
Abstract
To study the function and structure of membrane proteins, high quantities of pure and stable protein are needed. One of the first hurdles in accomplishing this is expression of the membrane protein at high levels and in a functional state. Membrane proteins are naturally expressed at low levels, so finding a suitable host for overexpression is imperative. Multidrug resistance protein 4 (MRP4) or ATP-binding cassette subfamily C member 4 (ABCC4) is a multi-transmembrane protein that is able to transport a range of organic anionic compounds (both endogenous and xenobiotic) out of the cell. This versatile transporter has been linked with extracellular signaling pathways and cellular protection, along with conferring drug resistance in cancers. Here we report the use of MRP4 as a case study to be expressed in three different expression systems: mammalian, insect, and yeast cells, to gain the highest yield possible. Interestingly, using the baculovirus expression system with Sf9 insect cells produced the highest protein yields. Vesicular transport assays were used to confirm that MRP4 expressed in Sf9 was functional using a fluorescent cAMP analogue (fluo-cAMP) instead of the traditional radiolabeled substrates. MRP4 transported fluo-cAMP in an ATP-dependent manner. The specificity of functional expression of MRP4 was validated by the use of nonhydrolyzable ATP analogues and MRP4 inhibitor MK571. Functionally expressed MRP4 in Sf9 cells can now be used in downstream processes such as solubilization and purification in order to better understand its function and structure.
Introduction
One of the limitations of membrane protein structural biology is expressing the membrane protein of interest. The challenge lies in not only expressing the protein of interest but also expressing it to a high level in its native conformation(s). Most membrane proteins are naturally expressed in low levels, and so obtaining sufficient amounts of the native membrane proteins to conduct functional and structural studies requires large amounts of resources and is really only realistic for proteins that are naturally abundant in certain cell types, such as rhodopsin in the retina. 1
To overcome the problem of low natural expression, recombinant overexpression can be performed, increasing the yield per cell. 2 Another advantage of recombinant expression is the ability to easily add tags to enable efficient separation of the target protein from the other membrane proteins. Common purification tags include histidine, strep, and flag tags, which can increase the purity and yield though affinity purification. 3 However, it is important that these tags do not interfere with the function of the protein. Recombinant membrane protein expression is also a means of producing more stable membrane proteins through the use of mutagenesis and protein engineering, but the native conformation will be altered and therefore the correct function and structure will not be discovered. 4
Effective recombinant membrane protein expression requires finding a suitable host. If the membrane protein is a prokaryotic protein, then Escherichia coli could potentially be used. The advantages of using E. coli for recombinant overexpression of membrane proteins is that it can be carried out quickly, as E. coli have a high growth rate, high quantities of cells are easily achieved, and it is cost-effective. 5 If the target protein is eukaryotic, such as human membrane proteins, a eukaryotic host such as yeast, insect, or mammalian cells can be used.
Insect cell expression is a commonly used expression system for recombinant mammalian membrane proteins. It requires the production of a recombinant baculovirus carrying the gene of interest, and infection of insect cells, such as Spodoptera frugiperda (Sf9), with this virus leads to protein expression. 6 Inclusion bodies are rarely formed with the baculovirus expression system in insect cells, unlike in E. coli. 7 This system has also been beneficial in the production of multiprotein subunit complexes.8–10
Two main strains of yeast have been used for membrane protein expression, Pichia pastoris and Saccharomyces cerevisiae. P. pastoris requires the integration of the recombinant gene of interest into the yeast genome, allowing a stable strain to be produced, but it is not possible to control the number of copies or location of the recombinant gene. On the other hand, S. cerevisiae expression tends to use plasmids containing the gene of interest, similarly to E. coli. However, the advantages of using P. pastoris are the high cells densities it can grow to, with exceptionally high yields of correctly folded protein, meaning a large amount of recombinant protein can be produced, 11 which is why P. pastoris was chosen for this study.
Mammalian cell expression offers potentially the most relevant cellular environment for human membrane proteins. Two of the most common mammalian cell lines used are human embryonic kidney (HEK) and Chinese hamster ovary (CHO) cells.6,12 The HEK cell line was chosen for this study as it has increasingly been used for membrane protein expression. 13 Proteins expressed in HEK cells are usually fully glycosylated compared with Sf9 cells. 8 HEK cells can be made to overexpress recombinant membrane proteins by producing either transient or stable cell lines. 14 While transient expression can give considerable batch-to-batch variability, creating stable cells often reduces the expression yield. Thus, transient transfections were utilized in this study.
