
Abstract
Physiological nucleosides are used for the synthesis of DNA, RNA, and ATP in the cell and serve as universal mammalian signaling molecules that regulate physiological processes such as vasodilation and platelet aggregation by engaging with cell surface receptors. The same pathways that allow uptake of physiological nucleosides mediate the cellular import of synthetic nucleoside analogs used against cancer, HIV, and other viral diseases. Physiological nucleosides and nucleoside drugs are imported by two families of nucleoside transporters: the SLC28 concentrative nucleoside transporters (CNTs) and SLC29 equilibrative nucleoside transporters (ENTs). The four human ENT paralogs are expressed in distinct tissues, localize to different subcellular sites, and transport a variety of different molecules. Here we provide an overview of the known structure–function relationships of the ENT family with a focus on ligand binding and transport in the context of a new hENT1 homology model. We provide a generic residue numbering system for the different ENTs to facilitate the interpretation of mutational data produced using different ENT homologs. The discovery of paralog-selective small-molecule modulators is highly relevant for the design of new therapies and for uncovering the functions of poorly characterized ENT family members. Here, we discuss recent developments in the discovery of new paralog-selective small-molecule ENT inhibitors, including new natural product-inspired compounds. Recent progress in the ability to heterologously produce functional ENTs will allow us to gain insight into the structure and functions of different ENT family members as well as the rational discovery of highly selective inhibitors.
Keywords
Introduction
In cellular systems, the separation of specialized chemistries is achieved mainly through compartmentalization by selectively permeable membranes that are intrinsically impermeable to most cellular molecules. Highly lipophilic small molecules can freely diffuse across bilayers, while large, hydrophilic, and charged molecules require specialized transport systems to facilitate import or export out of prokaryotic and eukaryotic cells. Naturally occurring purine (adenosine, guanosine, and inosine) and pyrimidine (uridine, cytidine, and thymidine) nucleosides act as metabolic precursors for the biosynthesis of fundamental biological molecules such as DNA, RNA, and ATP, but also serve as signaling molecules and neuromodulators that contribute to the regulation of a broad range of cellular events (
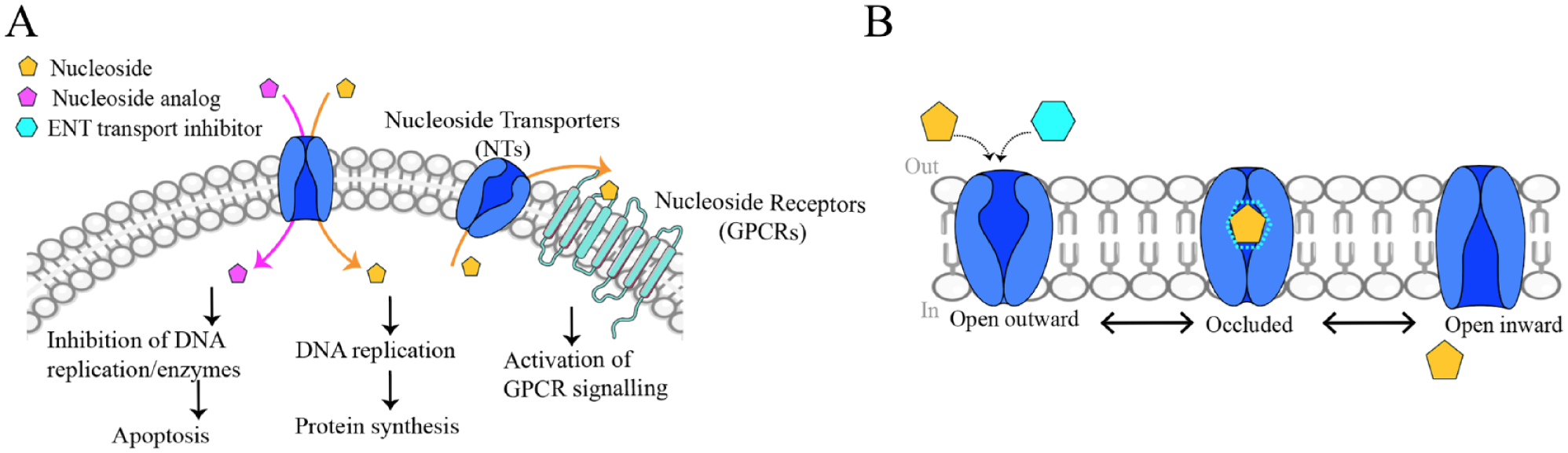
Physiological and pharmacological role of nucleosides and NAs mediated by ENTs. (
Because of their hydrophilic nature, both nucleosides and their chemical analogs (
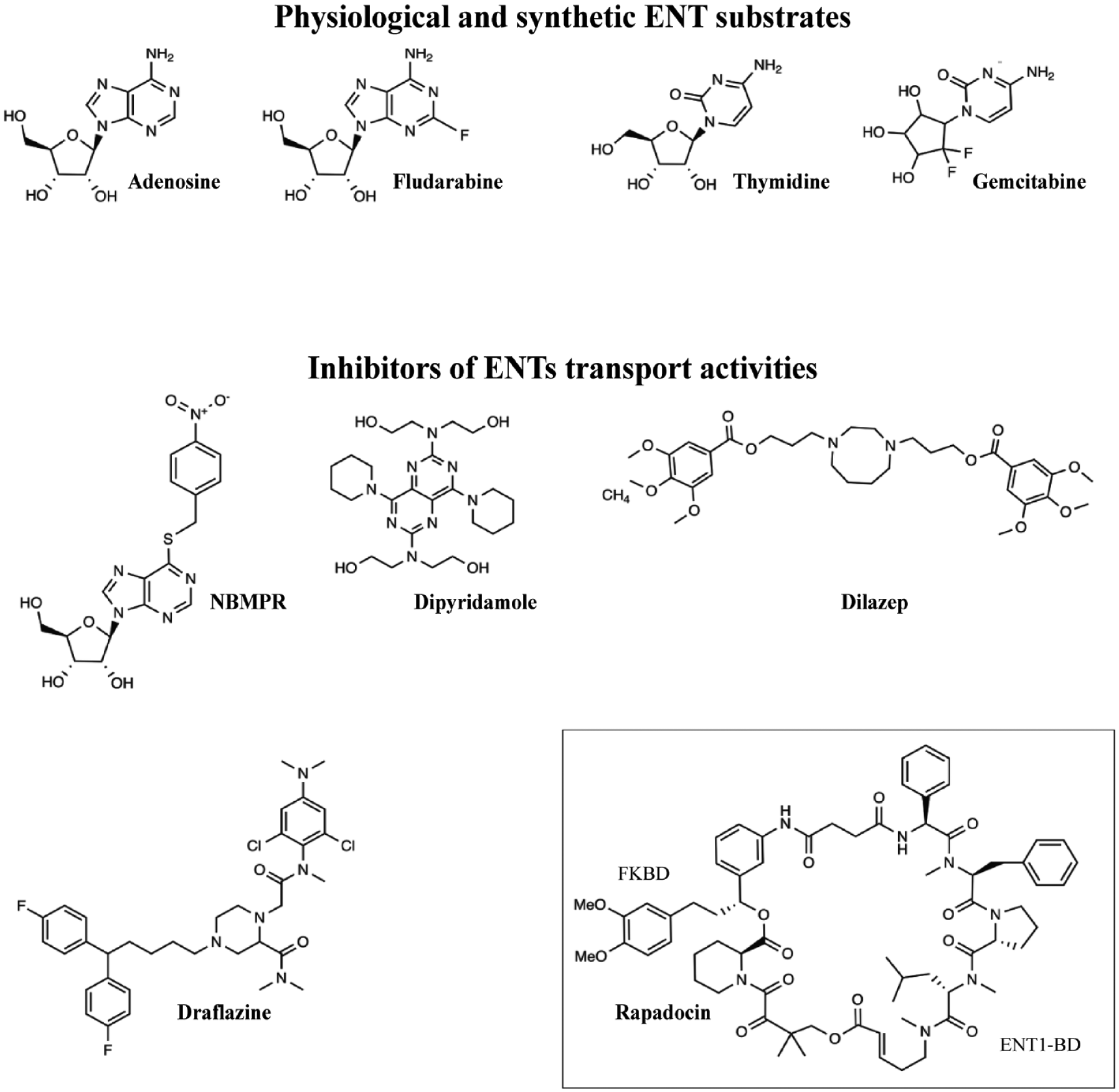
Representative chemical structures of nucleosides, their synthetic analogs, and inhibitors of ENT transport. Both physiological nucleosides, such as adenosine and thymidine, and their respective synthetic analogs, fludarabine and gemcitabine, are transported by ENTs. NBMPR, dilazep, dipyridamole, and draflazine block the nucleoside transport mediated by all ENT proteins, whereas rapadocin specifically inhibits only hENT1. Rapadocin inhibits hENT1 in a manner dependent on simultaneous binding to FKBP12 analogous to the way the natural product rapamycin binds its cellular target mTOR. All chemical structures were adopted from https://pubchem.ncbi.nlm.nih.gov.
