
Abstract
Recently, the F1FO-ATP synthase, due to its dual role of life enzyme as main adenosine triphosphate (ATP) maker and of death enzyme, as ATP dissipator and putative structural component of the mitochondrial permeability transition pore (mPTP), which triggers cell death, has been increasingly considered as a drug target. Accordingly, the enzyme offers new strategies to counteract the increased antibiotic resistance. The challenge is to find or synthesize compounds able to discriminate between prokaryotic and mitochondrial F1FO-ATP synthase, exploiting subtle structural differences to kill pathogens without affecting the host. From this perspective, the eukaryotic enzyme could also be made refractory to macrolide antibiotics by chemically produced posttranslational modifications. Moreover, because the mitochondrial F1FO-ATPase activity stimulated by Ca2+ instead of by the natural modulator Mg2+ is most likely involved in mPTP formation, effectors preferentially targeting the Ca2+-activated enzyme may modulate the mPTP. If the enzyme involvement in the mPTP is confirmed, Ca2+-ATPase inhibitors may counteract conditions featured by an increased mPTP activity, such as neurodegenerative and cardiovascular diseases and physiological aging. Conversely, mPTP opening could be pharmacologically stimulated to selectively kill unwanted cells. On the basis of recent literature and promising lab findings, the action mechanism of F1 and FO inhibitors is considered. These molecules may act as enzyme modifiers and constitute new drugs to kill pathogens, improve compromised enzyme functions, and limit the deathly enzyme role in pathologies. The enzyme offers a wide spectrum of therapeutic strategies to fight at the molecular level diseases whose treatment is still insufficient or merely symptomatic.
Introduction
The F1FO-ATP synthase is a ubiquitous enzyme complex localized in the inner mitochondrial membrane and thylakoid membranes of eukaryotic cells and in bacterial plasma membrane, endowed with bifunctional properties of adenosine triphosphate (ATP) synthesis and hydrolysis. because of the F1FO-ATP synthase’s prominent feature of “enzyme of life,” as a key participant in the cell bioenergetic machinery in aerobiosis, it is quite easy to imagine that enzyme inhibitors can be exploited as drugs to kill noxious cells. However, only in recent times has this potentiality been considered. The main difficulty in its therapeutic exploitation most likely relies in the consideration that the enzyme function and mechanism are similar in all taxa, despite some structural divergences, 1 while drugs should distinguish among targets. Basically, the F1FO-ATP synthase consists of an hydrophilic portion known as F1, which hosts the catalytic sites, and a membrane-embedded FO domain, which translocates H+. 2 The hydrophilic F1 domain protrudes in the mitochondrial matrix and is involved in the catalytic activity of ATP synthesis and hydrolysis. The embedded-membrane FO domain channels H+ driven by proton-motive force Δp and generates the torsion required for the F1 catalytic activity. The chemo-mechanical coupling between the two domains is ensured by a peripheral stalk and a central stalk, which connect F1 to FO. The F1FO-ATP synthase synthesizes ATP by exploiting the electrochemical energy produced by the respiratory chain in the form of Mitchell’s Δp across the inner mitochondrial membrane. Therefore, ATP generation results from a chemo-mechanical coupling mechanism. 3 Moreover, under physiopathological conditions, the enzyme can work in reverse as an ion pump. In this case, it hydrolyzes ATP to generate the torque that provides the Δp required for H+ uphill translocation. 4 All F1FO-ATP synthases have a conserved basic composition, which mirrors the relatively simple bacterial enzyme subunit stoichiometry of (αβ)3, γ, δ, ε in the F1 domain and of a, b2, and c10 subunits in the FO domain. In mammals, the hydrophilic F1 domain consists of five different globular subunits with (αβ)3, γ, δ, and ε stoichiometry, whereas the hydrophobic FO domain is formed from the subunits a, the c8-ring, two membrane-inserted α-helices of b subunits, and supernumeraries subunits, namely, e, f, g, A6L, DAPIT, and 6.8 kDa proteolipid, which are typical of mammalian mitochondria ( Fig. 1 ). Mitochondrial δ and bacterial ε subunits are homologous, and the mitochondrial oligomycin sensitivity conferral protein (OSCP) subunit is homologous to the bacterial δ subunit. Conversely, the mitochondrial ε subunit has no homologue in prokaryotes. 5 In addition, the mitochondrial enzyme shows OSCP, d, and F6 extra subunits on the stator subcomplex joined to the extrinsic α-helices of b subunits. 6 The a and A6L subunits of the F1FO-ATP synthase are codified by the mitochondrial DNA, whereas the other subunits are from nuclear DNA. Moreover, the nuclear gene of the c-subunits has three isoforms. Indeed, these subtle structural differences between the same enzyme complex in mammalian mitochondria and in bacteria are crucial in the perspective of exploiting the ATP synthase as a drug target to selectively fight pathogens without affecting the mammalian host.