ATP-binding cassette (ABC) transporters are integral membrane proteins that are found in all types of organisms, from prokaryotes to humans. They utilize energy from ATP binding and hydrolysis to transport a variety of substrates across the biological lipid bilayer. 15 In humans, the 48 different ABC transporters can be separated into 7 different subfamilies, ABCA–ABCG, of which multidrug resistance protein 4 (MRP4/ABCC4) is part of the C subfamily. 16
MRP4 can be found in a wide range of cells all over the human body, including blood cells, neurons, testis, ovaries, adrenal glands, prostate tubuloacinar cells, and renal proximal tubule cells. 17 Endogenously, MRP4 is able to transport substrates that are involved in inflammation, such as prostaglandins and leukotrienes 18 and cell signaling, including cyclic nucleotides such as cyclic AMP (cAMP) and cyclic GMP (cGMP). 19 It has also been shown to transport a wide range of drugs and their metabolites, including anticancer, antiviral, and antibiotic molecules. 20
How MRP4 is able to transport such a wide variety of substrates is not well known. In particular, how it can recognize, bind, and transport both relatively hydrophilic molecules like cAMP and hydrophobic molecules such as bile salts or drugs like methotrexate is unclear. This could be due to the lack of structural knowledge about the transmembrane domains (TMDs) of MRP4, which are responsible for transporting substrates. Therefore, functional and structural studies will help reveal the intricacies of this membrane protein.
In this study, we investigated the functional overexpression of MRP4 by examining which approach gave the best expression yield and then characterized the function with a fluorescent vesicular transport assay (VTA).
Materials and Methods
Sf9 Expression
Expression of the recombinant human MRP4-his6 within Sf9 cells was conducted using a baculovirus encoding for recombinant MRP4 generated from a pFastBac-MRP4-his6 construct as described previously. 21 Cells were grown in shaker cultures using Insect Xpress media (Lonza, Basel, Switzerland). To find the optimal expression conditions, cells at a density of either 1 or 2 million per milliliter were infected with baculovirus using a multiplicity of infection (MOI) of either 2 or 4, and cells harvested after 24, 48, or 72 h.
P. pastoris Expression
Growth media BMGY (buffered glycerol complex medium) and BMMY (buffered methanol complex medium) were made using 10 g of yeast extract, and 20 g of peptone was dissolved in 700 mL of water and autoclaved. After filter sterilization, 100 mL of 1 M potassium phosphate buffer, pH 6.0 (13.2 mL of 1 M K2HPO4 and 86.8 mL of 1M K2HPO4), 100 mL of 10× YNB (13.4% yeast nitrogen base with ammonium sulfate without amino acids), 2 mL of 0.02% biotin, and 100 mL of 10% glycerol for BMGY or 100 mL of 5% methanol for BMMY were added.
The recombinant pPICZαC-MRP4-his6 construct was created using a double digest of the pFastBac MRP4-his6 plasmid and pPICZαC with EcoRI, followed by ligation of MRP4-his6 into the pPICZαC plasmid, at a plasmid-to-insert molar ratio of 1:3, overnight at 16 °C. pPICZαC MRP4-his6 was linearized using PmeI and transformed into P. pastoris ×33 using electroporation. Colonies containing integrated MRP4 were grown essentially as described previously for Pichia expression of a membrane protein. 22 Briefly, colonies were grown in 25 mL of BMGY in sterile 250 mL flasks at 30 °C in a shaking incubator (250–300 rpm) until the culture reached an OD600 of 2–6. Cells were harvested by centrifugation at 3000g for 5 min, all BMGY was removed, and then they were washed with BMMY and resuspended in BMMY at an OD600 of 1.0 before being returned to the shaking incubator at 22 or 30 °C. Sterilized pure methanol was added every 24 h to a final concentration of 0.5% (v/v) methanol. Samples were taken every 24 h over a 72 h period.
HEK293T Expression
pcDNA3.1-MRP4-his6 plasmid was constructed by restriction digestion of MRP4-his6 out of the pFastBac plasmid and ligation into a pcDNA 3.1 Zeo + plasmid. pOPINE-MRP4–3C-flag-his8 was made by the Oxford Protein Production Facility (OPPF, Harwell, UK). pcDNA3.1-MRP4 without a his-tag was a kind gift from Professor Susan Cole (Queen’s University, Kingston, ON, Canada). HEK293T cells were seeded in a six-well plate with 300,000 cells/well in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin 24 h prior to transfection. Three hours prior to transfection, the media was replaced with low-serum DMEM containing 2.5% FBS and 1% penicillin/streptomycin. For transfection, 4 µg of plasmid DNA was combined with 18 µL of 10 mM linear polyethylenimine (PEI; Polysciences, Warrington, PA) and 100 µL of reduced serum media (OPTIMEM) and added to each well. Twenty-four hours after transfection the media was replaced with DMEM containing 10% FBS and 1% penicillin/streptomycin. Samples were taken every 24 h over a 72 h period.