Many earlier reviews focus on the identification and classification of nucleoside transport systems in various mammalian cells, the kinetic analysis of the transport mechanism, and the functional characteristics of different transport proteins.7,16,17 Ever since the first identification of NT proteins by the group of Prof. Steve Baldwin, 18 numerous independent groups have studied the roles of various residues and mutations and their implications on ENT transport and substrate selectivity. While these studies have been highly useful, the lack of 3D structural information has hampered the ability to gain a deeper understanding of ENT–ligand interactions in the physiological context.
In this review, we provide a new homology model for the human equilibrative nucleoside transporter-1 (hENT1), which provides a structural context for addressing the structure–function relationship data provided by earlier mutagenesis campaigns, and underline contributions of key ENT1 residues involved in gating, ligand binding, and substrate translocation. We believe this approach can identify key residues that may confer drug resistance in cancer or allow ENT engineering to increase the protein stability required for structural studies. Similarly, identifying point mutations that diminish the binding of physiological substrates without affecting inhibitor binding may allow conformation-selective trapping of the protein. Further, we highlight the conserved ENT regions and amino acid residues in several species from protozoa to mammals, which can allow the discovery of functional evolutionary linkages and structures of ENT homologs. Finally, we review data from recent attempts to heterologously express and purify human ENT1 for structural studies and discuss recent discoveries of new isoform-selective potent small-molecule ENT inhibitors, including new natural product-inspired compounds. 19
Residue Conservation and a Generic Residue Numbering Scheme for ENTs
We identified conserved residues and regions between different ENTs from 16 different species using ClustalW (https://www.ebi.ac.uk/Tools/msa/clustalw2/). We assigned sequence-based residue numbers for all ENTs similar to the numbering system developed for G-protein-coupled receptors (GPCRs).
20
Briefly, the X.50 format is used where X denotes the number of the TM helix (1–11) followed by a residue position relative to the most conserved residue between different species in the TM helix (50). In total there are 8, 14, 7, and 11 conserved residues found in ENT1, ENT2, ENT3, and ENT4, respectively. These conserved residues and regions are mostly clustered within the predicted TM helices of ENT transporters (
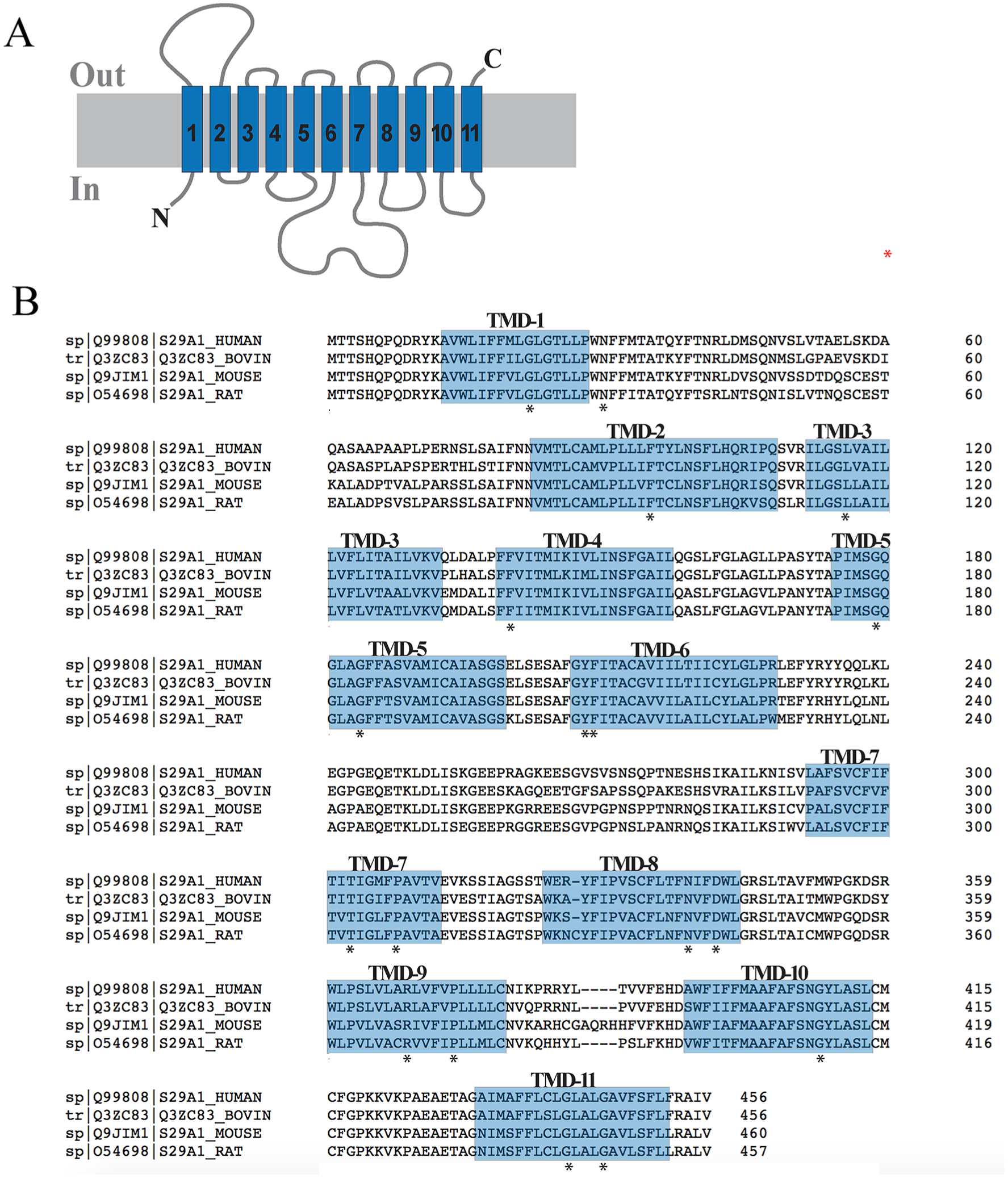
hENT1 topology and residue conservation. (
Equilibrative Nucleoside Transporter Family (SLC29).
