
Abstract
Retinitis pigmentosa (RP) is a degenerative retinal disease, often caused by mutations in the G-protein-coupled receptor rhodopsin. The majority of pathogenic rhodopsin mutations cause rhodopsin to misfold, including P23H, disrupting its crucial ability to respond to light. Previous screens to discover pharmacological chaperones of rhodopsin have primarily been based on rescuing rhodopsin trafficking and localization to the plasma membrane. Here, we present methods utilizing a yeast-based assay to screen for compounds that rescue the ability of rhodopsin to activate an associated downstream G-protein signaling cascade. We engineered a yeast strain in which human rhodopsin variants were genomically integrated, and were able to demonstrate functional coupling to the yeast mating pathway, leading to fluorescent protein expression. We confirmed that a known pharmacological chaperone, 9-cis retinal, could partially rescue light-dependent activation of a disease-associated rhodopsin mutation (P23H) expressed in yeast. These novel yeast strains were used to perform a phenotypic screen of 4280 compounds from the LOPAC1280 library and a peptidomimetic library, to discover novel pharmacological chaperones of rhodopsin. The fluorescence-based assay was robust in a 96-well format, with a Z′ factor of 0.65 and a signal-to-background ratio of above 14. One compound was selected for additional analysis, but it did not appear to rescue rhodopsin function in yeast. The methods presented here are amenable to future screens of small-molecule libraries, as they are robust and cost-effective. We also discuss how these methods could be further modified or adapted to perform screens of more compounds in the future.
Keywords
Introduction
Retinitis pigmentosa (RP) represents a group of hereditary diseases that cause progressive visual degeneration, affecting between 1:3000 and 1:5000 people worldwide. 1 Drug discovery approaches to this disease are particularly challenging due to the large number of RP-associated genes, diversity of mutations, and difficulty in characterizing the light-dependent function of proteins.2,3 Mutations in rhodopsin, a G-protein-coupled receptor (GPCR), account for 20%–30% of autosomal dominant RP cases, making rhodopsin a primary drug target. 1 Therefore, a goal of RP research is to discover compounds that function as pharmacological chaperones to correctly stabilize rhodopsin, which may prevent or even reverse vision loss.
The rhodopsin mutation P23H is responsible for the majority of autosomal dominant RP cases in North America. 1 P23H disrupts the extracellular-facing N-terminal cap, preventing rhodopsin from forming a stable complex with its natural chromophore 11-cis retinal. 4 Consequently, P23H rhodopsin does not efficiently activate the downstream G-protein transducin in response to light.4,5 In addition to this loss of rhodopsin function, overexpression of P23H rhodopsin in vitro causes the protein to accumulate in the endoplasmic reticulum (ER), 6 reducing plasma membrane localization while activating ER stress pathways. 1 Derivatives of 11-cis retinal, collectively called retinal analogs, have shown promise as pharmacological chaperones of P23H rhodopsin by improving biosynthesis, reducing ER retention, and increasing trafficking to the plasma membrane in vitro.7–9 Additional mutations in the rhodopsin N-terminal cap have also been rescued in vitro using retinal analogs, indicating potentially broad use of pharmacological chaperones to treat RP. 4 These studies have led to several clinical trials with retinal analogs, which have had mixed results, failing to significantly restore vision.10,11 Retinal analogs are also inherently light sensitive, making them difficult to deliver; moreover, they disassociate from rhodopsin following light exposure, and high concentrations are cytotoxic. 12
Based on the poor suitability of retinal analogs as drug candidates, nonretinal pharmacological chaperones have been investigated in several recent compound screening campaigns. A mammalian cell-based assay that quantifies rhodopsin trafficking to the plasma membrane 12 has contributed to the discovery of nonretinal compounds from diverse chemical families that rescue P23H rhodopsin trafficking, but not signaling function, in vitro.13–15 The diabetic drug metformin was also shown to improve both rhodopsin expression and trafficking in vitro, but counterintuitively increased photoreceptor cell death in a rat model. 16 The authors believed this failure was due to the inherent instability of P23H rhodopsin, resulting in destabilized photoreceptor outer segments. 16 This is consistent with other studies that indicate P23H instability, not ER retention, is the primary mechanism contributing to RP pathogenesis.17,18 Thus, improving the trafficking of P23H rhodopsin may not be sufficient for restoring vision.