Cell Lysis and Membrane Preparation
For both Sf9 and HEK293T, cells were harvested by centrifugation (5000g for 10 min) and cell pellets were resuspended in buffer 1 (50 mM Tris-HCl, pH 7.4, 250 mM sucrose, 0.25 mM CaCl2) containing protease inhibitors (1.3 µM benzamidine, 1.8 µM leupeptin, 1 µM pepstatin). Cells were disrupted through nitrogen cavitation at 500 psi for 15 min at 4 °C. The cell lysate was centrifuged at 750g for 10 min to remove cell debris; the supernatant was then ultracentrifuged at 100,000g for 20 min at 4 °C. The membrane pellet was resuspended in buffer 2 (50 mM Tris-HCl, pH 7.4, 250 mM sucrose) and stored at −80 °C.
P. pastoris cells were pelleted via centrifugation at 2500g for 30 min and then resuspened in buffer 3 (5.5% [w/v] glycerol, 2 mm EDTA, 100 mM NaCl, 50 mM NaH2PO4, 50 mM Na2HPO4) containing EDTA-free protease inhibitor tablets (Roche, Welwyn Garden City, UK). Cells were resuspended at a buffer (mL)-to-cell pellet weight (g) ratio of 3:1. Resuspended cell pellets were homogenized by passing them through the Emulsi Flex C3 machine (Avestin, Ottawa, Canada) three times. The homogenized cells were centrifuged at 5000g for 5 min, the supernatant was then centrifuged at 13,000g for 15 min, and finally the supernatant was centrifuged for 1 h at 100,000g. Membrane pellets were resuspended in buffer 4 (20 mM HEPES, pH 8, 50 mM NaCl, 10 % [w/v] glycerol) containing protease inhibitor (Roche) and stored at −80 °C.
Analysis of Expression
Expression of MRP4 was monitored by Western blot. The total protein concentration of membranes was measured using a bicinchoninic acid (BCA) assay kit (Pierce, Thermo Scientific, Waltham, MA). Specified amounts (µg) of total protein were loaded on 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinylidene fluoride (PVDF) membrane, and blocked with 5% (w/v) bovine serum albumin (BSA) in TBS-T (20 mM Tris, pH 7.5, 150 mM NaCl, 0.01% [v/v] Tween-20). Blots were probed with either a mouse anti-his antibody (R&D Systems, Abingdon, UK) at a dilution of 1:500 or a rat anti-MRP4 antibody (M4I-10, Enzo, Exeter, UK) at 1:100, followed by anti-mouse HRP (Cell Signaling, London, UK, 1:3000) or anti-rat HRP (Sigma, Gillingham, UK, 1:3000). All were visualized using chemiluminescence (Pierce) and a C-Digit Western blot scanner (Licor, Cambridge, UK).
Vesicular Transport Assays
VTAs were based on the study by Reichel et al. 23 and performed using the Sf9 control and Sf9 MRP4-expressing cell membrane vesicles from the optimized expression conditions (1 × 106 cells/mL, MOI of 2, 48 h incubation). Total protein membrane protein (10–100 μg) was incubated with 10 mM ATP (plus an ATP regenerating system: 100 μg/mL creatine kinase and 10 mM creatine phosphate) or AMP and 10 mM MgCl2 and 1–100 μM 8-(2-[fluoresceinyl]aminoethylthio)adenosine-3′,5′-cyclic monophosphate (fluo-cAMP) (Biolog, Bremen, Germany). VTAs were conducted in buffer 2 in a 50 μL volume and incubated at room temperature for 10 min. This time period was chosen since previous kinetic studies showed it to be within the linear range. 23 For vanadate inhibition, 500 μM sodium orthovanadate was added along with ATP. AMP-PNP inhibition was conducted by replacing the ATP with 10 mM AMP-PNP. MK571 (0.01–10 μM) was added along with ATP to measure MK571 inhibition.
After incubation, transport was stopped by the addition of 950 μL of ice-cold buffer 2. Samples were either filtered using a PVDF filter (Millipore 0.45 μM) or centrifuged at 14,000g for 5 min. The filter was washed with 5 mL of ice-cold buffer 2 or the pelleted vesicles washed with 1 mL of ice-cold buffer 2. The filter or pellet was solubilized with 1 mL of SDS/HEPES buffer (1% [w/v] SDS, 7.5 mM HEPES) for 15 min. The amount of fluo-cAMP transported was measured by the fluorescence signal (RFU) of the solubilized sample measured on a PerkinElmer LS55 Fluorescence Spectrometer (excitation 480 ± 5 nm, emissions 500–600 ± 20 nm). Samples were run in triplicate and an average of five scans was taken for each sample.