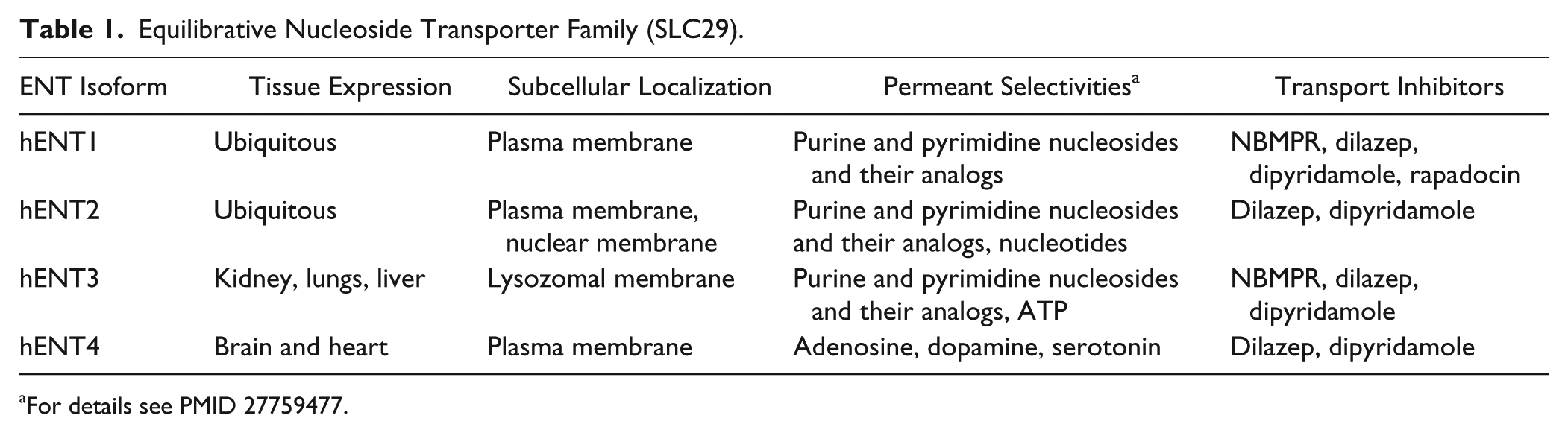
For details see PMID 27759477.
Conserved Amino Acids, Their Positions, and TM Domains in Respective ENT1 Proteins from Various Species.
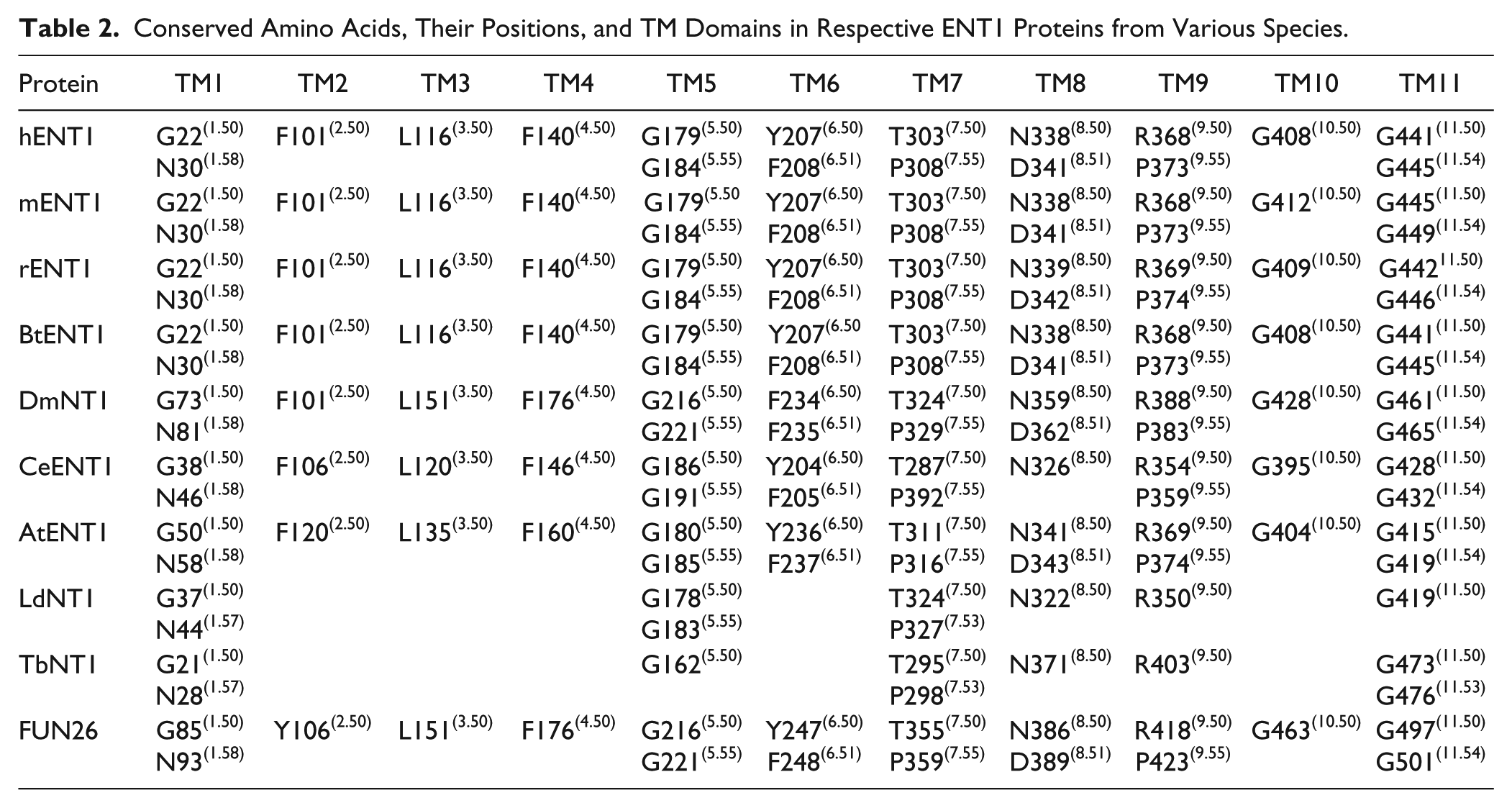
Conserved Amino Acids, Their Positions, and TM Domains in Respective ENT2 Proteins from Various Species.