Pharmacological chaperones that rescue both the trafficking and signaling function of other GPCRs have been discovered by using cell-based screening assays of downstream G-protein activation.19–21 Many of these compounds were originally discovered as antagonists but have shown remarkable versatility in rescuing multiple pathogenic variants of the same GPCRs.22,23 The most successful pharmacological chaperones of GPCRs, such as those of gonadotropin-releasing hormone receptor (GnRHR) and vasopressin 2 receptor (V2R), are peptidomimetics. 24 This is likely because the natural ligands of these GPCRs are peptides and peptidomimetics can diffuse into cells and mimic protein–protein interactions. Pharmacological chaperones acting on V2R, which neither are peptidomimetics nor act as antagonists, have also been discovered. 25 Thus, there may be pharmacological chaperones yet to be discovered that rescue mutant GPCR function without disturbing natural ligand interactions.
Rhodopsin differs from most GPCRs as it is covalently bound to its retinal chromophore, and is activated by light rather than a ligand, which presents unique challenges in discovering compounds that rescue rhodopsin signaling function. We previously developed a yeast-based system that functionally links human rhodopsin to the yeast mating pathway. 26 This system has the advantages of being rapid to screen and more cost-effective compared with mammalian cell-based methods, while enabling quantification of rhodopsin-dependent G-protein signaling in response to light by using a fluorescent reporter gene.
In this study, we extended yeast-based assays of rhodopsin function to screen for pharmacological chaperones that rescue rhodopsin’s ability to activate a downstream G-protein signaling pathway, acting as a proxy for rescued light-dependent function. Similar yeast-based phenotypic assays have been used to discover compounds acting on human GPCRs, 27 and various yeast-based models of human diseases have been used for compound screening assays.28–30 We show that light-dependent signaling of yeast-expressed P23H rhodopsin can be partially rescued with high concentrations of retinal chromophore, a known pharmacological chaperone. Furthermore, we describe engineered yeast strains that enable rapid quantification of G-protein activation by rhodopsin in a cellular context. Our work outlines a simple method of screening for compounds that rescue rhodopsin signaling function without requiring light exposure.
Materials and Methods
Yeast Strain Engineering
Parent yeast strain BS017 was used for all strain engineering.
Yeast Microscopy
Yeast strains expressing human rhodopsin C-terminally tagged with GFP were grown to log phase in SD-Complete media (BioShop Canada) and then plated on glass slides treated with 1 mg/mL concanavalin A (L7647, Sigma-Aldrich, St. Louis, MO) in sterile phosphate-buffered saline (AM9625, Invitrogen, Waltham, MA) supplemented with 1 mmol/L CaCl2 and 1 mmol/L MgCl2. Images were acquired using a Leica TCS SP5 confocal microscope.
Yeast Light Activation Assay and Pharmacological Rescue of Plasmid-Expressed Human P23H Rhodopsin
This protocol is similar to the “yeast light activation assay” previously described. 26 Yeast strain BS017 transformed with a human rhodopsin mutant gene in the pRS316 pTDH3 vector was incubated overnight in 5 mL of SD-URA media (BioShop Canada) at 30 °C, while shaking. To rescue P23H rhodopsin function, 20 μmol/L 9-cis retinal in ethanol (Sigma-Aldrich) was added to the overnight culture media. The following morning, cells were diluted to OD600 0.05 in 5 mL of fresh SD-URA media containing 5 μmol/L 9-cis retinal, in a LightSafe 50 mL centrifuge tube (Sigma-Aldrich), and incubated for 2 h protected from light at 30 °C, while shaking. Cultures were then exposed to light using a Fiber-Lite MI-150 high-intensity illuminator (Dolan-Jenner, Boxborough, MA) set to full intensity for 15 min at room temperature. After 100 μL samples were taken for analysis, cultures were placed back in the 30 °C shaking incubator. An additional 5 μmol/L 9-cis retinal after each light bleach was added to cultures in order to rescue the signaling function of P23H rhodopsin.
Flow Cytometry Analysis of Rhodopsin-Expressing Yeast Strains
Yeast strain BS017 transformed with a plasmid was cultured in SD-URA media, while yeast strains stably expressing rhodopsin (BS023, BS024, BS025, BS026) and GFP-tagged rhodopsin (BS027, BS028, BS032, BS033) were cultured in SD-Complete media. Cells were treated with the protein synthesis inhibitor cycloheximide (BioShop Canada), to a final concentration of 10 μg/mL. The mCherry signal, indicative of G-protein activation, of at least 6000 cells was measured for each sample with a Miltenyi Biotec MACSQuant VYB flow cytometer. In independent experiments, the GFP signal of at least 6000 cells was used to quantify rhodopsin expression levels, measured with an Invitrogen Attune NxT flow cytometer. The mean fluorescence was determined using FlowJo.
