
Abstract
Phosphodiesterase type 2A (PDE2A) has received considerable interest as a molecular target for treating central nervous system diseases that affect memory, learning, and cognition. In this paper, the authors present the discovery of small molecules that have a novel modality of PDE2A inhibition. PDE2A possesses GAF-A and GAF-B domains and is a dual-substrate enzyme capable of hydrolyzing both cGMP and cAMP, and activation occurs through cGMP binding to the GAF-B domain. Thus, positive feedback of the catalytic activity to hydrolyze cyclic nucleotides occurs in the presence of appropriate concentrations of cGMP, which binds to the GAF-B domain, resulting in a “brake” that attenuates downstream cyclic nucleotide signaling. Here, we studied the inhibitory effects of some previously reported PDE2A inhibitors, all of which showed impaired inhibitory effects at a lower concentration of cGMP (70 nM) than a concentration effective for the positive feedback (4 μM). This impairment depended on the presence of the GAF domains but was not attributed to binding of the inhibitors to these domains. Notably, we identified PDE2A inhibitors that did not exhibit this behavior; that is, the inhibitory effects of these inhibitors were as strong at the lower concentration of cGMP (70 nM) as they were at the higher concentration (4 μM). This suggests that such inhibitors are likely to be more effective than previously reported PDE2A inhibitors in tissues of patients with lower cGMP concentrations.
Keywords
Introduction
Cyclic nucleotide phosphodiesterases (PDEs) are a superfamily of enzymes that regulate the cellular levels of second messengers (cyclic adenosine monophosphate [cAMP] and cyclic guanosine monophosphate [cGMP]) by controlling their rates of hydrolysis. There are 11 different PDE families, based on gene sequence, substrate specificity, regulation, and pharmacology,1–3 and each family typically has several different isoforms and splice variants.4,5 Among them, phosphodiesterase type 2A (PDE2A) family members are predominantly expressed in the forebrain and their action affects cognitive processes.6,7 The genes of three splice variants, PDE2A1–PDE2A3, have been cloned, and the protein expression of PDE2A3 has been further demonstrated to be specific to the brain. 8 Thus, we conducted high-throughput screening of our small-molecule compound libraries to identify novel inhibitors of human PDE2A3. Human PDE2A3 is a protein of 941 amino acids (aa) that is organized into four domains—the N-terminal (1–214), GAF-A (215–372), GAF-B (393–541), and catalytic (579–941) domains—and functions as a homodimer. 9 The GAF-B domain of PDE2A binds to cGMP, thereby enhancing the enzyme’s PDE activity. 10 It has also been demonstrated that several cyclic nucleotide analogs bind to the GAF domains with higher affinity than to the catalytic site. 11 Thus, these cyclic nucleotide analogs have facilitated investigations into the mechanisms by which GAF domains enhance catalytic activity of PDEs.11–13 It has been demonstrated that binding of cGMP to the GAF-A domain of PDE5A is necessary and sufficient for achieving full activation of catalytic activity. 14 The same group also demonstrated that an activated form of PDE5 had higher sensitivity toward a PDE5A inhibitor, sildenafil, than that of PDE5 in a nonactivated form, and this conversion could be obtained through the GAF-A domain-dependent and -independent mechanisms. 15 However, there have been no reports to date describing a change of PDE2A sensitivity to inhibition through GAF domain-dependent regulatory mechanisms. The purpose of this study was to identify new PDE2A inhibitors with a novel mode of inhibition.