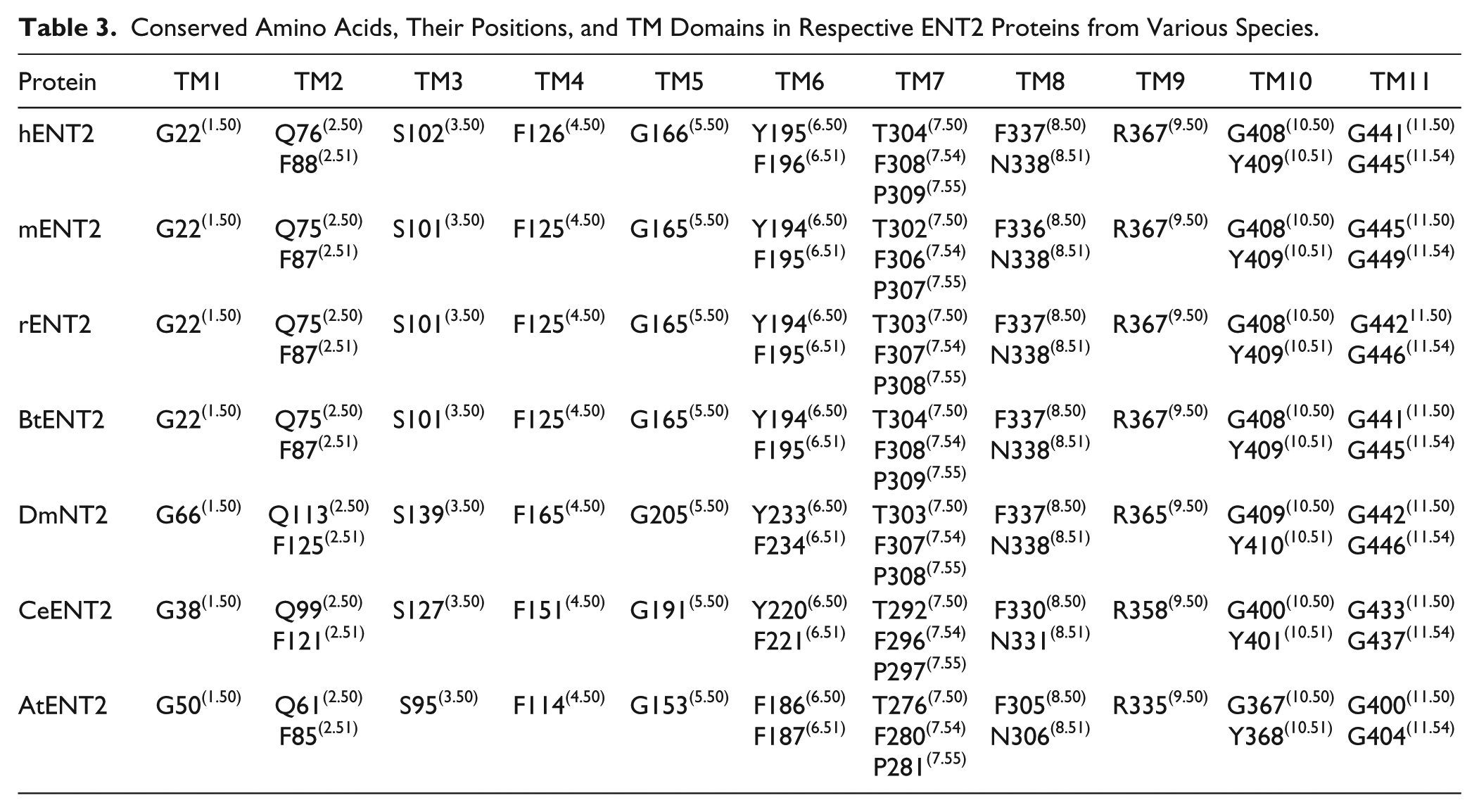
Conserved Amino Acids and Their Positions in Respective ENT3 Proteins from Various Species.
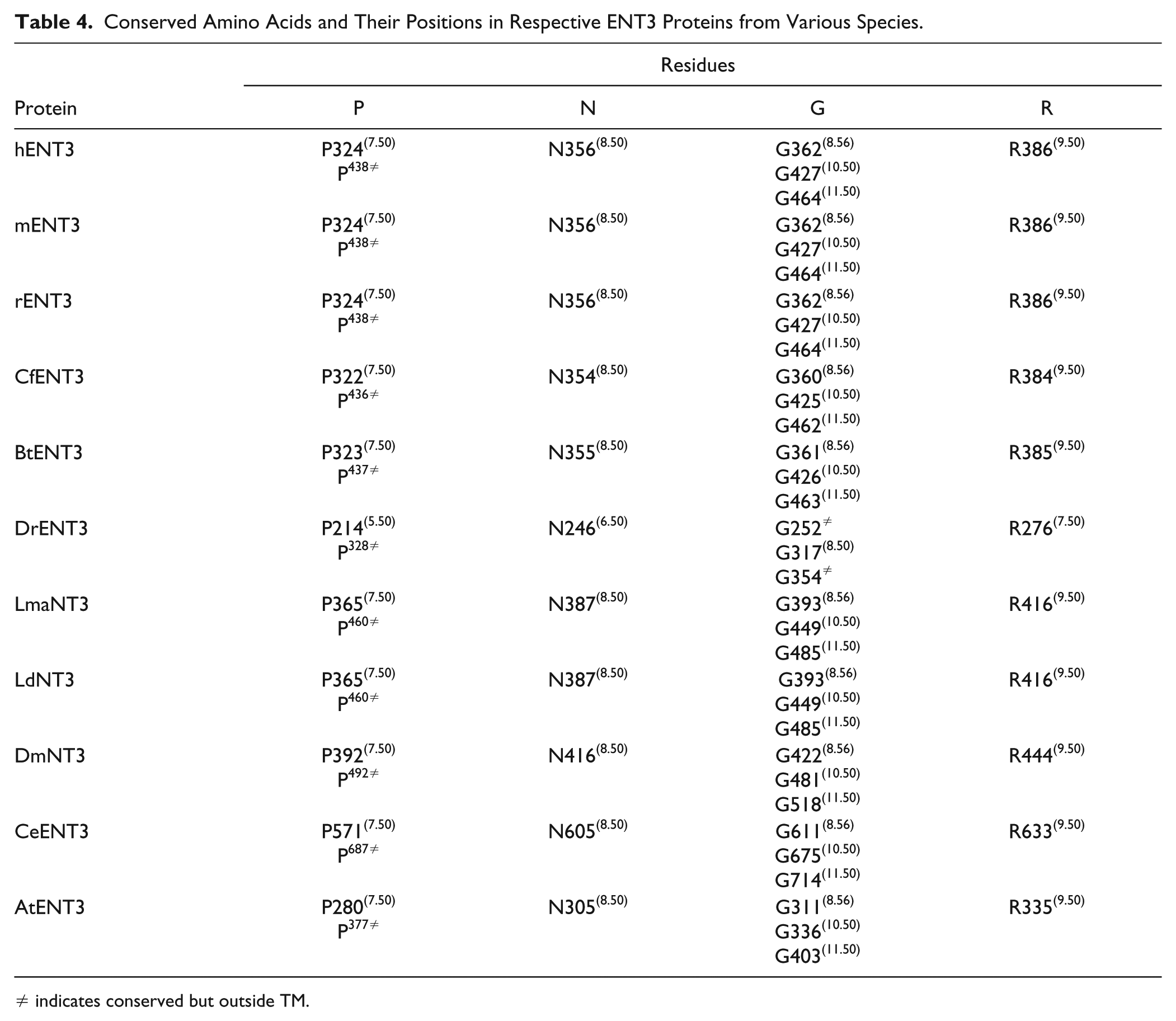
≠ indicates conserved but outside TM.
Conserved Amino Acids and Their Positions in Respective ENT4 Proteins from Various Species.