Compound Library Screening by 96-Well Fluorescence Plate-Based Assay
The LOPAC1280 library (Sigma-Aldrich) and 3000 compounds from the Ontario Institute for Cancer Research (OICR) peptidomimetic library OICR-L110 (ChemDiv, San Diego, CA) were selected for screening (4280 total compounds). Overnight cultures of yeast strains BS025 (EM rhodopsin) and BS026 (P23H EM rhodopsin) were diluted to an initial OD600 0.3 with SD-Complete media, and 500 μL of BS025 or BS026 dilution was dispensed to column 1 or columns 2–12, respectively, of a 96-well deep-well block (89237-526, VWR, Randor, PA). A total of 720 nL of DMSO vehicle or small-molecule compound was dispensed with an Echo 550 (Labcyte, San Jose, CA) into columns 1 and 12 or columns 2–11, respectively, of a V-bottom 96-well plate (3363, Corning, Corning, NY). A total of 120 μL of SD-Complete media (BioShop Canada) was added to each well and mixed with a Bravo Automated Liquid Handling Platform (Agilent). Then 100 μL from each well was added to the corresponding well in the deep-well block containing yeast, for a total volume of 600 μL per well and final concentration of 10 μmol/L LOPAC1280 (0.1% DMSO) or 5 μmol/L peptidomimetic (0.5% DMSO) compound, with yeast at a final OD600 of 0.25. Each well contained a 6 mm borosilicate glass bead (Sigma-Aldrich) to maintain yeast in suspension while culturing. Cultures were grown in the deep-well blocks for 6 h at 30 °C, while shaking. Then 100 μL from each well was transferred to a 96-well, black, clear-bottom plate (Costar 3603, Corning), and mCherry fluorescence (585 nm excitation/615 nm emission) and cell density (600 nm absorbance) were quantified using a Synergy Neo plate reader (BioTek Instruments, Winooski, VT). Potential hit compounds were tested as above in triplicate. False positives were identified by incubating compound with yeast strain BS017, which contains the mCherry reporter but does not express rhodopsin, and then measuring the fluorescence and cell density as above.
Data Analysis
The mCherry fluorescence of column 1 (DMSO, EM rhodopsin, strain BS025) of each plate served as the High Control, while the mCherry fluorescence of column 12 (DMSO, P23H EM rhodopsin, strain BS026) served as the Low Control. Assay results were normalized on a per-plate basis using mCherry fluorescence measurements, with the following equation:
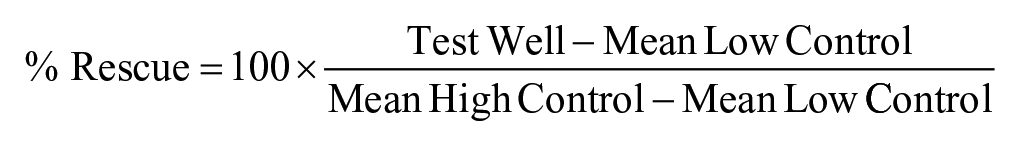
Wells with cell density >3 standard deviations (SD) below the mean Low Control cell density were eliminated from further characterization. Potential hit compounds were identified using a cutoff of three times the SD of the mean Low Control percent rescue, on a per-plate basis. The plate Z′ factor was calculated using the following equation, 31 where SDH and SDL are the SDs of the mCherry fluorescence for the plate High Control and Low Control, respectively:
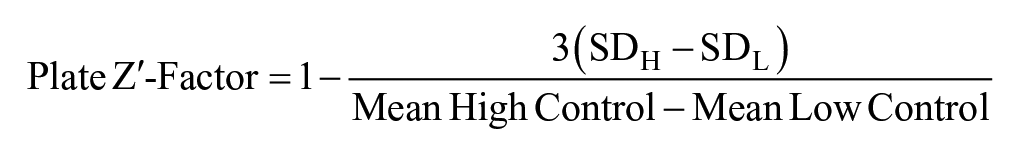
The signal-to-background ratio (S/B) of mCherry fluorescence was calculated as the ratio between the mean High Control and mean Low Control in the experiment. During assay development, the Low Control for S/B was either the yeast strain BS017 expressing P23H EM rhodopsin from a plasmid, or the yeast strain BS026 expressing P23H EM rhodopsin from the yeast genome.