Materials and Methods
Reagents
Cyclic nucleotide analogs (5,6-DM-cBIMP, 1-NOcAMP, and 2′-dcGMP) were purchased from Biolog Life Science Institute (Bremen, Germany). cAMP and cGMP were purchased from Sigma-Aldrich (St. Louis, MO). The 3H-labeled [
3
H]cGMP (5–14 Ci/mmol) and scintillation proximity assay (SPA) yttrium silicate beads were purchased from PerkinElmer (Waltham, MA). Our identified hit compounds, 1-(2,6-dichlorophenyl)-4-(trifluoromethyl)[1,2,4]triazolo[4,3-a]quinoxaline (CAS no. 343372-37-2/compound-1), N-butyl-3-((3-oxo-3,4-dihydroquinoxalin-1(2H)-yl)carbonyl)-N-phenylbenzenesulfonamide (CAS no. 878448-06-7/compound-2), and N-(1-benzylpiperidin-4-yl)-2-(4-methoxyphenyl)-1H-imidazo[4,5-b]pyridine-7-carboxamide (compound-3), were synthesized in-house (
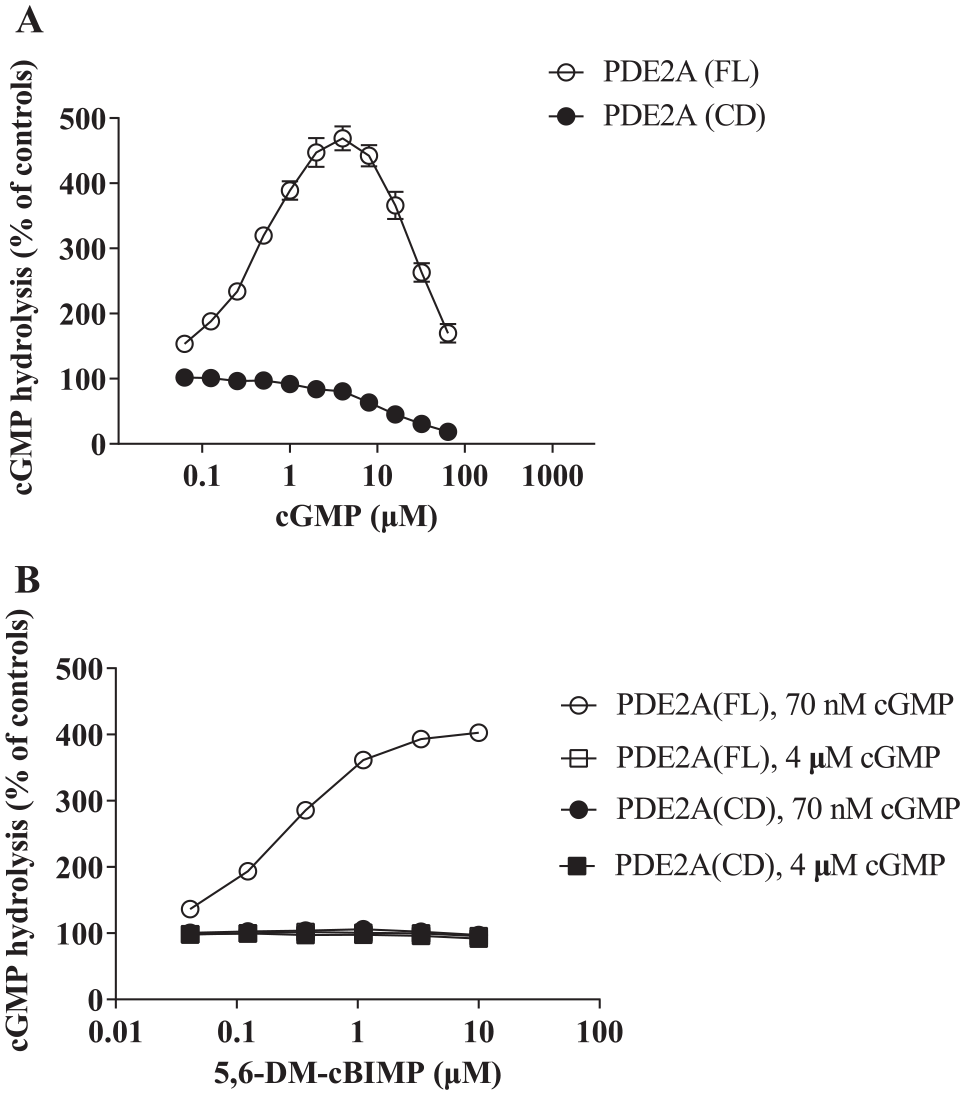
Effects of cGMP and a cyclic nucleotide analog on catalytic activities of the full-length (FL) and the catalytic domain (CD) of PDE2A. (
Preparation of Human PDE2A3 Catalytic and GAF Domains
cDNA for human PDE2A3 (GenBank: U67733) was used as a template for the construction of His-tagged full-length PDE2A3 and His-tagged PDE2A3 catalytic domain (residues 578–919 aa). Both constructs were cloned into the pcDNA3.1(+)neo vector (Thermo Fisher Scientific Inc., Waltham, MA) and transfected into COS-7 cells (ECACC, Salisbury, UK). Membrane fractions were used for the enzyme assays. To assess binding of cyclic nucleotide analogs and PDE2A inhibitors to the GAF domains, a His-SUMO-tagged GAF domain of PDE2A3 (215–573) was transfected into COS-7 cells. The enzymes and the GAF domain were stored at −70 °C until use.
Human PDE2A Enzyme Assay
PDE activities were measured using SPA beads according to the manufacturer’s instructions with some following modification. In this assay, the product of the PDE reaction from the substrate [ 3 H]cGMP can bind directly to the yttrium silicate PDE SPA beads, resulting in light emission. The enzyme assays were conducted in an assay buffer (50 mM HEPES-NaOH, 8.3 mM MgCl2, 1.7 mM EGTA, and 0.1% bovine serum albumin [pH 7.4]) in 96-well half-area plates (Corning Inc., Corning, NY). To determine the effects of compounds or cyclic nucleotide analogs on PDE enzyme activity, 10 μL of the dilution was incubated with 20 μL of PDE2A enzyme in an assay buffer at room temperature. To start the reaction, 10 μL of a substrate ([ 3 H]cGMP or a mixture of [ 3 H]cGMP and unlabeled cGMP to give a final concentration of 4 μM or 2 mM) was added to the wells for compounds, 0% controls, or 100% controls, followed by incubation at room temperature. The final assay volume per well was 40 μL, and the final concentration of DMSO in the assays was 1%. After 90 min (for assays in the presence of 2 mM unlabeled cGMP) or 30 min (for other assays), SPA yttrium silicate beads containing zinc sulfate were added (20 μL of 18 mg/mL) to terminate the PDE reaction. After resting for 60 min at room temperature, the assay plates were counted in a scintillation counter (PerkinElmer) to determine enzyme activity. The hydrolysis rate of cGMP was calculated by setting the control wells without enzyme at 0% and control wells with enzyme but without PDE2A inhibitors at 100% in each condition of concentrations of [ 3 H]cGMP and unlabeled cGMP. All experiments were conducted in triplicate and repeated three times.