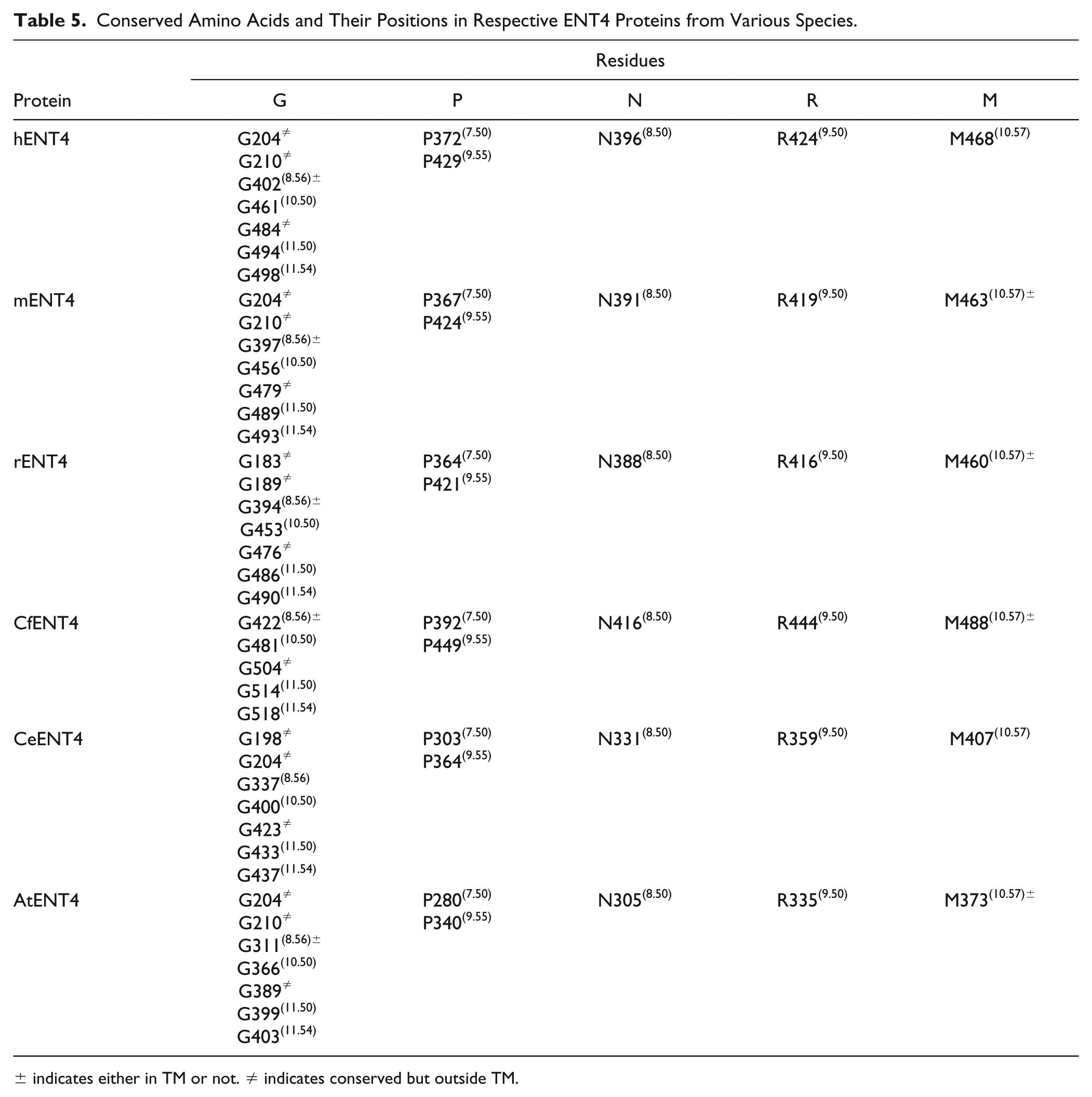
± indicates either in TM or not. ≠ indicates conserved but outside TM.
Structure–Function Relationship of hENT1
Investigation of the 3D structure of ENTs has been hampered by low endogenous expression levels in many host organisms and difficulty in isolating them in a functional form. However, a wealth of information about the structure–function relationships of hENT1 has been produced using site-directed mutagenesis and generation of chimeras between human and rat NTs, with ligand binding assays (
Summary of Mutagenesis Studies in hENT1 Protein.
To analyze the positions of hENT1 predicted to have an active role in ligand binding and/or substrate transport, we created a homology model of the hENT1 structure (see Supplemental Information). Since homology searches using the hENT1 sequence (UniProtKB Q99808) against the Protein Data Bank (PDB) entries at the National Center for Biotechnology Information (NCBI) (http://blast.ncbi.nlm.nih.gov/) failed to reveal suitable structural templates for homology modeling, we used I-TASSER structure prediction to produce a 3D model for hENT1. In this approach,21,22 threading using Local Meta-Threading-Server (LOMETS)
23
is first used to identify structural templates from PDB, which are then used to assemble template fragments into a complete 3D model of the full-length target protein. The results of the protein homology/analogy recognition engine Phyre2
24
and the homology detection and structure prediction server HHPred25,26 showed a good correlation with the model of hENT1 obtained using I-TASSER. The templates used for modeling by each server are listed in
Implications of Different Mutations on hENT1 Structure and Function
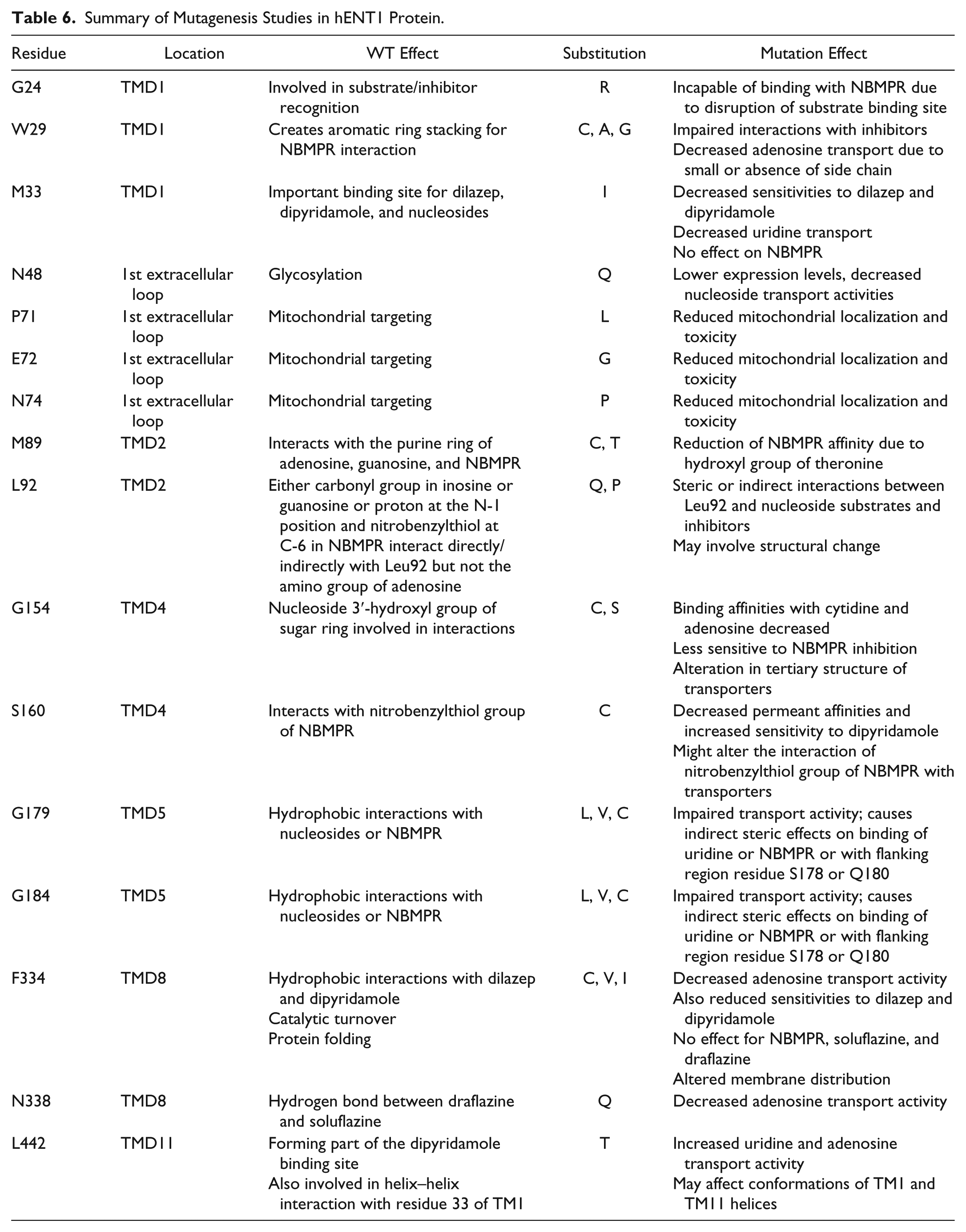
Many site-specific mutational studies have addressed the contribution of individual residues for ligand binding and transport within ENT1.32–35 The putative locations for these residues are depicted in the 3D homology model of hENT1 in an inward-facing conformation (
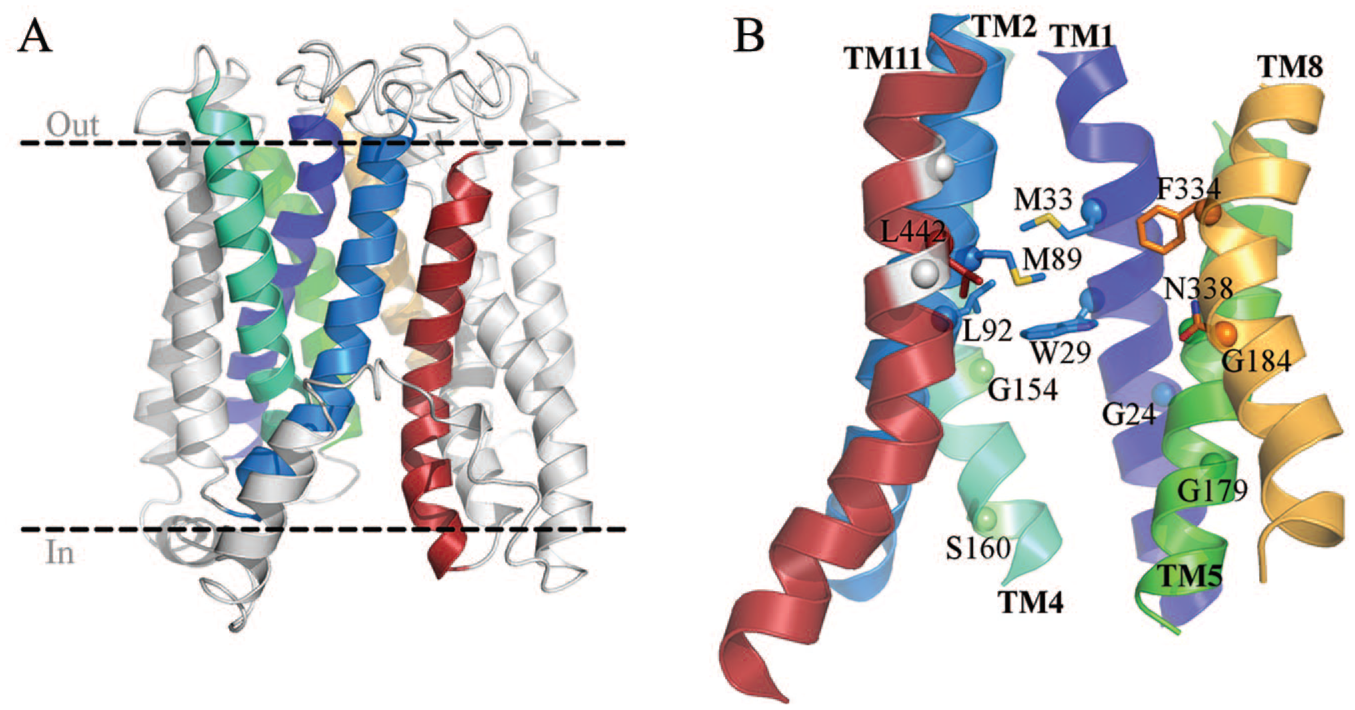
3D model for hENT1 showing key residues involved in protein function. (