Dose–Response Analysis and Light Activation Test with Compound L251-0001
Yeast strain BS026 (P23H EM rhodopsin) was incubated overnight in SD-Complete media. Yeast cells were diluted to OD600 0.05 into 600 μL of fresh media, containing up to 320 μmol/L of peptidomimetic compound L251-0001 in DMSO (MolPort, Beacon, NY; compound named by MolPort cat. #; also MolPort-010-860-710; original library identifier, OICR0005849A01). At the highest compound concentrations, the cultures contained up to 0.3% DMSO. Cultures were then incubated for 6 h at 30 °C, while shaking. Pharmacological rescue with 9-cis retinal was performed as described above, with the additional step of adding 50 μmol/L L251-0001 after diluting overnight cultures to OD600 0.05 in 5 mL of fresh SD-Complete media, and utilizing yeast strains BS023 (WT rhodopsin) and BS024 (P23H rhodopsin) instead of expressing rhodopsin from a plasmid. Following light activation, the mCherry signal of at least 6000 cells was measured for each sample with a Miltenyi Biotec MACSQuant VYB. The mean mCherry fluorescence was determined using FlowJo.
Results and Discussion
Rescue of Plasmid-Expressed P23H Rhodopsin
We first sought to determine if incubation with 9-cis retinal, a well-characterized pharmacological chaperone of rhodopsin, would rescue the light-dependent activation of P23H rhodopsin. Previous studies have shown that incubating cells with retinal during P23H rhodopsin biosynthesis is crucial to stabilizing the protein.4,5,12 Thus, yeast strains were incubated with 20 μmol/L 9-cis retinal overnight, and then 5 μmol/L 9-cis retinal after refreshing the media. To rescue P23H rhodopsin, 5 μmol/L 9-cis retinal was also added after each time the samples were exposed to light, a step that is required to activate rhodopsin. This treatment rescued approximately 20% of P23H rhodopsin’s signaling activity in response to light (
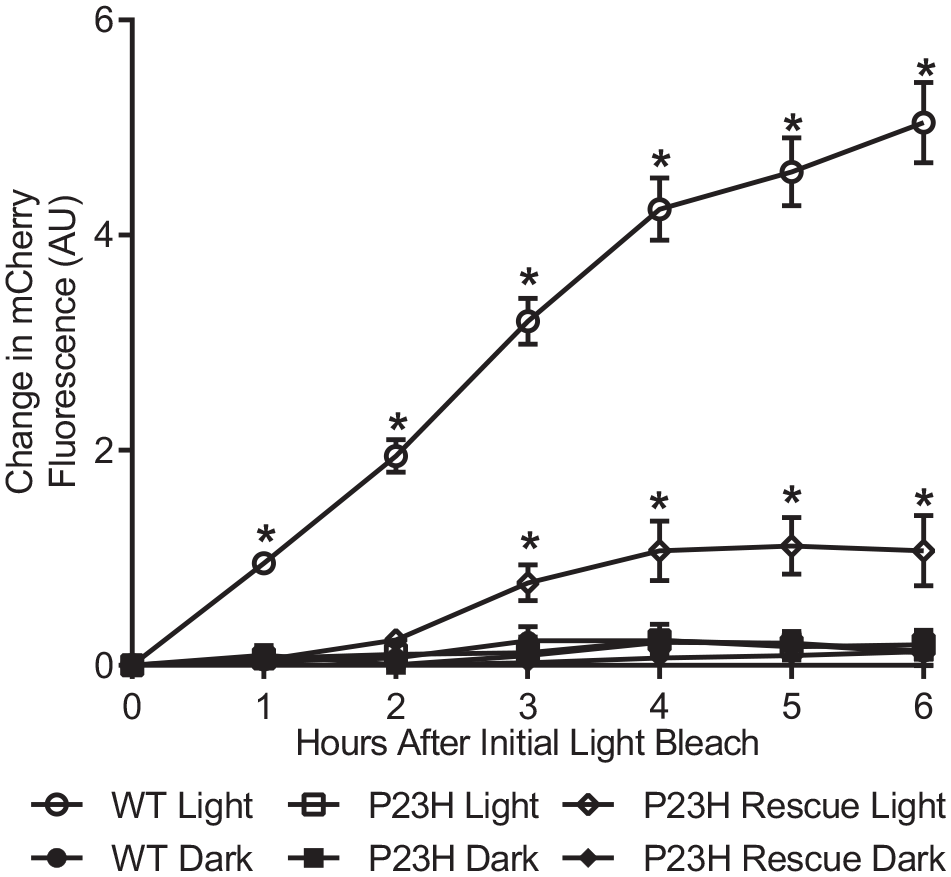
Flow cytometry-based assay to measure light-dependent activation of rhodopsin in yeast. Partial rescue of P23H rhodopsin activation in response to light was observed, following incubation with 20 μmol/L 9-cis retinal overnight, followed by 5 μmol/L 9-cis retinal after each light bleach (P23H Rescue Light). The initial mCherry fluorescence of each sample (0 h time point) was subtracted from each, to compare the relative change in fluorescence. Data points represent averages of four individual colonies, each from a 5 mL culture. Error bars represent SEM; *P < 0.05 as determined by a two-way ANOVA.
