 6(c)). Within these sponges, several of the targets showed qualitative and semi-quantitative improvements in broad matrix deposition, mineralization in the alizarin red staining of histological sections from the 3D collagen sponges, and micro-CT imaging (Figure 7(a)–(d)). For all cell types that displayed mineralization by alizarin red staining, the majority of the mineralization occurred in peripheral regions of the sponges, possibly indicating that the mineralization nucleation site was at or near the surface of each sponge. The top-performing cell lines based on monolayer ALP activity and histology from the 3D cultures were selected for further mineralization confirmation and quantification via micro-CT scanning. Micro-CT analysis indicated mineralization patterns and quantities that closely matched the results of the alizarin red staining and semi-quantitative analysis (Figure 7(e)). SPRED2 and ATXN7L3B upregulation resulted in increased calcium deposition and mineralization in 3D culture on type-I collagen sponges, with SPRED2 cells achieving micro-CT attenuation values similar to those in the NT + BMP2 group (Figure 7(e)). RUNX2, the top-performing target from the monolayer ALP activity assay, showed no changes in mineralization as compared to the NT.

(a) H&E, (b) Alizarin red-stained section, and (c) micro-CT images of the collagen sponges (scalebar = 2 mm). (d) Quantified Alizarin red staining and (e) quantified data from the micro-CT scans of the sponges after 4 weeks of culture in osteogenic medium. (f) Target gene expression of top-performing sgRNAs from the genome-wide screen targets in dCas9-VPR ASCs after 2 weeks of monolayer culture in growth medium.
The CRISPRa screen identified effective sgRNAs for target gene upregulation
qRT-PCR was performed on dCas9-VPR ASCs transduced with sgRNAs for the selected top targets (RUNX2, SPRED2, and ATXN7L3B) from the ALP-activity and alizarin red assays to verify upregulation of the targeted genes. Each of the three cell lines demonstrated upregulation of its respective target gene (Figure 7(f)). SPRED2 gene expression was upregulated 1.31 ± 0.09 fold, ATXN7L3B was upregulated 3.42 ± 0.16 fold, and RUNX2 was upregulated 4.23 ± 0.94 fold. Taken together with the results of the osteogenesis validation experiments, these upregulation values indicate that the genome-wide screen was effective not only for selecting functionally effective gene targets, but also for selecting sgRNAs capable of upregulating the genes of interest.
Discussion
Due to their proliferative capacity, multipotency, and immunomodulatory characteristics, ASCs are a promising cell source for cell and tissue engineering applications treating musculoskeletal disorders. However, ASCs require external signaling cues to guide their differentiation. Herein we present a CRISPR-guided gene modulation technique that identifies novel gene targets that can be used to engineer ASCs for bone healing and regeneration outcomes.
Initially, we optimized our targeted CRISPRa upregulation by comparing different CRISPRa effector molecules to select the most potent system to drive endogenous gene upregulation in ASCs. We targeted BMP2 and RUNX2 due to their relevance in osteogenesis as well as their differences in baseline expression levels. In naïve ASCs, BMP2 is expressed at much lower levels than RUNX2. 50 Therefore, including both of them in the comparison allowed us to observe the upregulation capacity of each system for genes with high and low endogenous expression levels. All three systems that we compared utilize dCas9 from S. pyogenes and are compatible with standard sgRNAs, allowing us to compare sgRNAs and effector molecules directly. Consistent with the literature, we found that dCas9-VP160 and dCas9-VPR robustly upregulated both of our target genes, while dCas9-p300Core achieved only modest upregulation for one of the two targets. 64 The stronger upregulation and higher consistency we observed in the dCas9-VPR system led to its selection over the dCas9-VP160 system for our genome-wide CRISPRa screen.
The use of an effective phenotypic reporter system is paramount to the success of any genome-wide functional screen. In this study, we generated an endogenous reporter cell line by transducing dCas9-VPR ASCs with a lentivirus containing an OCN-dTomato expression cassette. Since OCN is a reliable marker for late-stage osteogenesis in MSCs, activity at the OCN promoter is indicative of osteogenic differentiation. This reporter system allowed us to identify osteogenic cells by observing dTomato expression. An analogous reporter system has been previously used to observe osteogenesis in bone marrow MSCs. 65 By comparing the fluorophore expression of reporter ASCs cultured in growth medium to ASCs cultured in osteogenic medium, we have demonstrated that this system can be used to indicate osteogenesis in ASCs. There was a marked uniform increase in dTomato expression in the reporter ASCs cultured in osteogenic conditions, however we elected to include a gap between the dTomato (+) and dTomato (−) thresholds to minimize noise and obviate the presence of each sgRNA in each population.