ENT1 contains a highly conserved tryptophan residue at position 29(1.57) of TM1 facing the ENT1 central cavity, which likely plays an important role in all ENTs. Several substitutions at this position (W29C, W29T, W29A, and W29G) result in impaired binding of NBMPR and other inhibitors, whereas the conservative W29F mutation exhibits wild-type-like affinity, suggesting that the identity of the side chain at this position is critical for inhibitor binding. W29(1.57) may facilitate interactions with the nitrobenzyl group of NBMPR by aromatic ring stacking. Substitution of W29 with a smaller amino acid (such as glycine, alanine, cysteine, or valine) resulted in decreased nucleoside binding. Replacement with tyrosine caused a small increase in the transport of adenosine and other purine nucleosides. Strikingly, replacing W29(1.57) with a threonine showed a drastic decrease in the transport of uridine but not adenosine. It has been suggested that W29T may form a hydrogen bond with another residue that is specific and critical for pyrimidine nucleoside interactions without dramatically influencing purine nucleoside transport, which is altered by mutation of the nearby L442 mutant (L442I). 32 Together, these results suggest that W29(1.57) is involved in permeant selection and translocation. 37 Another highly conserved residue among ENTs is G24(1.52) in helix TM1. Arginine replacement of G24 has been shown to affect substrate binding, protein folding, and correct targeting of hENT1 to the plasma membrane. Furthermore, G24R impairs binding of NBMPR, even when the protein is localized to the plasma membrane, suggesting a key role for this residue in substrate binding, in addition to promoting protein stability and correct plasma membrane trafficking. 38
Random mutagenesis and adenosine complementation assays have also identified the central roles of TM2 residues M89 and L92 and TM4 residue S160 in inhibitor binding and/or nucleoside transport.
34
Consistent with the prediction from the helical wheel analysis in our 3D model, both M89 and L92 face the same side of TM2 and point to the ligand binding cavity. ENT1 M89 substitutions result in increased (M89L or M89Q) or severely diminished (M89C or M89T) NBMPR binding
34
but have no effect on the ability of ENT1 to bind dipyridamole. S160 is located at the end of TM2 facing away from the ligand binding cavity (
Another important residue at the extracellular end of hENT1 TM1 is M33(1.61), which has been shown to be important for binding of dilazep, dipyridamole, and physiological nucleosides.