However, this repetitive addition of light-sensitive retinal followed by exposing cultures to light was not conducive to a compound screening assay. Thus, we sought to determine if the function of constitutively active rhodopsin could be rescued by incubating with 9-cis retinal. The previously characterized constitutively active synthetic mutant was generated by site-directed mutagenesis,
32
with or without the pathogenic P23H mutation (
(
Genomic Expression of Rhodopsin Improves Downstream Signaling
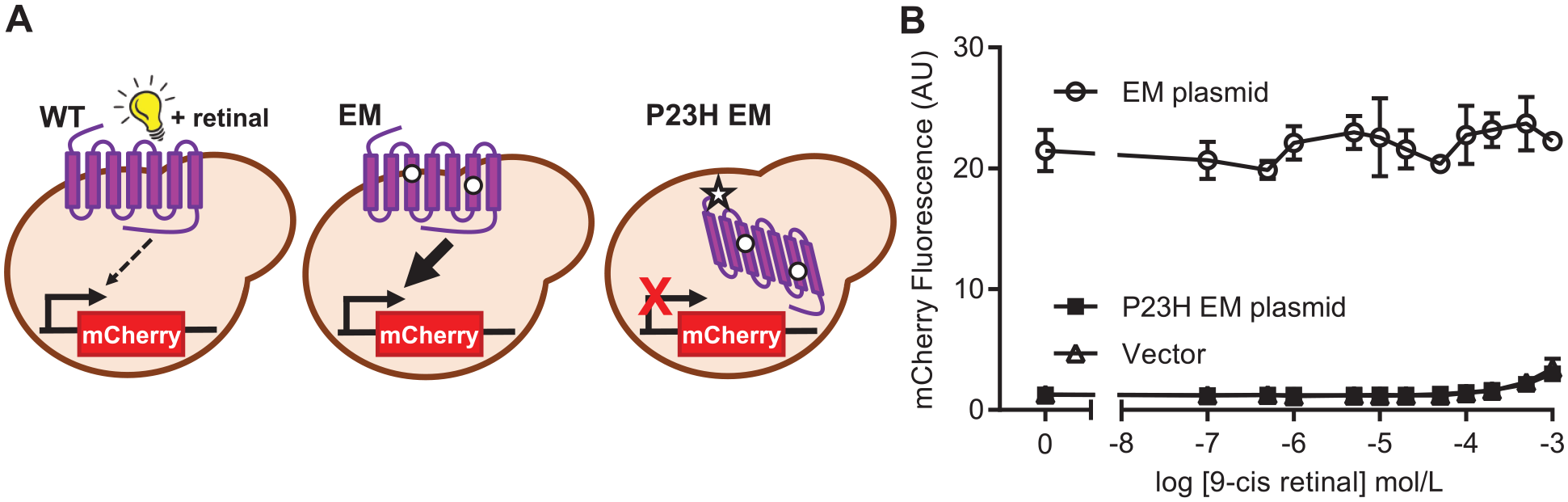
As plasmid-based expression is more variable than the expression of genomically integrated genes in yeast,
33