Data fitting for concentration curves in the VTA was performed by fitting a Michaelis–Menten, and for MK571 inhibition used a dose–response curve. Statistical analysis was performed using an unpaired two-tailed t test or one-way analysis of variance (ANOVA). Data fitting and statistical analysis were carried out using GraphPad Prism.
Results
MRP4 Expression
The first step of the investigation was to determine the optimal conditions for MRP4 expression in each of the three expression systems, Sf9 insect cells, P. Pastoris yeast cells, and HEK293T mammalian cells. For Sf9 insect cell expression, the cell density, MOI, and infection period were altered. Western blots in
Figure 1A
show that the expression of MRP4 within Sf9 cells was successful. As reported previously, MRP4 expressed in Sf9 cells migrated at approximately 150 kDa.
24
After 48 h, an increased expression was seen compared with that at 24 h (
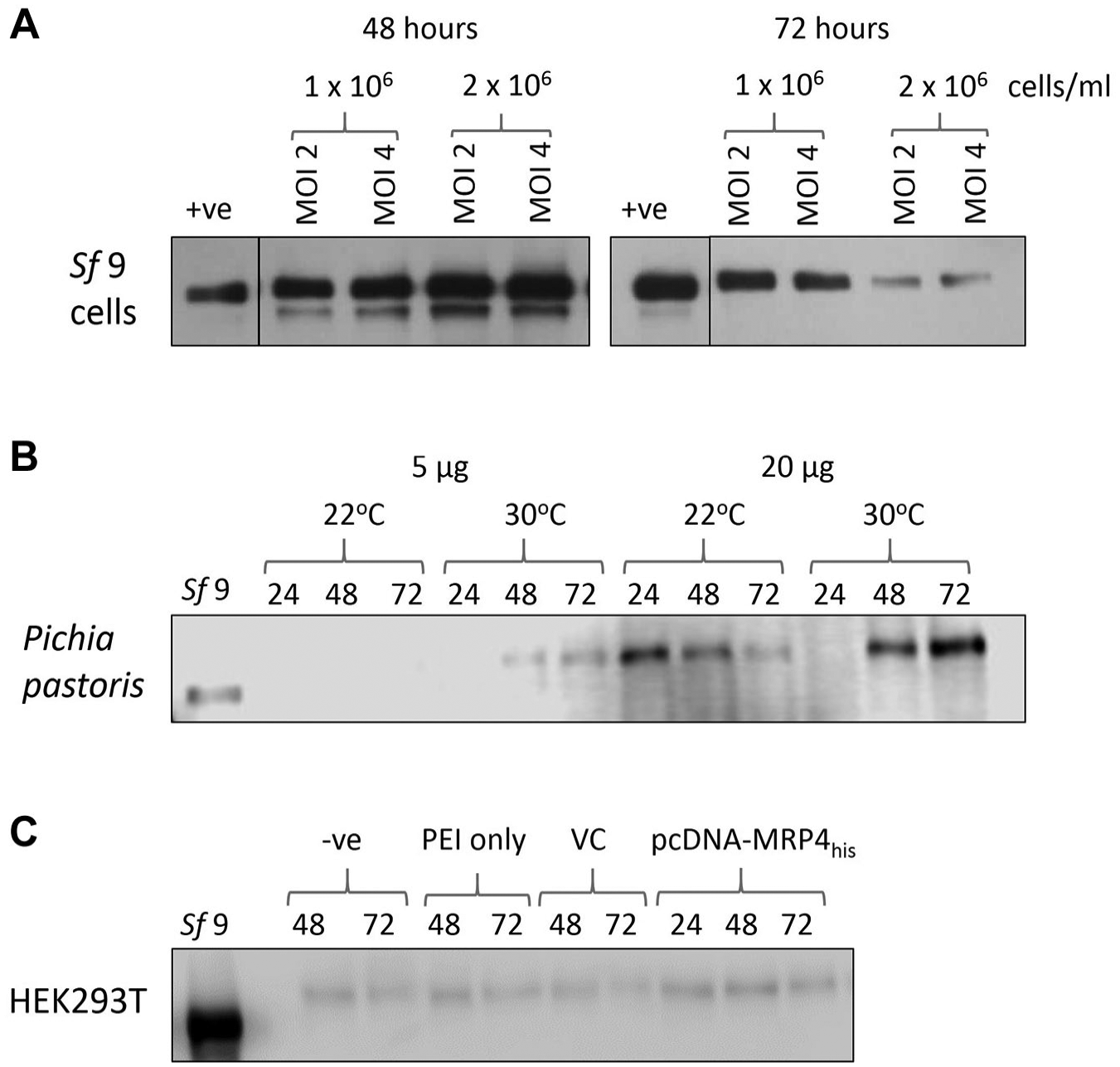
Overexpression trials for MRP4 in Sf9 insect cells, P. pastoris yeast cells, and HEK293T mammalian cells. (
After successful integration of MRP4 into P. pastoris, the temperature and time were altered to gain the highest yield possible in shaker flasks.