39
In the 3D model of hENT1 M33 lies above W29(1.57) in TM1 and interacts with TM2 residues M89 and L92I (
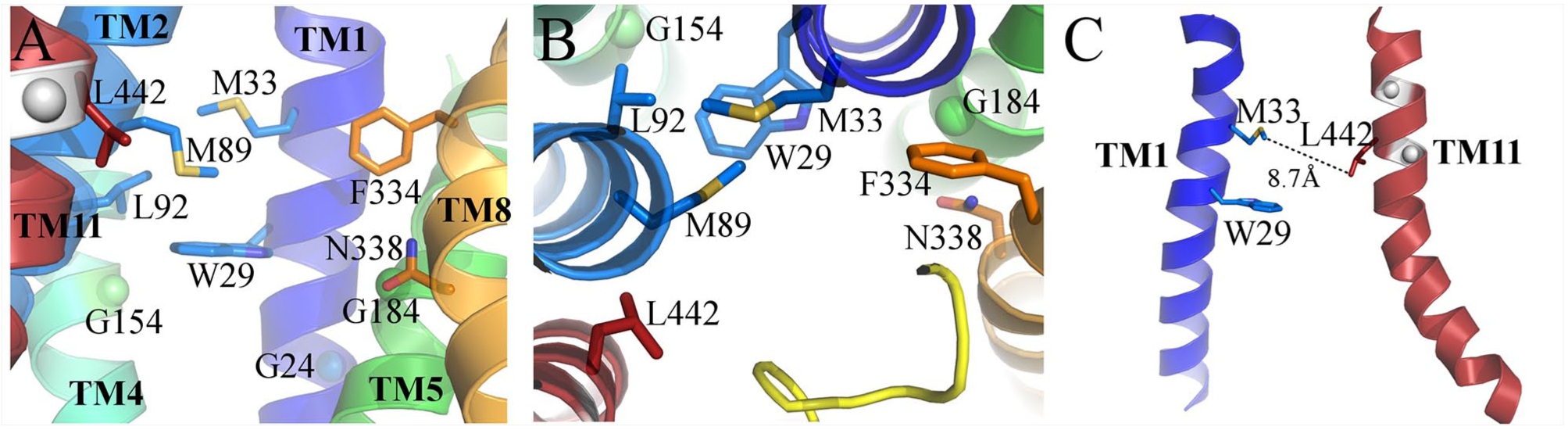
Close-up views of the predicted ligand binding site in hENT1. (
It was shown that TM1 and TM11 are critical for dipyridamole binding within hENT1, CeENT1, and hENT2.
39
This is indicated by the finding that the mutation L442I in hENT1 and M33 and L442(11.51) substitution in hENT2 make the protein sensitive to dipyridamole compared with wild-type protein. Based on the assumption that TM1 and TM11 are in close proximity, a mechanistic model of dipyridamole interaction was proposed: increased dipyridamole binding affinity is suggestive of a functional interaction between L442(11.51) and M33, which may be favored by the proximity of TM1 and TM11 induced by enhanced interactions resulting from the introduction of the L442I mutation.
39
Because the M33 of TM1 and leucine and isoleucine residues of TM11 contain the highly conserved GXXXG helix–helix interaction motif, it is likely that the mutation of these residues affected the conformations of both TMs. Studies have shown that the mutation L442T changes the conformation of TMs such that the helices move farther apart.
39
In the 3D model of hENT1, M33 and L442(11.51) are well positioned and far enough from each other to be involved in dipyridamole binding (
Mutations in TM1 and conservative substitutions of the hydrophobic leucine and isoleucine residues at the TM11 position in hENT1 had only minor effects on ENT1 uridine transport. Analysis of uridine and adenosine transport by the TM11 mutants revealed that L442T at the TM11 position resulted in greatly increased uridine and adenosine transport by hENT1, whereas the L442I mutation showed impaired adenosine transport without effects on uridine transport, suggesting specific alterations to the shape of the permeant binding pocket.
39
Similar substrate-selective effects affecting the binding of NBMPR, dilazep, and inosine, but not dipyridamole, adenosine, or pyrimidines, were observed with mutations of L92.
44
Because of the structural differences between the purines adenosine, inosine, and guanosine, it was suggested that L92 would directly interact with the C-6 and N-1 positions of the purine group,
44
but not the amino group of adenosine. Substituting L92 with polar (glutamine) or nonpolar (proline) residues may hinder hENT1 interactions with inhibitors or substrates due to structural changes influencing neighboring or other residues directly involved in substrate and inhibitor interaction. As L92 is adjacent to P91, this position of the α-helix may be sterically stressed, and sensitive to neighboring mutations.
44
However, the observed selective effects of L92 mutations on the affinity of inosine and guanosine, but not adenosine, suggest that these mutations do not cause gross alterations to the transporter structure. In line with the notion that L92 together with M89 and W29 would promote the structural integrity of the ligand binding site, all of these residues cluster together in the central cavity of the hENT1 homology model (
The conserved residue G154 in hENT1 is equivalent to S141 in hENT2, and the corresponding hENT1 mutation G154S significantly decreases hENT1 sensitivity to NBMPR and reduces its affinity for other nucleosides.
45
In the 3D model of hENT1 (
TM5 of hENT1 contains two highly conserved residues, G179(5.50) and G184(5.55), which may contribute to helix packing in ENTs from different organisms since bulky amino acid substitutions (G179L, G179V, G179C) impair nucleoside transport, whereas smaller substitutions (G179A, G179S) do not affect nucleoside binding. 48 Earlier work suggested that G179(5.50) would be located in a hydrophobic region of TM5, along with other nonpolar residues (A183, F186, A190). 35 However, in our hENT1 model, G179(5.50) (Fig. 4B and 5A) and the nearby non-polar residues clearly face the membrane–TM1 interface, a notion that is also supported by further mutational studies. In contrast to G179(5.50), mutations in G184(5.55) appear to prevent correct plasma membrane localization of hENT1, indicating that this residue may be important for correct folding or trafficking. 35 Substitution of G184(5.55) with a cysteine revealed six TM5 residues on one face of an amphipathic helix, where the G184C residue is accessible for sulfhydryl modification, indicating that this residue in hENT1 is solvent exposed and may form part of the nucleoside translocation pathway. In the hENT1 model, G184(5.55) is located next to N338 in TM8 (see below) and N30(1.58) in TM1, and a larger residue in the mutants might sterically hinder ligands interacting with N338.
Sequence alignment of ENTs from different organisms shows high evolutionary conservation at residues F334 and N338 (
Collectively, these results suggest that large hydrophobic substitutions at F334 favor the binding of dipyridamole and dilazep, whereas hydrophilic mutations at N338 promote hENT1 interactions with soluflazine and draflazine. Our structural model indicates that both F334 and N338 are located close to one another on the same side of TM8 (
Proposed Mechanism of Ligand Binding and Translocation
To date, no experimental structures for any ENT orthologs are available. The closest homolog for which structural data exist, a CNT from Vibrio cholerae, 51 shares only moderate sequence identity with ENT1 (9.5%), has a different membrane topology, and couples nucleoside transport with proton transfer, and is therefore not useful for investigating ENT family members. Computational models such as ligand-based quantitative structure–activity relationships (QSARs) built on in vitro affinity data have provided information about ligand requirements for interactions with ENT. Most of these studies have focused on the analysis of the NBMPR class of compounds and have revealed a critical role for the NBMPR nitrobenzyl moiety required for high-affinity ENT binding. In particular, NBMPR analogs with electron-withdrawing groups such as a nitro group at the 6-position of the benzyl substituent contribute strongly to ligand binding affinity. 52 The size of the NBMPR molecule and enhanced affinity with a nitro group at the 6-position suggest that the ENT binding pocket is quite large and can accommodate the inhibitor in a region that tolerates the presence of a bulky charged moiety. Further studies revealed that substituents in the NBMPR C2-purine position negatively affect hENT1 binding affinity. 53 Also, the C-3 hydroxyl of the NBMPR sugar moiety was revealed to be essential for interaction with hENT1. 54
Evolutionarily, NTs share common features with the major facilitator superfamily (MFS) membrane transporters.