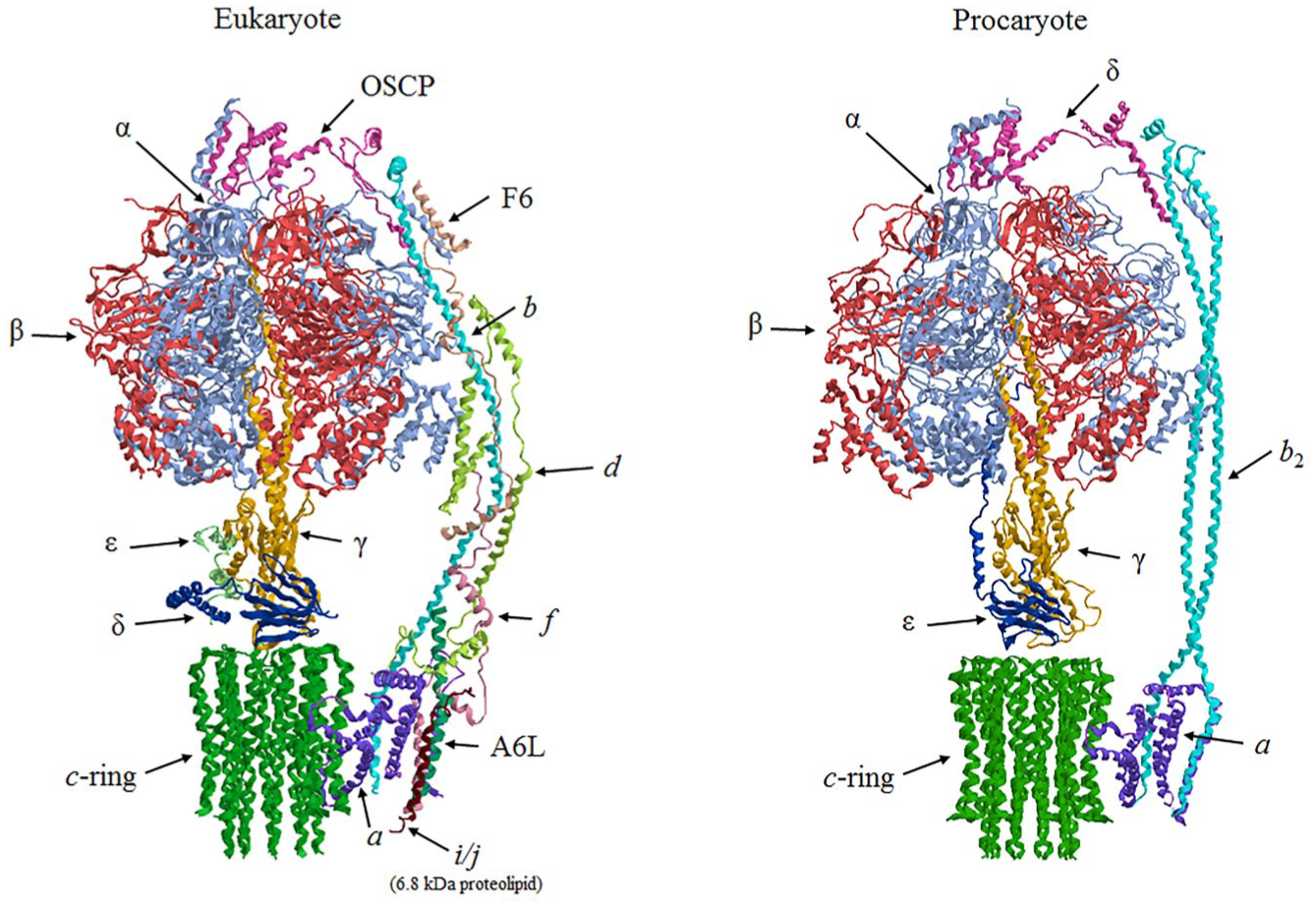
Eukaryotic and prokaryotic structures of the F1FO-ATP synthase. Protein subunits are drawn as ribbon representations obtained from modified PDB ID codes: 6B8H of yeast mitochondrial F1FO-ATP synthase monomer and 5T4O of bacteria F1FO-ATP synthase. In the eucaryotic structure, the 6.8 kDa proteolipid (i/j subunit in yeast), the truncated DAPIT (k subunit in yeast), e and g subunit are not represented.