GAF Domain Binding Assays
GAF domain binding assays were conducted as previously described. 11 Briefly, the recombinant His-tagged GAF domain was incubated with a PDE2A inhibitor or a cyclic nucleotide analog and 30 nM [ 3 H]cGMP in an assay buffer (50 mM HEPES-NaOH, 8.3 mM MgCl2, 1.7 mM EGTA, and 0.1% bovine serum albumin [pH 7.4]) for 30 min, followed by mixture with copper His-tag YSi SPA Scintillation beads (PerkinElmer). After resting for 60 min at room temperature, the assay plates were counted in a scintillation counter (PerkinElmer). A dissociation constant (Kd) for binding of cGMP to the His-tagged GAF domain was calculated as 44.9 nM (data not presented), which is in reasonable agreement with the previously reported Kd of 66 nM. 11
Statistics
The calculations were carried out using GraphPad PRISM version 8.2.0 (GraphPad Software Inc., San Diego, CA).
Results
Activation of PDE2A by cGMP
To confirm cGMP-dependent activation of PDE activity of PDE2A through the GAF domains, cGMP hydrolyses by the full-length and catalytic domain of PDE2A were conducted in the presence of various concentrations of unlabeled cGMP and 35 nM [ 3 H]cGMP. As illustrated in Figure 1A , 4 μM unlabeled cGMP caused a maximum of ~4.7-fold activation of full-length PDE2A, which declined at higher cGMP concentrations, in accordance with previous reports.11,20 By contrast, obvious cGMP-dependent activation was not observed for the catalytic domain alone, as we expected ( Fig. 1A ). These results demonstrate that cGMP at a concentration of ~4 μM severely affects the GAF domain-dependent positive regulation compared with cGMP at lower concentrations. Additionally, we determined the effects of a cyclic nucleotide analog, 5,6-DM-cBIMP, on activities of the full-length and catalytic domain, as its selectivity for the GAF domain over the catalytic site is as much as 7580-fold higher. 11 As illustrated in Figure 1B , 5,6-DM-cBIMP markedly increased the activity of full-length PDE2A in the presence of 70 nM cGMP (~4.0-fold over the basal levels), as described previously,11,13 whereas this effect was completely abrogated both in the full-length PDE2A in the presence of 4 μM cGMP and in the catalytic domain of PDE2A in the presence of 70 nM cGMP. These results indicate that the effect of cGMP on the positive regulation was mimicked by the cyclic nucleotide analog that did not interfere with the binding of cGMP to the catalytic site and that this positive regulation was saturated in the presence of 4 μM cGMP.
Identification of Small-Molecule Inhibitors of PDE2A
Because the PDE SPA system is well established, this system was applied for the PDE2A enzyme assays in this report. A KM value of PDE2A for cGMP was calculated as 18.9 μM from the Michaelis–Menten plot (data not presented). The screening of our small-molecule compound libraries (approximately 800,000 compounds) was performed in the presence of 77 nM cGMP, sufficiently lower than the KM value, to obtain substrate-competitive inhibitors dominantly. Through the screening campaign, hit compounds with IC50 values of less than 1 μM including compounds 1–3 were identified ( Fig. 2A ). IC50 values for the inhibitory activities of these three compounds were 120, 800, and 120 nM, respectively, in the presence of 4 μM cGMP as a substrate ( Fig. 2B ). The previously reported compounds (compounds a–c and BAY60-7550) had IC50 values of 48, 12, 5.8, and 0.13 nM, respectively ( Fig. 2B ).