we postulated that genomic integration of rhodopsin could improve a rescue phenotype. Following successful integration of EM rhodopsin into the yeast genome, mCherry-positive cells increased from 42% to 87% of the total (
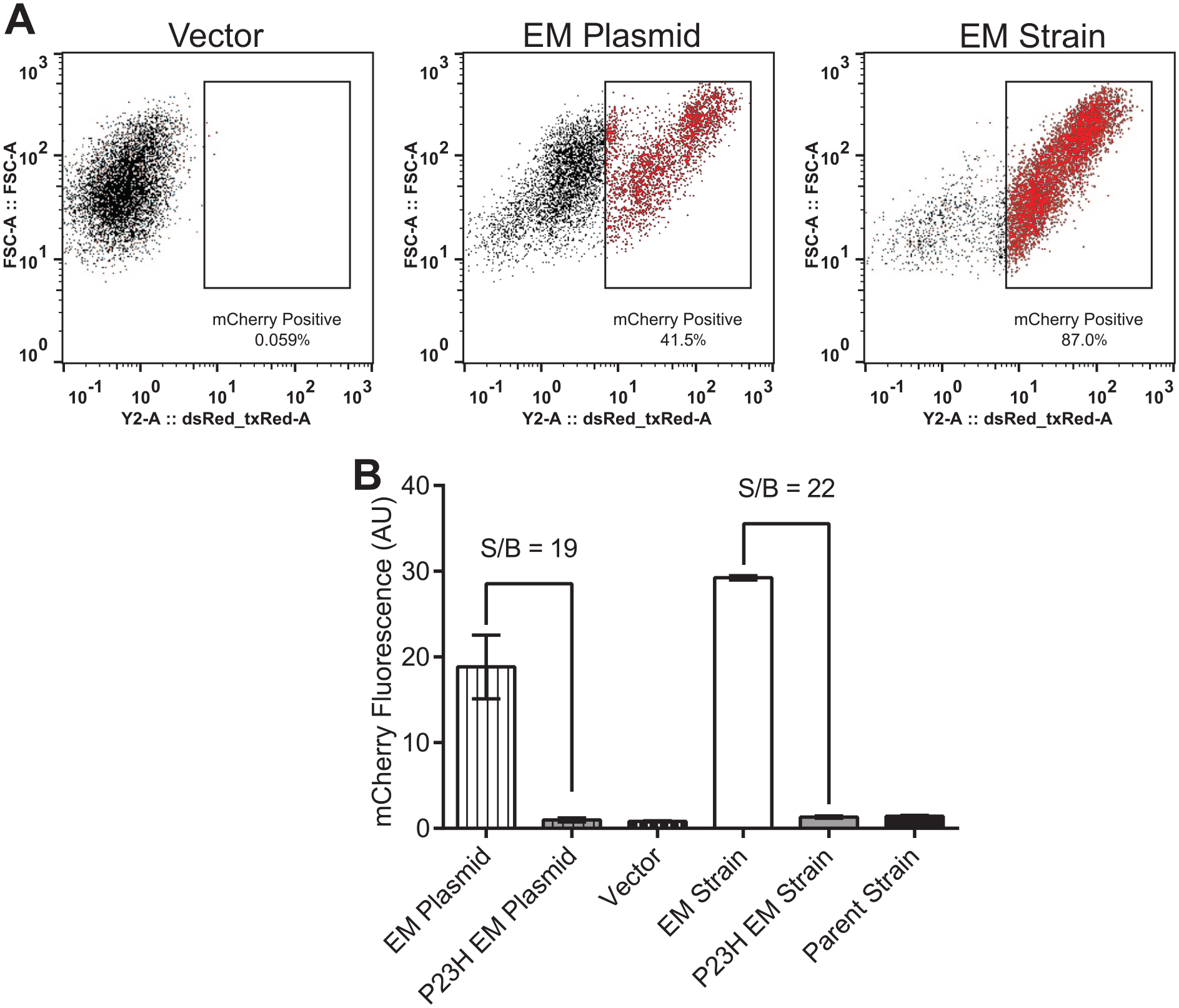
(
Although the mCherry S/B was improved by using genome-integrated rhodopsin, the mCherry signal from the P23H EM strain was not improved by incubation with up to 20 μmol/L 9-cis retinal for either 6 or 16 h (data not shown). Combined with the results from plasmid-expressed EM rhodopsin, this suggests that in yeast, 9-cis retinal does not act as a pharmacological chaperone of rhodopsin when it also contains the EM mutations.