55
The MFS transporters function by an alternating access mechanism,
56
and hENT1 is believed to act by a similar ligand translocation mechanism because of shared topology with the MFS transporters. This model would indicate that hENT1 could exist in at least two primary conformations exposing either extracellular or intracellular substrate binding sites. In this model, these conformations alternate regardless of ligand binding in a way that only exposes a single ligand binding site at a time (
Recent Advances in Discovery of New ENT Modulators
Because of the well-characterized protective role of extracellular adenosine, 60 preventing adenosine uptake by pharmacological ENT inhibition has been postulated to provide beneficial effects in pathological conditions such as stroke, ischemia-reperfusion injury, and diabetes. In this context, classical ENT inhibitors such as NBMPR, and coronary vasodilators dilazep and dipyridamole and their congeners have demonstrated the feasibility of inhibiting ENTs to promote protective adenosine signaling. Historically, NBMPR has been used as a probe compound to study adenosine transporter function and biology and to quantify transport elements of various cell types. However, these classical inhibitors lack selectivity for different ENT isoforms, which may limit the potential for their further therapeutic development. 16 Some ENT isoforms such as ENT4 are expressed in a highly restricted tissue pattern, suggesting that small molecules targeting only selected ENT isoforms could have less adverse effects arising from inhibition of unwanted target proteins. Additionally, highly isoform-specific inhibitors would serve as valuable probe compounds for studying the roles of individual ENT isoforms in the regulation of nucleoside transport in complex physiological environments and different animal models of human disease. Many kinase inhibitors also share structural features with adenosine and have been shown to inhibit ENT1 transport, raising the possibility of off-target effects of classical ENT modulators. 61
Despite problems associated with excess toxicity or poor availability of existing ENT1 inhibitors, progress in the design of new chemically distinct isoform-selective ENT inhibitors has been lacking in recent years. A recent study coauthored by us now suggests a potential new strategy for discovering potent macrocyclic inhibitors of different ENT isoforms. This method relies on the reengineering of rapamycin, a mechanistically unique inhibitor of mTOR. 62 In this study, a series of structural rapamycin variants were developed with an altered mTOR binding domain in order to discover new rapamycin-like macrocyclic inhibitors with the ability to expand the repertoire of inhibited protein targets. Screening of this library of “rapafucins” revealed a single compound that binds to and inhibits hENT1 with single-digit nanomolar potency in a manner dependent on simultaneous binding to FKBP12, mimicking the mode of action of rapamycin itself. 19 In demonstration of the therapeutic potential for ENT-targeting rapafucins, the authors demonstrated the beneficial efficacy of inhibiting ENT1 in an animal model of kidney reperfusion injury. The development of new ENT-targeting rapafucins will demonstrate the applicability of enhancing adenosine receptor signaling by inhibiting adenosine uptake in other diverse models of human disease. Interestingly, the identified rapafucin molecule, named rapadocin, inhibited ENT1 with high specificity over the related ENT2 isoform, suggesting the possibility to develop new highly isoform-selective ENT inhibitors. Importantly, because of their unique chemical structure, these macrocyclic inhibitors are unlikely to suffer from undesired pleiotropic off-target effects arising from the inhibition of nucleoside binding cellular targets. Similar to rapamycin, the ENT-targeting rapafucins will likely benefit from the unique properties offered by their unique mode of action. These properties arise from complexation with proteins of the FKBP family (in particular FKBP12) and include increasing the effective size of the inhibitor, allowing allosteric blockade of ENT substrate molecules and enhanced stability and efficient in vivo delivery. Future work will demonstrate the applicability and therapeutic benefits of isoform-selective ENT inhibition by rapafucin macrocycles.
Isolation of ENTs for Structural Studies
Despite their importance for the import of therapeutic NAs and as direct drug targets,11,12 practically no structural information exists for hENT1 and other ENTs. Mostly this lack of structural information reflects the inherent difficulty to produce different ENTs in heterologous systems in an active and stable form and in sufficient quantities for purification and structural studies. Several groups have successfully expressed hENT1 in yeast systems,39,63,64 and this has enabled detailed functional studies, of which many are summarized in this review; however, purifying ENTs from yeast has not allowed structural studies. Likewise, our attempts to express hENT1 in Escherichia coli, Saccharomyces cerevisiae, and Pichia pastoris failed to produce properly folded and targeted functional protein (unpublished data). In our experience, robust heterologous expression of hENT1 is most readily achieved in baculovirus-infected insect cells, which allows the isolation of milligram amounts of functional hENT1 when solubilized with lauryl maltose neopentyl glycol (LMNG) detergent. Using the guidelines we established, 49 several other groups were also able to isolate functional hENT1.38,65 Studies for the heterologous expression of other hENT family members are ongoing and will allow study of their biochemical differences, such as differences in transported substrates,16,66–68 and structure-guided development of inhibitors, in particular for ENTs, from disease-relevant parasites.
Perspectives
Our current structural understanding of nucleoside transport and the regulation of ENT family transporters still remains at a juvenile stage. Most of the current molecular understanding of cellular nucleoside transport is derived from experiments using cell lines and animal models with contributing background activity from several ENT and CNT subtypes. To circumvent this problem, heterologous expression of individual ENTs and functional studies with isolated and reconstituted proteins is required to gain a better understanding of substrate selectivities of physiological nucleosides and their analog drugs with their corresponding ENT isoforms. Additionally, overexpressed proteins may be used to identify chemical probes by high-content screening approaches. One of the most challenging yet exciting areas of future research will be unraveling the complex network of functional interactions between ENTs, CNTs, and adenosine receptors (and possibly other unidentified purinergic receptors), metabolic enzymes, and signaling pathways. In addition, 3D structures for all ENT transporter types, complexed with their substrates and/or known inhibitors, would be desirable for creating more reliable models of their biological functions. With new tools in the form of FKBP12–rapadocin–ENT complexes, cryo-EM may provide a suitable route for future structural investigations. Approaches to stabilize distinct ENT conformations by use of small molecules, camelid antibodies, 69 or recently introduced synthetic nanobodies 70 may also be needed for the successful preparation of conformationally homogenous ENT samples suitable for structure determination by crystallization or single-particle electron microscopy approaches.
Supplemental Material
Supplementary_material_current_progress_on_equilibrative_nucleoside_transporter_funtion_and_inhibitor_design_Rehan,_et_al – Supplemental material for Current Progress on Equilibrative Nucleoside Transporter Function and Inhibitor Design
Supplemental material, Supplementary_material_current_progress_on_equilibrative_nucleoside_transporter_funtion_and_inhibitor_design_Rehan,_et_al for Current Progress on Equilibrative Nucleoside Transporter Function and Inhibitor Design by Shahid Rehan, Saman Shahid, Tiina A. Salminen, Veli-Pekka Jaakola and Ville O. Paavilainen in SLAS Discovery
Footnotes
Acknowledgements
We are thankful for the bioinformatics (J. V. Lehtonen), translational activities, and structural biology infrastructure support from Biocenter Finland and Instruct-FI at the Structural Bioinformatics Laboratory, Åbo Akademi University. We thank Dr. Dale Tranter and Dr. Juho Kellosalo for their valuable comments on the manuscript.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: V.-P.J. is an employee of Novartis Institutes for BioMedical Research.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was made possible by the following funding sources. V.O.P. is supported by the Academy of Finland (grants 289737 and 314672), V.O.P. and T.A.S. by the Sigrid Juselius Foundation, and T.A.S. by Tor, Joe, and Pentti Borg’s Foundation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.