Figure 1B
shows the expression of MRP4 within P. pastoris. Notably, the MRP4 from P. pastoris runs at a higher molecular weight than the Sf9-expressed MRP4. At a lower temperature (22 °C), the highest expression level was achieved after 24 h and then decreased over the 72 h period. At higher temperature (30 °C), the expression level increased over time, reaching the highest expression level after 72 h. The use of a 2 L bioreactor for P. pastoris expression was also investigated (
Transient transfections were performed in HEK293T cells using PEI as a transfection reagent. As shown in
Figure 1C
, in contrast to Sf9 and P. pastoris, HEK293T cells express MRP4 endogenously. Transfection of the HEK293T cells with pcDNA3.1-MRP4his gave only marginally increased levels of MRP4 expression. Similarly, transfection with pOPINE-MRP4-3C-flag-his8 led to very little overexpression of MRP4 (
MRP4 was successfully overexpressed in all three expression systems. However, in HEK293T cells it was only achieved in the absence of a his-tag, which would make downstream purification challenging. The yield obtained with Sf9 cells was higher than that achieved with P. pastoris. In addition, the MRP4 from Sf9 cells migrated at a lower molecular weight than in the other two expression systems, possibly related to the degree of glycosylation. Extensive glycosylation can be problematic for downstream structural biology; thus, this was perceived as another benefit of the Sf9 cell system. Therefore, the Sf9 expression system was taken forward to assess if the MRP4 expressed was functional.
Vesicular Transport
Finding the balance between overexpression and quality needs to be obtained. Therefore, it is vital to ascertain that the protein is functional following overexpression. To facilitate this, a fluorescent VTA was used. cAMP is a known substrate for MRP4, 25 and this assay utilizes a fluorescent analogue of cAMP: fluo-cAMP. This substrate had previously been reported to be transported by MRP4 within renal proximal tubules and by MRP4 overexpressed in Sf9 membrane vesicles. 23 By measuring the amount of substrate transported into the vesicle when ATP is present compared with AMP, the specific transport activity can be determined ( Fig. 2 ).
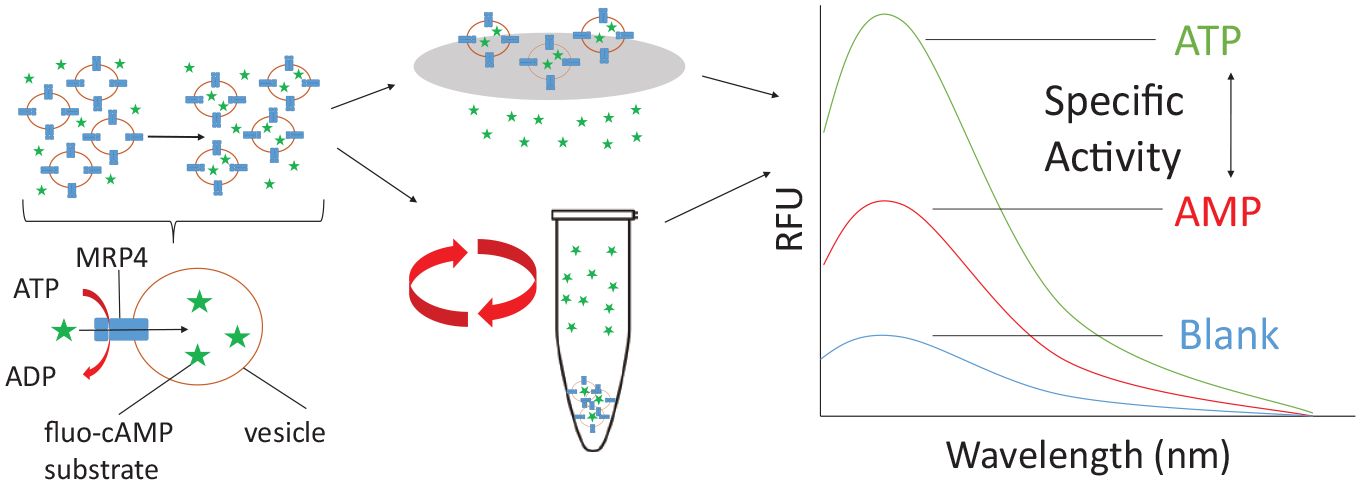
Schematic of the steps in the VTA. The first step is the incubation of the fluorescent cAMP substrate (green stars) with the membrane vesicles (orange circles) in the presence of AMP or ATP (along with an ATP regenerating system). The fluorescent cAMP is transported into the membrane vesicles via ATP hydrolysis. The vesicles are then either filtered or centrifuged to remove all excess fluo-cAMP. The vesicles are then solubilized, and the amount of fluorescent cAMP transported into the vesicles is measured on a fluorescent spectrometer. The difference between ATP and AMP is calculated giving the specific transport activity.