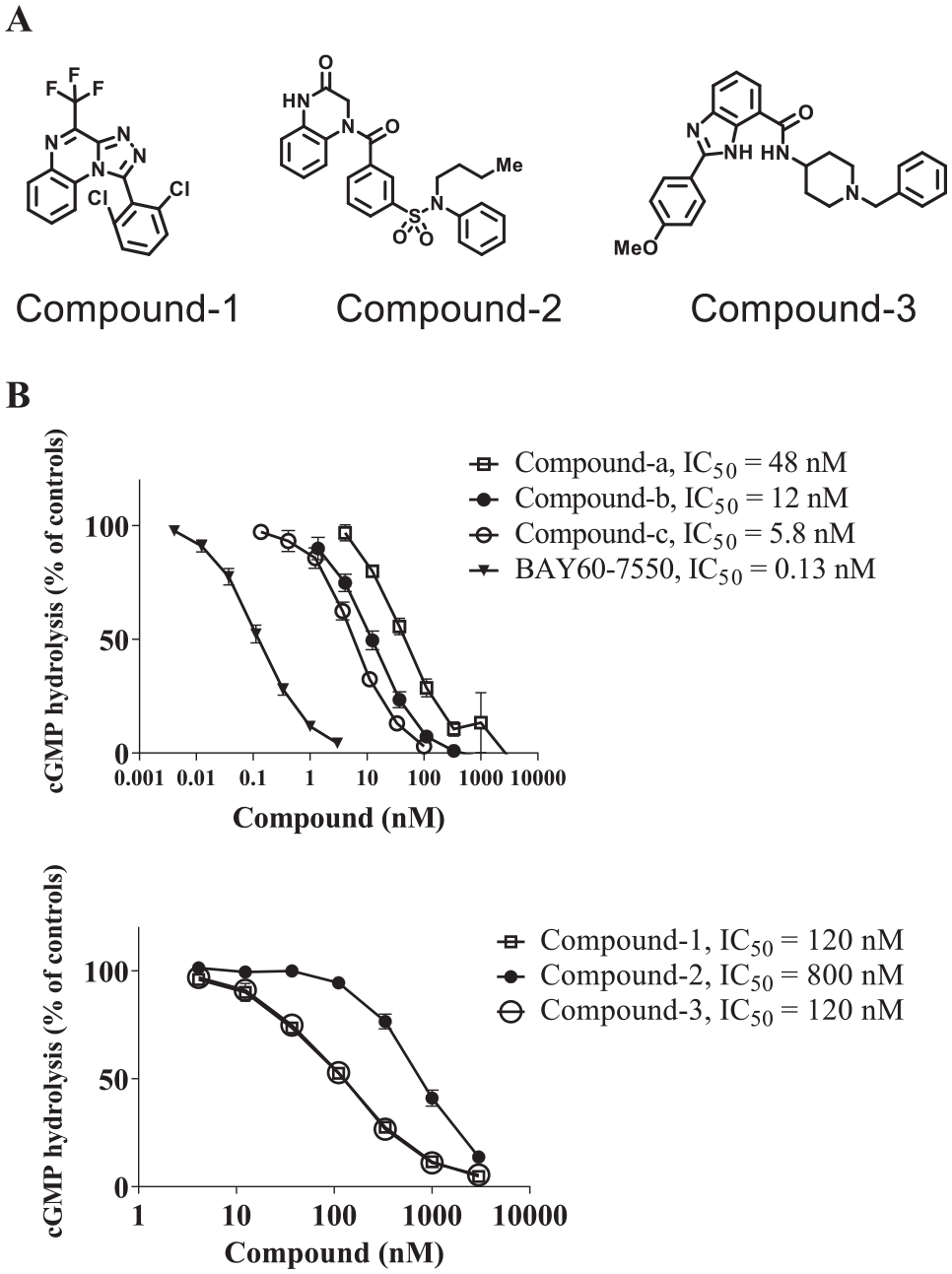
The PDE2A inhibitor hit compounds and PDE2A inhibition assays. (
Substrate Competition Analysis
To determine whether the PDE2A inhibitors are substrate competitive, we evaluated the inhibitory potency of PDE2A inhibitors on PDE activity in a cGMP competitiveness assay and compared the levels of inhibition in the full-length and catalytic domain of PDE2A. Because the KM value of PDE2A for cGMP was calculated as 18.9 μM (data not presented), we compared the inhibition curves in the presence of 4 μM (lower than the KM value) and 2 mM (around 100-fold greater than the KM value) cGMP. As illustrated in Figure 3 , all hit compounds and the previously reported compounds exhibited much lower inhibition in the presence of 2 mM cGMP than with 4 μM cGMP when using full-length PDE2A. In addition, all of the compounds had similar inhibitory activities when tested against the catalytic domain and the full-length protein. These results indicate the possibility of competitive inhibition dependent on the catalytic domain.