Because of its function as the main ATP maker, the inhibition of F1FO-ATP synthase is incompatible with life under aerobic conditions. However, the recent implication of the F1FO-ATP synthase in cell death extends the potential exploitation of the enzyme as a drug target to kill unwanted cells or, by blocking its lethal role, to prevent cell death associated with some still poorly treatable pathologies. Accordingly, under pathophysiological conditions, the enzyme complex can favor or even directly produce an increase in permeability of the inner mitochondrial membrane, an event that dissipates Δp and ion homeostasis and ultimately leads to programmed cell death. 7 Interestingly, cell death in diseases featured by mitochondrial dysfunctions and bioenergetic failure is accompanied by a common event, namely, the formation of the mitochondrial permeability transition pore (mPTP). 8 The mPTP is an open high-conductance channel in the inner mitochondrial membrane that opens when Ca2+ concentration rises in the mitochondrial matrix.9,10 Recent works strongly suggest that the F1FO-ATP synthase, in its dimeric state or by exploiting the c-ring,11,12 could form the mPTP.
On considering the emerging double role of this intriguing enzyme complex, as an aerobic builder of ATP, the main energy currency molecule, and as an energy-dissipating engine, the perspective of exploiting it as a drug target ( Table 1 ) is not remote, and some promising attempts have already been described. 24 New-generation inhibitors may be addressed to selectively kill bacterial pathogens, overcoming the increasing threat of antibiotic resistance, whereas mPTP modulators targeting the F1FO-ATP synthase may constitute innovative therapeutic interventions28–30 to prevent cell death in diseases associated with mitochondrial dysfunctions or, conversely, to initiate the deathly pathway to lessen the proliferation of malignant cells. Although the possibility of exploiting FO subunits as drug targets has been strongly supported by bioinformatic insights, 31 the possible modulation of the drug potency, on one side by acting on drug design or structural modifications of natural compounds18,24 and, on the other, through amino acid substitutions or posttranslational modifications on both the F1 and FO domains, 32 still poses some technical difficulties and interrogatives.
Main Known Inhibitors of the F1FO-ATP Synthase.
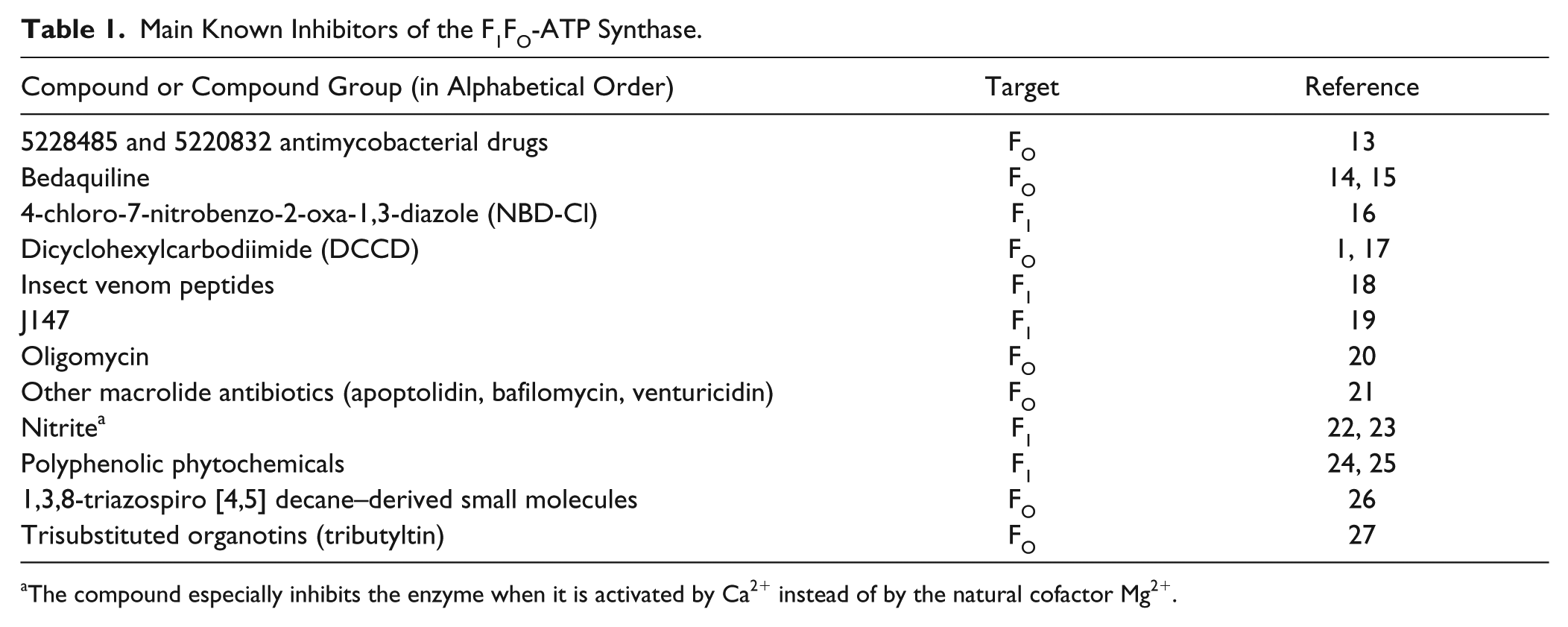
The compound especially inhibits the enzyme when it is activated by Ca2+ instead of by the natural cofactor Mg2+.