Figure 3A shows a significant increase in the transport of fluo-cAMP in the presence of ATP compared with AMP in Sf9 MRP4 vesicles, showing ATP-dependent transport of fluo-cAMP. MRP4 was shown to be responsible for the transport of fluo-cAMP, as there is an increase in ATP-dependent specific activity of Sf9 MRP4 vesicles compared with Sf9 control vesicles ( Fig. 3B ). There was a positive correlation of ATP-dependent specific activity in Sf9 MRP4 vesicles with increased total membrane protein content, again indicating that MRP4 was responsible for the transport of fluo-cAMP, whereas the Sf9 control vesicles had a steady background fluorescence with increasing total membrane protein content. Figure 3C demonstrates a concentration-dependent transport of fluo-cAMP with a Km of 5.8 μM, which is comparable to previously reported values. 23
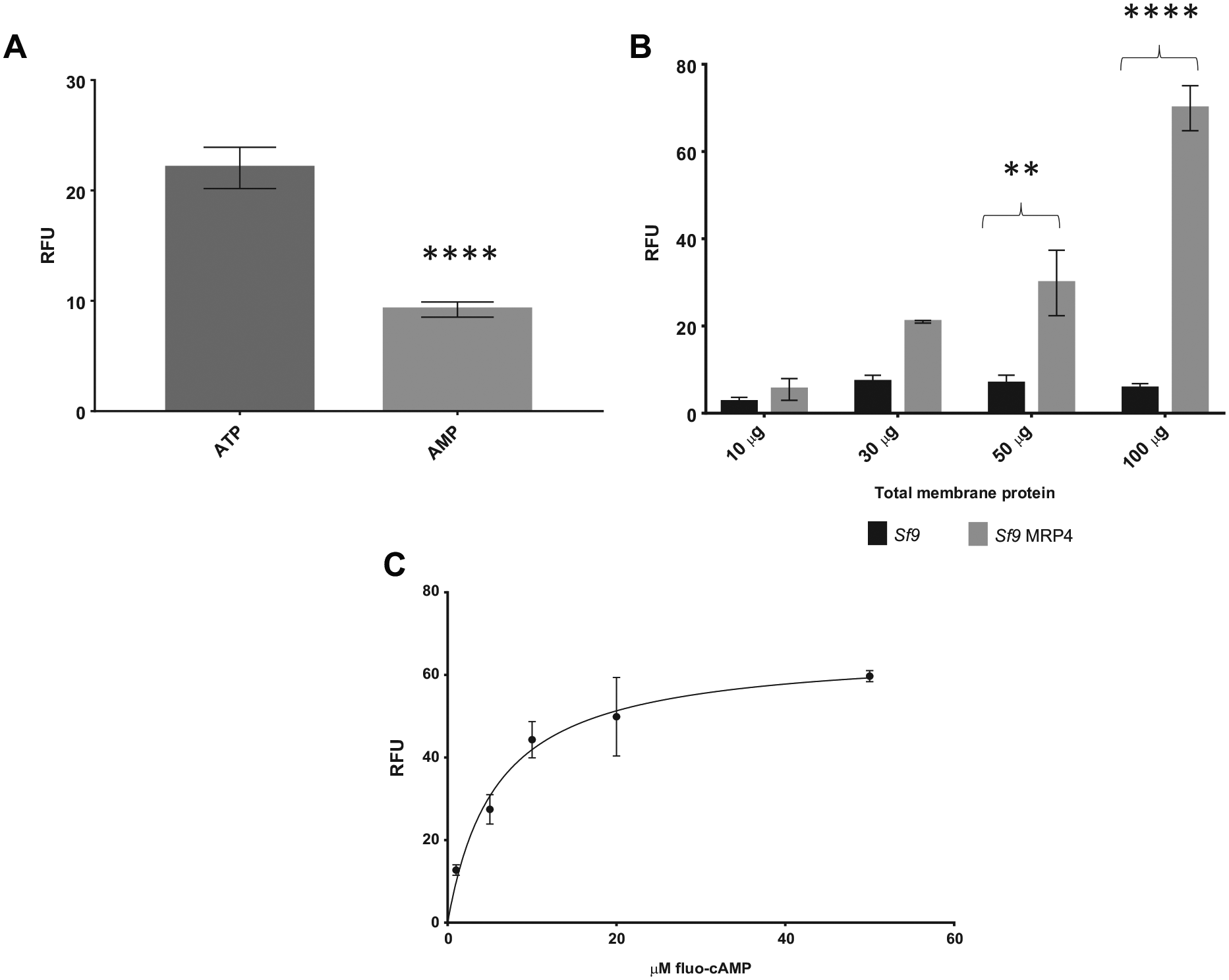
Vesicular uptake of fluo-cAMP is both ATP and MRP4 dependent. (