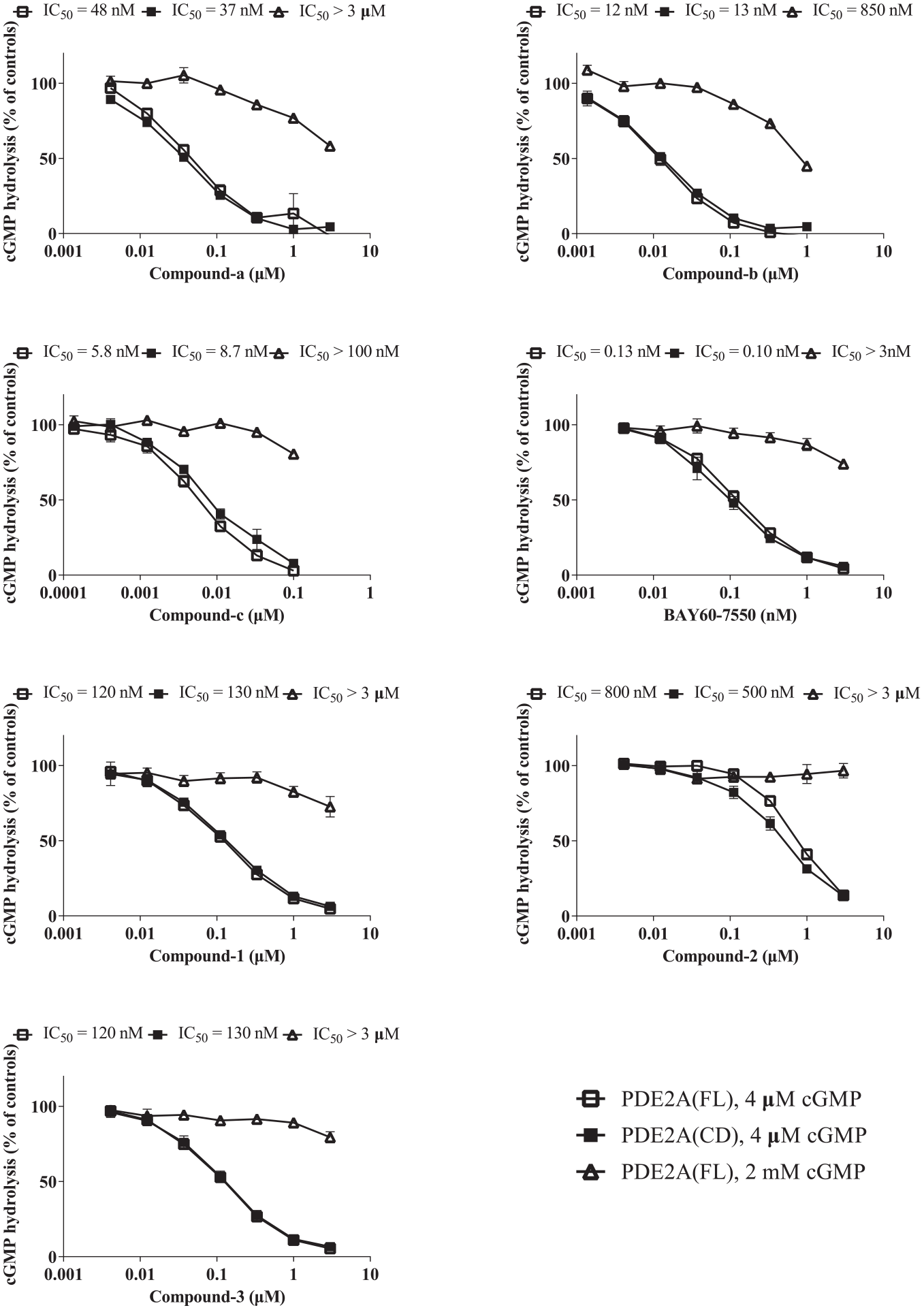
Substrate competition analyses. Previously reported PDE2A inhibitors and the hit compounds were tested in PDE2A enzyme assays in the presence of full-length human PDE2A and 4 μM or 2 mM cGMP or the catalytic domain of human PDE2A and 4 μM cGMP to predict the competitiveness of the compounds with the substrate. Each data point represents the mean ± SEM for three independent experiments.
Inhibition Mode of LAL-Type and SI-Type Inhibitors
To investigate the modes of inhibition for these PDE2A inhibitors, we compared the inhibitory effects of these compounds in the presence of 4 μM and 70 nM cGMP. Those concentrations were used because 70 nM cGMP had little effect on GAF domain-dependent PDE2A activation but was greater than the detection limit, whereas 4 μM cGMP saturated the GAF domain-dependent regulatory mechanism ( Fig. 1A,B ). Surprisingly, the inhibitory effects against full-length PDE2A of all the previously reported PDE2A inhibitors in the presence of 70 nM cGMP were 4–10 times less potent in terms of IC50 values than those in the presence of 4 μM cGMP ( Fig. 4 , compounds a–c and BAY60-7550). Contrastingly, the inhibitory effects against the catalytic domain did not decrease in the presence of 70 nM cGMP ( Fig. 4 ). We defined this type, which included all of the previously reported PDE2A inhibitors tested in this study, as “low activity in low substrate concentration type” (LAL-type). Furthermore, we studied the inhibitory effects of the previously reported PDE2A inhibitors against full-length hPDE2A in the presence of 70 nM cGMP and 10 μM 5,6-DM-cBIMP, the latter of which saturated the positive regulatory mechanism. Notably, this cyclic nucleotide analog essentially eliminated the decrease in inhibitory effects in the presence of 70 nM cGMP ( Fig. 4 ). Next, we conducted the same PDE2A assays for the newly identified hit compounds to determine their modes of PDE2A inhibition and found that one of them was a LAL-type inhibitor, similar to the previously reported PDE2A inhibitors ( Fig. 4 , compound-1). However, the other two hit compounds had a different behavior, in which inhibitory activities were similar in the presence of 4 μM and 70 nM cGMP ( Fig. 4 , compounds 2 and 3). We defined this behavior as “similar inhibition type” (SI-type).