As expected, the positive shift in dTomato expression was much more muted during the CRISPRa screen than during reporter validation, since we were not driving all the cells toward osteogenesis. Instead of a uniform increase in dTomato expression across the whole population, there was a subtle shift in a subset of the population with an increase in dTomato (+) cells of only 2.9%. From the screen we identified 8678 sgRNAs that were enriched in the dTomato (+) group, with only 204 genes being significantly enriched. This suggests that a small minority of sgRNAs in the screen drove osteocalcin expression and presumed osteogenesis but the majority of them did not.
sgRNA enrichment and subsequent gene-level analysis by the MAGeCK algorithm ranked RUNX2 as the top target in the screen. RUNX2 is a well-known marker of osteogenesis and established inducer of OCN expression. 66 Its presence at the top of the list strongly substantiates the screen as an effective method for identifying osteogenic upregulation targets. Some top-ranked targets further corroborate the validity of the screen, while others present themselves as novel upregulation targets. SPRED2 and HVCN1 have been previously associated with osteogenesis in other cell types, however their effect on osteogenesis in ASCs has yet to be explored.67,68 The remaining targets have not been implicated in osteogenesis before now, thus presenting novel biology and indicating gene function that has been otherwise unknown.
Analysis of the 204 significantly enriched genes further corroborates that the screen identifies genes associated with osteogenesis and bone formation. Initially, significantly regulated molecular functions include G protein-coupled receptor activity, regulation of ion channel activity, transcriptional regulation, and kinase binding. 61 The identification of such processes, which have been implicated in osteogenesis, RUNX2 expression, and OCN-expression, further validates our findings. 61 When evaluating the transcription factor protein-protein interactions (PPIs) there is an interconnected network containing SMAD and YAP signaling, as well as P300, which are all transcriptional co-activators and have been associated with osteogenesis.59–61 The continued evaluation of the biological processes and phenotypes associated with these genes identifies significant increases in calcium ion transport which can be implicated with OCN and RUNX2 expression, where its expression mediates calcium influx through ion channels and protein kinases. 61 Such functions are expected after examining the top genes due to the high number of significantly enriched transcription factors. Additionally, genes associated with osteogenesis would predictably increase ion channel activity due to increased calcium transport. The result of the influx of calcium ions would result in osteoblast differentiation, increased bone strength, mineral content, and density, which are predicted phenotypes associated with the significantly enriched genes identified from the CRISPRa genome-wide screen. 61 Taken together, the OCN-reporter system and the CRISPRa genome-wide screen were highly effective at identifying genes associated OCN-expression, osteogenesis, and bone mineralization.
While the objective of the screen was to identify pro-osteogenic upregulation targets, the screen itself only selects upregulation targets that would drive OCN expression. Validation was therefore essential to verify that the selected targets drive not only OCN expression, but also produce an osteogenic effect in ASCs. In monolayer cultures grown in non-osteogenic growth medium, sgRNAs targeting 6 of the top 10 genes from the screen demonstrated early signs of osteogenesis as indicated by increased ALP activity. Considering that the initial library for the screen contained 102,645 sgRNAs targeting 18,915 genes, this shows that the screen was able to identify osteogenic targets with remarkable efficiency.
To further test osteogenic capacity and evaluate the performance of the cells in a bone tissue-engineering application, the cells were cultured on 3D type-I collagen sponges. These are the same types of sponges that are currently used in BMP2-based clinical bone healing applications. In culturing the cells on 3D type-I collagen sponges, we further tested their osteogenic capacity. Substrate stiffness has been shown to be an important environmental factor for osteogenic differentiation in ASCs, with stiffer substrates promoting osteogenesis and softer substrates promoting adipogenesis. 69 The sponges, therefore, present a more difficult osteogenic environment than does the tissue culture plastic that was utilized in the monolayer cultures. Additionally, the use of osteogenic medium instead of growth medium, the altered spatial format, and the differences in cell morphology elicited by the substrate create an entirely different environment than that of the initial screen. With such drastic environmental differences and the associated signaling changes they undoubtedly produce, the cells may not respond in the same manner. Nevertheless, multiple targets from the screen showed improved osteogenesis in the 3D-cultures as evident from the alizarin red staining of the histological sections and the micro-CT scanning. Semi-quantitative analysis of the alizarin red staining showed that sgRNAs targeting SPRED2 and ATXN7L3B both displayed a statistically significant increase in calcium deposition. The improvements in mineralization were also evident in the quantified micro-CT data that revealed that sponges cultured with SPRED2-upregulated ASCs had higher signal attenuation than their NT counterparts, and similar levels to the positive control that was cultured in the presence of exogenous BMP2. These results present SPRED2 and ATXN7L3B as novel upregulation targets for driving osteogenesis in ASCs. Furthermore, they show the capabilities of CRISPRa in affecting changes in cell phenotype, and demonstrate that this method can be as effective as high doses of exogenous growth factors for driving osteogenesis. As previously discussed, these genes have not been directly implicated in osteogenesis in ASCs, however their known functions provide insight into their mechanisms for regulating osteogenesis. SPRED2 regulates the mitogen-activated protein kinases (MAPK) signaling cascade 70 and fibroblast growth factor receptor-3 (FGFR3) signaling networks, 71 which have both been implicated in cell differentiation, proliferation, and patterning, as well as ion channel regulation. 71 Moreover, FGF’s mediate metabolic functions, tissue repai,r and regeneration in adult tissues and have been linked to regulating BMP signaling pathways. 69 ATXN7L3B directly regulates ATXN7L3 which is a known to be a transcription regulatory histone acetylation (HAT) complex that is a component of a multiprotein complex that directly regulates transcription, DNA replication, and RNA polymerase II. 72 With ATXN7L3 being a direct regulator of ATXN7L3B, these studies and interactions indicate this complex may be a novel transcription factor that regulates osteogenesis, though additional studies will need to be performed confirm this hypothesis.