Development of 96-Well Fluorescence Plate Reader Assay
In instances where a positive control compound cannot be found for a compound screening assay, an artificial rescue phenotype (i.e., EM rhodopsin) can be used as a positive control instead. 34 The high S/B between strains expressing EM rhodopsin versus P23H EM rhodopsin provided a foundation to develop an unbiased phenotypic screening assay, requiring only that G-protein signaling is rescued by a compound, which is predicated on correct folding and trafficking of rhodopsin to the plasma membrane. A general goal for pharmacological chaperone screens is to discover compounds that do not compete for the native ligand binding pocket; 19 thus, the inability of retinal to bind EM rhodopsin is not expected to negatively impact screening applications of our assay.
Up to this point, all fluorescence measurements were conducted with flow cytometry-based methods, which can be difficult to adapt to a rapid compound screen when lacking access to a high-throughput sampling flow cytometer. We therefore investigated the use of a fluorescence plate reader-based assay, which could measure fluorescence in up to 96 wells at once. When using flow cytometry, mCherry expression can be detected just 1 h after rhodopsin has been activated by light, and measurement at 6 h gives a robust S/B, 26 which is comparable to other fluorescent reporters of yeast mating pathway activation. 35 Based on this, we reasoned that 6 h would be sufficient time to detect rescued rhodopsin function during a compound screen.
However, fluorescence plate reader measurements are typically less sensitive than flow cytometry, and the fluorescence signal is influenced by cell density. Thus, various initial cell densities were tested to ensure the S/B remained high, balanced with the desire to have a high compound-to-cell ratio during a 6 h incubation. An initial cell density of OD600 0.25 followed by culturing the yeast for 6 h led to an S/B of >12 in these initial tests, while maintaining a relatively low number of cells in the well (
Pilot Screen of 4280 Compounds
To investigate the feasibility of our phenotypic screening strategy, a pilot screen of 4280 compounds was performed. The yeast strain containing P23H EM rhodopsin integrated into the genome was incubated with compounds from chemical libraries, where observed mCherry fluorescence would indicate successful rescue of rhodopsin’s signaling function. The LOPAC1280 library was chosen based on its well-characterized suite of chemicals, with ~50% of compounds targeting human GPCRs. 36 A peptidomimetic compound library compiled by OICR was also selected based on previous reports of peptidomimetics functioning as pharmacological chaperones of GPCRs. 24 The 4280 compounds were screened over the course of 3 days, utilizing 96-well deep-well blocks to culture the yeast in the presence of compound. mCherry signal was measured using a fluorescence plate reader, and percent rescue was normalized to the High Control in rows of the same plate (EM strain). The scatterplot of the normalized results of the pilot screen is shown in Figure 4 . An average Z′ factor of 0.65 ± 0.11 (SD) for controls in the complete library screen was achieved, indicating a robust screening assay. 31 A cutoff for determining potential hits was set at a percent rescue 3SD above the plate Low Control. Forty-eight compounds were found above this cutoff; however, they were each very close to the noise of the assay.
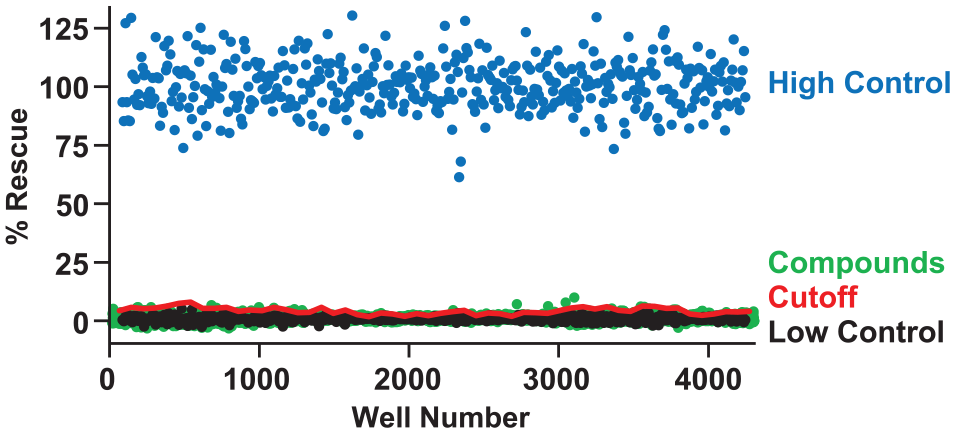
Compound screening to discover pharmacological chaperones of P23H EM rhodopsin. A total of 4280 compounds from LOPAC1280 and the OICR peptidomimetic libraries were screened in singlicate, at 10 and 5 μmol/L, respectively. Data are shown normalized to the High Control, a strain expressing constitutively active EM rhodopsin. A cutoff value of three times the SD of the mean plate Low Control (P23H EM + DMSO only) was used to determine hits. The screen had an average plate Z′ score of 0.65 ± 0.11 with an average plate S/B of 14.7 ± 4.7.