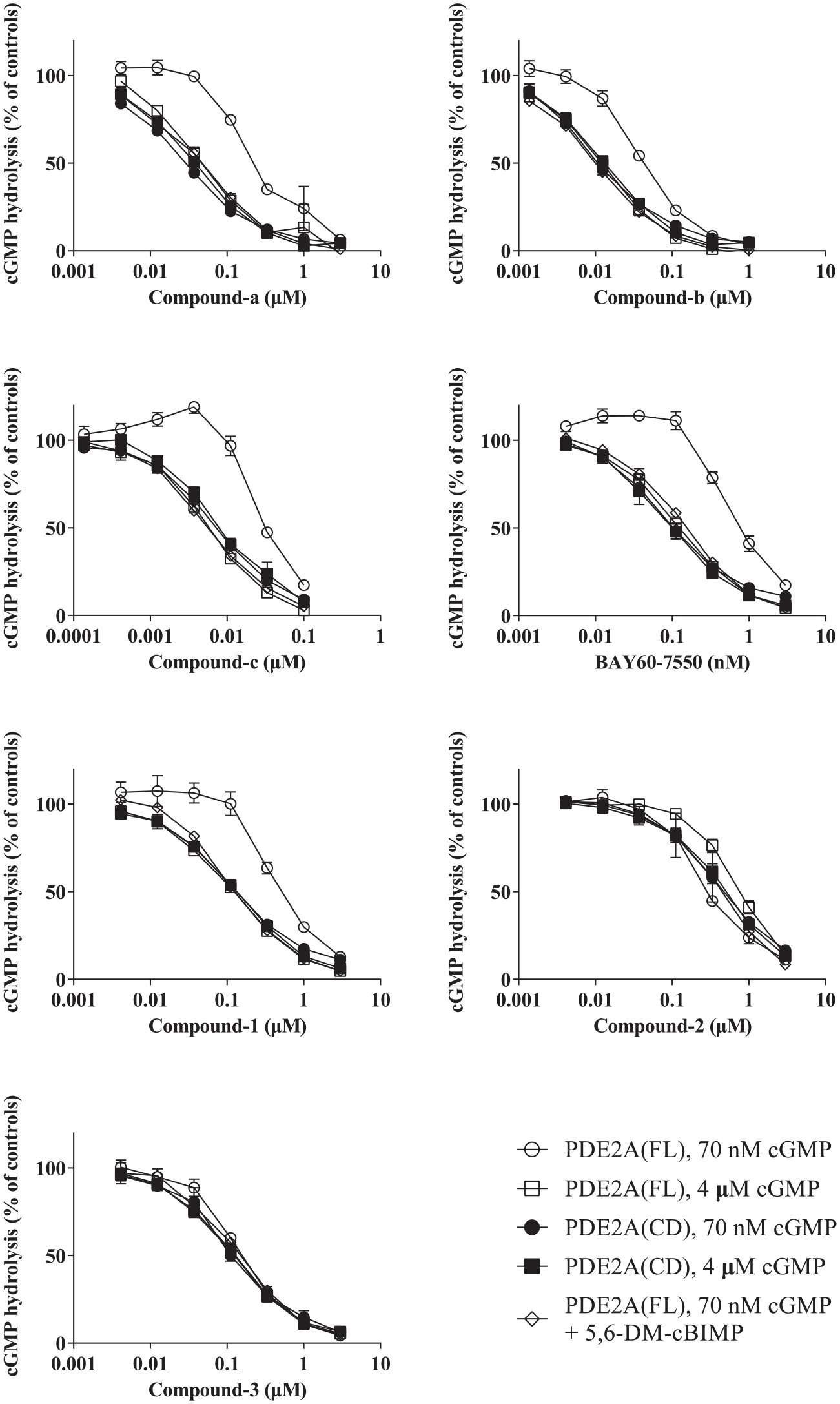
Inhibition curves of LAL-type and SI-type inhibitors. Inhibitory effects were compared in the full-length and catalytic domain of PDE2A in the presence of 70 nM and 4 μM cGMP. The inhibitory effect of the inhibitors on full-length hPDE2A in the presence of 70 nM cGMP and 10 μM 5,6-DM-cBIMP was also compared for the previously reported PDE2A inhibitors (compounds a–c and BAY60-7550) and identified hit compounds (compounds 1–3). Each data point represents the mean ± SEM for three independent experiments. Each EC50 value is listed in Supplemental Table S1.
Binding Assays of PDE2A Inhibitors to the GAF Domain
We next conducted binding assays in an attempt to elucidate the mechanisms distinguishing LAL-type and SI-type PDE2A inhibitors. Specifically, we wanted to determine whether the LAL-type inhibitors associated with the cGMP-binding sites in the GAF domain, as it had been hypothesized that direct interaction of LAL-type inhibitors with the GAF domain as well as the catalytic domain might lead to their decreased effects in the presence of low concentrations of the substrate. It was confirmed that cAMP, cGMP, and cyclic nucleotide analogs (1-NO-cAMP, 5,6-DM-cBIMP, and 2′-dcGMP) exhibited comparable inhibition of [ 3 H]cGMP binding to the GAF domain ( Fig. 5A ), as reported previously. 11 It is notable that not only the SI-type inhibitors but also the LAL-type inhibitors did not compete with cGMP binding to the GAF domain at concentrations up to 10 μM ( Fig. 5B ), which is higher than the concentrations at which LAL-type inhibitors typically present their specific features (i.e., 0.001–1 µM) ( Fig. 4 ). This result implies that the mechanism underlying the difference between the LAL- and SI-type inhibitors does not depend on binding of the inhibitors to the site in which cyclic nucleotides bind in the GAF domain.