ATP hydrolysis is needed for the transport of substrates by MRP4, and inhibiting ATP hydrolysis should therefore inhibit transport. As shown in Figure 4A , in the presence of vanadate or the nonhydrolyzable ATP analogue AMP-PNP, the uptake is reduced to the same level as with AMP, indicating that ATP is the driving force behind the transport of fluo-cAMP. MK571, a known inhibitor of MRP4, was also used to demonstrate the functionality of MRP4. MK571 inhibits the transport of substrates by binding within the TMDs rather than the nucleotide binding sites like vanadate and AMP-PNP. 26 MK571 inhibited the transport of fluo-cAMP in a concentration-dependent manner with an IC50 of 0.39 μM.
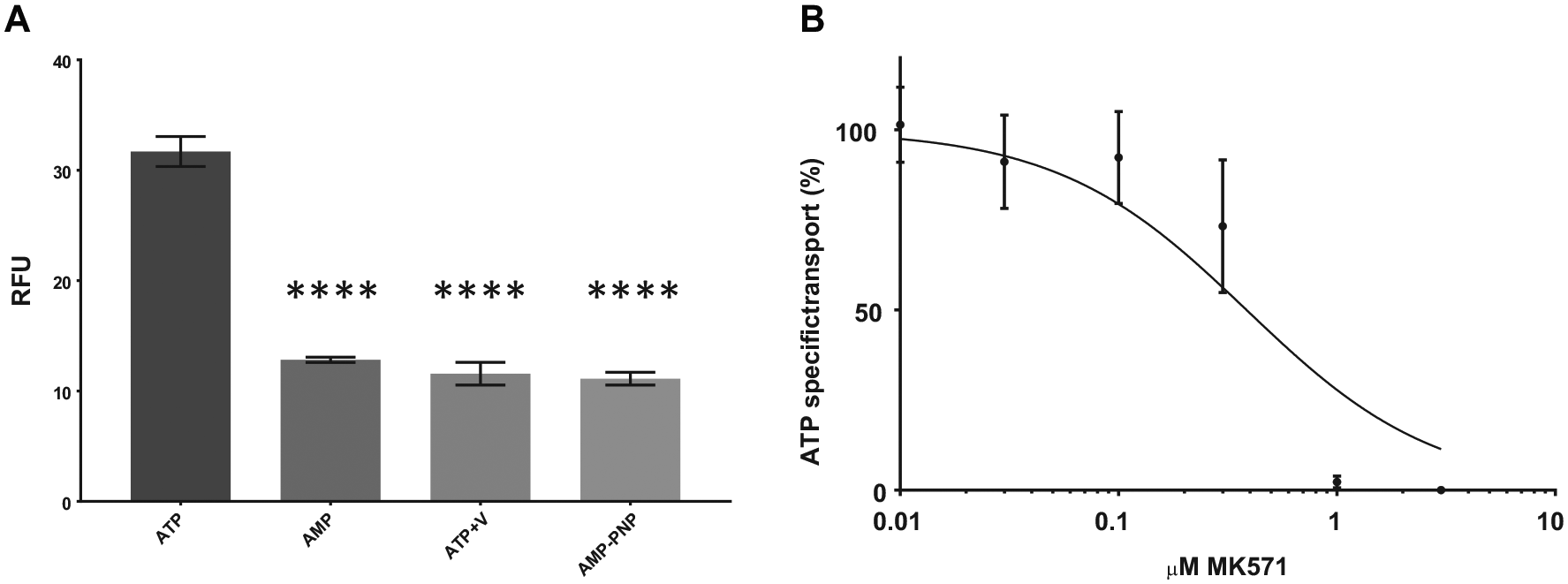
Vesicular uptake of fluo-cAMP is inhibited by nonhydrolyzable ATP analogues and MK571. (
These results verify that MRP4 expressed in Sf9 cells is functional as it is responsible for the transport of fluo-cAMP in a concentration- and ATP-dependent manner and transport was prevented by inhibiting either ATP hydrolysis or substrate binding.