These 48 compounds and 1 additional compound of interest (13-cis-retinoic acid) were tested again for their ability to rescue P23H EM rhodopsin function in our plate reader-based assay, this time in triplicate. Four peptidomimetic compounds had one replicate above 3% rescue (
Characterization of L251-0001
To better determine if compound L251-0001 could rescue rhodopsin’s function, a dose–response assay was performed using the P23H EM strain, followed by measurement of mCherry fluorescence by flow cytometry (
Future Refinement and Applications
Although the pilot screen we performed did not discover a compound that rescues the function of P23H rhodopsin, these yeast-based methods were simple to perform, robust, and maintained a relatively low cost ($10–$15 per 96-well plate). However, a larger high-throughput screening campaign, which typically involves 10- to 100-fold the number of compounds screened here, will require refinement of these methods. Miniaturizing the assay to 384-well plates may be possible, 37 facilitated by replacing the fluorescent reporter gene with an auxotrophic marker to enable a growth-based assay, although the lack of aeration can pose a challenge. 30 Plate reader-based assays also enable the continuous monitoring of yeast growth and reporter gene expression during culturing, which could be explored in future experiments. It may also be advantageous to incubate yeast for up to 48 h with compound and monitor phenotype, a strategy used to identify promising compounds in other yeast-based assays.28–30,37
There is also the yeast cell wall to consider, which mammalian cells lack, which may impede the diffusion of compounds into the cell. The cell wall has a size exclusion of ~4500 Da, 38 but the libraries screened here were under this size cutoff (OICR average, 418 Da; LOPAC1280 average, 337 Da). It remains possible that compounds that naturally diffuse into mammalian cells may bind the cell wall, limiting their diffusion. Evaluating the efficacy of the recently published nonretinal pharmacological chaperones of rhodopsin would help to reveal such differences between yeast and mammalian cell phenotypes.13–15
Constitutively active GPCRs have been used in compound screens to discover antagonists or inverse agonists of GPCRs, 39 and pharmacological chaperones often act as an antagonist or inverse agonist of the GPCR they rescue. Thus, rather than screening for the rescued signaling function of P23H EM rhodopsin, the EM rhodopsin yeast strain could instead be used to discover novel antagonists and inverse agonists of rhodopsin, by screening for reduced constitutive activity. Such a screen would extend applications of the yeast-based screening assay described here, and hit compounds may additionally have applications as novel pharmacological chaperones of rhodopsin.
Supplemental Material
Supplemental_Material_for_Yeast-Based_Model_of_Retinal_Disease_by_Scott_et_al – Supplemental material for Screening of Chemical Libraries Using a Yeast Model of Retinal Disease
Supplemental material, Supplemental_Material_for_Yeast-Based_Model_of_Retinal_Disease_by_Scott_et_al for Screening of Chemical Libraries Using a Yeast Model of Retinal Disease by Benjamin M. Scott, Leanne E. Wybenga-Groot, C. Jane McGlade, Elise Heon, Sergio G. Peisajovich and Belinda S. W. Chang in SLAS Discovery
Footnotes
Acknowledgements
The small-molecule compound screening assay was developed and performed at SPARC BioCenter, The Hospital for Sick Children, Toronto, Ontario. Microscopy was performed with assistance from Dr. Joy Dunkers, at the National Institute of Standards and Technology (NIST), Gaithersburg, Maryland. NIST notes that certain commercial equipment, instruments, and materials are identified in this paper to specify an experimental procedure as completely as possible. In no case does the identification of particular equipment or materials imply a recommendation or endorsement by NIST, nor does it imply that the materials, instruments, or equipment are necessarily the best available for the purpose. The opinions expressed in this article are the authors’ own and do not necessarily represent the views of NIST.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported in part by an NSERC Discovery grant (BSWC), and the SickKids HTS subsidy program (CJM, EH).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.