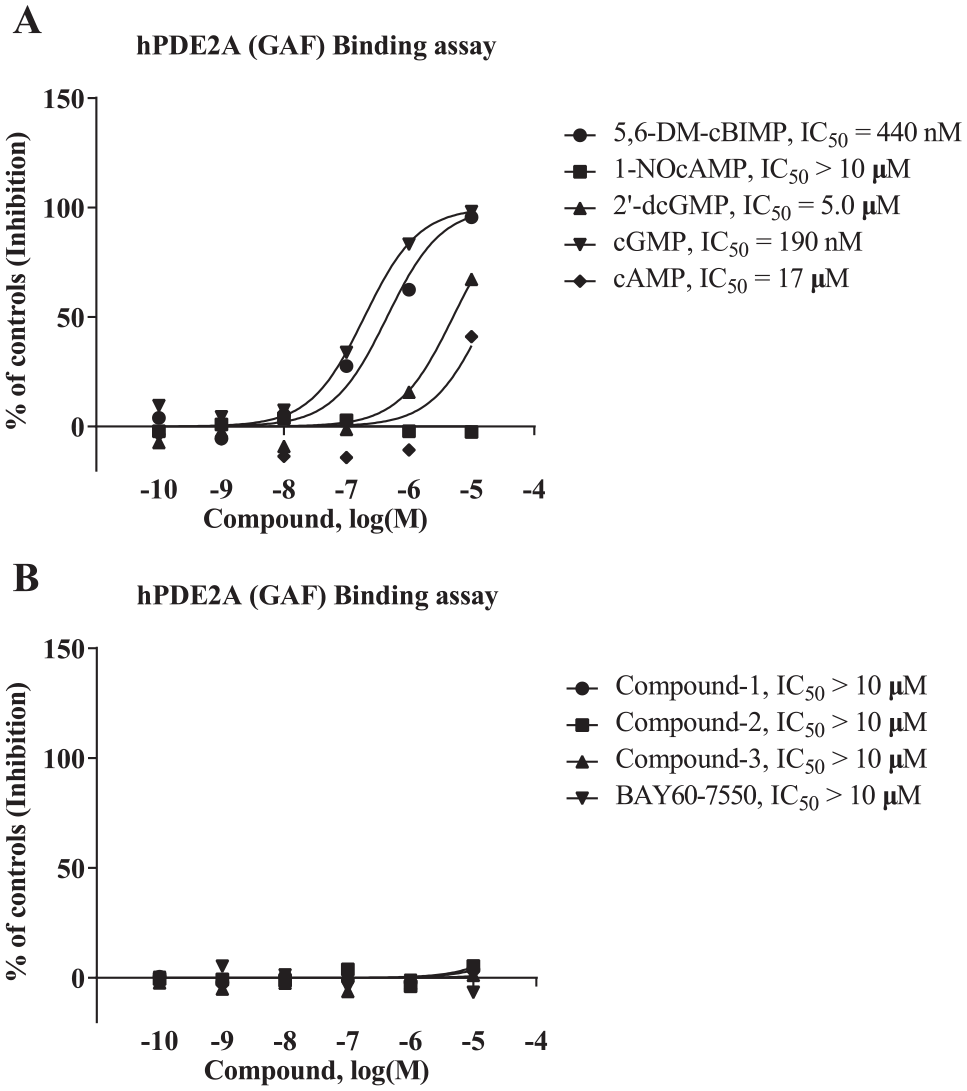
Binding of PDE2A inhibitors to the GAF domain of PDE2A. (
Discussion
There is a large body of literature indicating that PDE2A exhibits a low level of basal hydrolytic activity (“active” state) that is further stimulated when its GAF-B domains bind cAMP or cGMP (“super active” state).10,11,13,20–23 We confirmed that 4 μM cGMP saturated GAF domain-dependent PDE2A activation, whereas 70 nM cGMP, approximating the concentration in human cerebrospinal fluid,24,25 had little effect on GAF domain-dependent PDE2A activation (
We identified PDE2A inhibitors by screening in the presence of 77 nM cGMP, because this concentration is thought to be close to physiological conditions in human cerebrospinal fluid.24,25 In addition, it is sufficiently lower than the KM value to favor the identification of substrate-competitive inhibitors. We conducted PDE2A PDE assays to potentially identify a novel type of PDE2A inhibitors that have consistent inhibitory activity even in the presence of a lower concentration of cGMP. Among the hit compounds, we notably found PDE2A inhibitors that did not have decreased inhibition in the presence of a lower concentration of cGMP ( Fig. 4 , compounds 2 and 3). We here defined two types of PDE2A inhibitors as LAL-type and SI-type.