Notable among the results of the validation experiments is the performance of the cells with upregulated RUNX2. In monolayer cultures, the RUNX2-upregulated cells had much higher ALP activity than all other groups. Yet, in the 3D cultures, there was no evidence of calcification. RUNX2 is widely known as a reliable marker of osteogenesis and has been shown to induce osteogenesis when overexpressed transiently in ASCs.38,73 However, in agreement with the results shown here, sustained overexpression of RUNX2 has been reported to prevent the maturation of osteoblasts.74,75 The ability to increase RUNX2 expression for a short duration while dosing with BMP2 or increasing expression of other pro-osteogenic genes could provide an avenue for enhancing osteogenesis without preventing maturation of osteoblasts, as has been previously shown.74,75 Furthermore, developing a transient expression system for RUNX2 regulation could be an avenue to investigate avenues for overcoming the inhibitory effects of sustained expression of RUNX2. Using the CRISPR or Tet system for controlling transient expression is a potential avenue for continued research in developing strategies and methods to improve bone healing and fracture repair treatments. Notably, despite the success of most of the screen hits, the second top target, ZNF865, was a false positive and did not produce bone during our validation screening. However, we have since identified it as an important regulator of protein production, senescence, and DNA damage.36,37 The false positive was driven by the underlying biology of the gene and the identification and characterization of ZNF865 has led to important discoveries and novel biology itself.
Future work will investigate the ability to multiplex the top enriched sgRNA genes and investigate improvements in osteogenesis and mineral deposition. Previously, we have shown that multiplex CRISPRa upregulation of specific genes can have a synergistic effect on ASC differentitation, 31 thus investigating the multiplexing of different gene targets could enhance osteogenesis and mineralization to further improve the osteogenic outcomes of ASCs. Furthermore, orthogonal CRISPR/Cas9 species can be used for simultaneous up- and down-regulation could enhance differentiation outcomes. Previous work has shown the ability to downregulate the gene noggin, a known antagonist to BMP2 signaling, and drive osteogenesis with endogenous levels of BMP2.76,77 The ability to downregulate antagonists to BMP2 signaling while simultaneously upregulating genes associated with osteogenesis could further improve and enhance ASC osteogenic differentiation for use in cell and tissue engineering applications.
This study has identified novel upregulation targets that drive osteogenesis in ASCs and demonstrated their efficacy in multiple culture environments. However, on a broader scale, these experiments have shown that genome-wide CRISPRa screens can be effectively used for engineering specific attributes of cell phenotype. Here, we utilized such a screen to engineer ASCs to improve their osteogenic capacity. However, similar strategies could be applied in many other cell types to engineer diverse attributes for differing therapeutic applications across the fields of cell/tissue engineering and regenerative medicine.
Supplemental Material
sj-xlsx-1-tej-10.1177_20417314251390038 – Supplemental material for CRISPRa genome-wide screen identifies novel gene targets for osteogenic cell engineering
Supplemental material, sj-xlsx-1-tej-10.1177_20417314251390038 for CRISPRa genome-wide screen identifies novel gene targets for osteogenic cell engineering by Jacob D. Weston, Hunter Levis, Brandon Lawrence, Rodrigo Somoza and Robby D. Bowles in Journal of Tissue Engineering
Supplemental Material
sj-xlsx-2-tej-10.1177_20417314251390038 – Supplemental material for CRISPRa genome-wide screen identifies novel gene targets for osteogenic cell engineering
Supplemental material, sj-xlsx-2-tej-10.1177_20417314251390038 for CRISPRa genome-wide screen identifies novel gene targets for osteogenic cell engineering by Jacob D. Weston, Hunter Levis, Brandon Lawrence, Rodrigo Somoza and Robby D. Bowles in Journal of Tissue Engineering
Footnotes
Acknowledgements
We would like to acknowledge the DNA/Peptide Synthesis Core, Genomics Core, and Pre-Clinical Imaging facilities at the University of Utah for use of their equipment.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research reported in this publication was supported by the Office Of The Director of the National Institutes of Health under Award Number AR074998, AR083990, S10OD026959 and NCI Award Number 5P30CA042014-24. This work was funded in part by a grant from the National Institutes of Health (EB021911).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.