Discussion
The need for good starting material is paramount in elucidating the function and structure of membrane proteins. To address this, we investigated MRP4 expression in three different systems, Sf9 insect cells, P. pastoris yeast, and HEK293T mammalian cells. All three of these systems have been successfully utilized in the past for overexpression of mammalian ABC transporters for functional and structural studies.
P. pastoris has been successfully used for the overexpression of mouse MRP1/ABCC1,27,28 mouse P-glycoprotein/ABCB1,
29
and human TAP1/2.
30
In this study, we found that human MRP4 could also be successfully overexpressed using P. pastoris. Surprisingly, the level of expression achieved was lower when using a bioreactor rather than shaker flask cultures (
The expression of MRP4 within Sf9 cells has been reported previously;21,23,24,32,33 however, this has predominantly been utilized for functional assays to date, rather than with the aim to develop an expression system for future purification. Here we showed that MRP4 with a his-affinity tag could be successfully overexpressed in Sf9 cells, and the expression level could be optimized by changing the time of infection ( Fig. 1A ). Insect cells have previously been utilized for the expression, purification, and structural study of human P-glycoprotein/ABCB1, 34 although this used High Five (Trichoplusia ni) cells rather than Sf9 cells. Insect cells have also been proven to be especially useful for structural studies on G-protein-coupled receptors (GPCRs), which have shown a preference for Sf9 cells. 35
The overexpression of MRP4 in HEK cells has also been reported many times previously,32,36–38 but again, to date this has mainly been for the purposes of functional studies. Transient transfection of HEK cells has been carried out using the transfection reagent Lipofectamine.
36
In this study, we have successfully shown overexpression of MRP4 in HEK cells using the much cheaper reagent PEI (
Notably, both the P. pastoris and HEK-expressed MRP4 migrated at higher molecular weights than the Sf9 MRP4 ( Fig. 1B,C ). It is known that MRP4 is glycosylated 36 and Sf9 cells are only able to carry out simple mannose glycosylation, 6 so this difference is likely due to differential glycosylation in the three systems. Glycosylation can be problematic for structural studies since it adds heterogeneity.
Taken together, the higher yield of MRP4, the potential lower levels of glycosylation, and the ease of scale-up led us to choose Sf9 cell expression to proceed with.
The next step was to check that the Sf9 overexpressed MRP4 was functional. Typically, function is assessed by VTAs using radiolabeled substrates; however, a fluorescent-based assay can be both cheaper and easier. It was previously shown that MRP4 can transport the fluorescent analogue of cAMP, fluo-cAMP. 23 We found that crude membranes of Sf9 cells expressing MRP4 were able to transport fluorescent cAMP in an ATP-dependent manner ( Fig. 3 ). The Km of fluo-cAMP was found to be very similar to that found in the previous study, 23 showing that this method is a robust way of determining the functionality of MRP4 using fluorescent analogues. Transport was also inhibited by MK571 and ATP analogues, confirming its functionality ( Fig. 4 ).
During this study, both rapid filtration and a centrifugation technique were tested for separating free fluo-cAMP from the vesicles ( Fig. 2 ). Rapid filtration is typically used with radiolabeled substrates; however, with the fluorescent assay particles from the filters caused an increase in light scattering, which decreased the signal-to-noise ratio. PVDF filters were better than glass fiber filters; however, the centrifugation method improved this even further, as well as increasing the efficiency of the transport assay.
In conclusion, we have successfully demonstrated functional overexpression of MRP4 in Sf9 cells that can now be taken forward for solubilization and purification to enable mechanistic and structural studies.
Supplemental Material
DS_DISC867070 – Supplemental material for Functional Expression of Multidrug Resistance Protein 4 MRP4/ABCC4
Supplemental material, DS_DISC867070 for Functional Expression of Multidrug Resistance Protein 4 MRP4/ABCC4 by David Hardy, Roslyn Bill, Anass Jawhari and Alice Rothnie in SLAS Discovery
Footnotes
Acknowledgements
We would like to thank the Oxford Protein Production Facility for generating the pOPINE-MRP4-3C-flag-his8 construct. Thanks to Professor Susan Cole, Queen’s University, Kingston, ON, for the pcDNA3.1-MRP4 construct. Thanks to the CALIXAR team for help and advice with experiments. The underlying data for this publication can be found at ![]()
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.J. is an employee of CALIXAR, which has patent applications that cover some of the CALX detergents described in this paper.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by a Biotechnology and Biological Sciences Research Council Industrial Case Studentship (BB/L015846/1). A.J.R. was also the recipient of a Royal Society Research Grant (RG110156).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.