The lower inhibitory activity of LAL-type inhibitors was observed only for full-length PDE2A but not for the catalytic domain of PDE2A ( Fig. 4 ). In addition, all tested compounds had much lower inhibitory activity in the presence of 2 mM cGMP, which is around 100-fold higher than the KM value ( Fig. 3 ). In each compound, the calculated shift in IC50 values from 4 μM to 2 mM cGMP condition was no less than 17-fold, except for compound-2. Compound-2 did not suppress the cGMP hydrolysis at up to 3 μM at all in the presence of 2 mM cGMP, while it showed inhibition with an IC50 value of 800 nM in the presence of 4 μM cGMP. These results are not inconsistent with the thought that these inhibitors are competitive, and it is doubtful that the lower inhibitory activity of the LAL-type inhibitors in the presence of 70 nM cGMP, compared with 4 μM, is due to uncompetitive inhibition. The LAL-type inhibition has still not been reported in the literature. Many previously reported PDE2A inhibitors have been tested in assay conditions in the presence of 1 μM cGMP,16,18,26 probably to obtain a high signal-to-background (S/B) ratio. This is one possible reason why the LAL-type inhibition has not been reported previously.
cGMP activates PDE2A activity by binding to the GAF-B domain and, in addition, competes at the catalytic domain with radiolabeled substrates and thus inhibits their degradation.11,20 As a result, assessment of PDE enzyme activity modulation mediated by cGMP binding to the GAF-B domain is challenging. The cGMP analog 5,6-DM-cBIMP activates PDE2A catalytic activity through binding to the GAF domain but does not interfere with binding of the substrate to the catalytic domain at a concentration of 10 μM. 11 Thus, we used this cyclic nucleotide analog to induce the super active state of PDE2A.
In the presence of 10 μM 5,6-DM-cBIMP, the decrease in the inhibitory effects by the LAL-type inhibitors in the presence of 70 nM cGMP was essentially eliminated ( Fig. 4 ), implying that the feature of LAL-type inhibitors was involved in regulation controlled by the GAF domain bound to cGMP. A PDE5 inhibitor, sildenafil, was reported to inhibit PDE5 with different affinities depending on its conformation, regulated by mechanisms involving cGMP/GAF-A. 15 However, there are no reports suggesting that different affinities of PDE2A inhibitors regulate catalytic activity through GAF domains. We attempted to elucidate the mechanisms that distinguish LAL-type from SI-type inhibitors. It had been hypothesized that direct interaction of LAL-type inhibitors with the GAF domain might be responsible for their behavior. However, no obvious associations of either LAL- or SI-type inhibitors with GAF-binding sites were observed in our binding assays at concentrations up to 10 μM, which is high enough to reveal the specific features of the LAL-type ( Fig. 5B ). This implies that the mechanism underlying the difference between the LAL- and SI-type inhibitors does not depend on binding of the inhibitors to the cyclic nucleotide-binding site in the GAF domain. Differences in binding kinetics or other binding mechanisms might contribute to this difference. Full-length PDE2A has been reported to form homodimers, and the catalytic sites in a dominant formation are closed in the inner sides of both subunits when the cGMP concentration is low. 9 One possible explanation is that the LAL-type inhibitors can bind to a catalytic site in one subunit and stabilize the other catalytic site in an open configuration, although further studies are warranted to clarify the underlying mechanisms.
It should be noted that two of the newly identified PDE2A inhibitors were found to have an SI-type feature, whereas none of the previously reported PDE2A inhibitors tested in this study were of the SI-type. This suggests that these newly identified PDE2A inhibitors might be superior to previously known inhibitors for use in treating conditions such as schizophrenia and Alzheimer’s disease, in which patients have lower cGMP concentrations than healthy controls.24,25
In this article, we report the identification of small-molecule compounds as PDE2A inhibitors. We found that two of these compounds had an inhibition modality that was different from previously reported PDE2A inhibitors. These compounds’ inhibitory activities were equally potent in the presence of low and high concentrations of cGMP, while all of the previously reported PDE2A inhibitors that we tested were impaired in the presence of a low concentration of cGMP. The newly identified PDE2A inhibitor hit compounds, with a novel modality of inhibition, may be superior to the previously reported inhibitors for certain patients.
Supplemental Material
Supplemental_Material_for_ANovelInhibitionModalityforPhosphodiesterase2A_by_Nakashima,etal – Supplemental material for A Novel Inhibition Modality for Phosphodiesterase 2A
Supplemental material, Supplemental_Material_for_ANovelInhibitionModalityforPhosphodiesterase2A_by_Nakashima,etal for A Novel Inhibition Modality for Phosphodiesterase 2A by Kosuke Nakashima and Hideki Matsui in SLAS Discovery
Footnotes
Acknowledgements
The authors would like to express their great appreciation to Mitsuyo Kondo for helpful discussions regarding the experiments and for the His-PDE2A GAF domain protein preparation used for the binding assays. We would like to thank Hideyuki Oki, Yusuke Kamada, and Eri Shiraishi for valuable discussions and experiments to elucidate the inhibitory mechanism. We would also like to thank Masaaki Nishimura for constructing the plasmids carrying the full-length and catalytic domain of PDE2A. Special thanks go to Hiroki Iwashita, Takahiko Taniguchi, and Satoshi Mikami, who significantly contributed to the quality of this report through their valuable and well-aimed comments.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employed by Takeda Pharmaceutical Company Limited, and their research and authorship of this article was completed within the scope of their employment with Takeda Pharmaceutical Company Limited.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.