F1FO-ATP Synthase as Target of New Antimicrobial Drugs
The use of the mitochondrial F1FO-ATP synthase as a drug target has been extensively considered in drug design,32–35 especially to selectively kill noxious cells, 36 because of the immediate link between ATP deprivation and cell death. The possibility of exploiting the F1FO-ATP synthase as a target of innovative drugs to kill pathogens is based on the prerequisite that drug molecules can selectively bind and inhibit the bacterial enzyme and do not affect the mammalian F1FO-ATP synthase. In the worrying context of increased antibiotic resistance, the enzyme luckily looks to be the Achille’s heel of multidrug-resistant micro-organisms. 37 To fight pathogens, slight structural differences between microbial and mammalian enzymes are crucial. Inhibitors of the F1FO-ATP synthase bind to the enzyme and often produce posttranslational modifications of enzyme proteins, namely, they chemically modify amino acid residues that are essential for enzyme catalysis or, indirectly, for driving protons across the inner mitochondrial membrane.32,38 Moreover, the role of the F1FO-ATP synthase as a key molecular and enzymatic switch between cell life and death34,39 increases its attractiveness in pharmacology. Any natural or synthetic compound that targets the F1FO complex and/or modulates its catalytic activity can potentially be used in therapy to counteract pathogens,16,29 assuming that it is able to discriminate between eukaryotic and prokaryotic F1FO-ATP ATP synthases.
The tight connection between antibiotics and F1FO-ATP synthase is long known. Accordingly, the membrane-embedded rotor FO takes its name from the antibiotic oligomycin, which specifically inhibits both F1FO-ATP synthesis and hydrolysis by blocking proton translocation through FO. In the past decades, many natural compounds produced by micro-organisms, insects, and amphibians were shown to bind to and inhibit the F1FO-ATP synthase. Some of them exhibit multiple properties, namely, antimicrobial/anticancer activities.
Drugs Targeting F1
The immediate connection between the block of ATP production and arrest of cell proliferation addressed most studies to inhibitors that bind to the catalytic portion F1. 40 Many phytochemicals with antimicrobial properties inhibit bacterial F1FO-ATP synthase by interacting with a phytochemical-binding region on F1 ( Fig. 2 ). 24 Most have a polyphenolic or polycyclic structure. Thymoquinone, safranal, piceatannol, and baicalein 100% inhibit the Escherichia coli wild-type F1FO-ATP synthase. The inhibition power depends on the type and positioning of the functional groups of these molecules and offers helpful hints in drug design. Accordingly, the addition, deletion, and rearrangement of functional groups can enhance the degree of inhibition. Natural resveratrol causes about 40% inhibition of F1FO-ATP synthase with IC50 at about 94 µM, but its chemical modification by removal, addition, or repositioning of its functional groups can result in much more powerful drugs. 25 Another phytochemical, hydroxytyrosol from olives, caused about 60% inhibition of E. coli membrane-bound F1FO-ATP synthase, but the repositioning of its –OH groups resulted in almost complete enzyme inhibition. 24 Some venom peptides 18 from wasp, spider, bee, and scorpion that target the F1FO-ATP synthase also block bacterial proliferation. These peptides bind to the βDELSEED-motif residues of F1. Modified venom peptides with C-terminal amide (–NH2) groups enhanced both the enzyme inhibition and E. coli cell death. These peptides show different degrees of hydrophobicity and hydrophilicity and interact with different amino acid residues in the common region of F1.
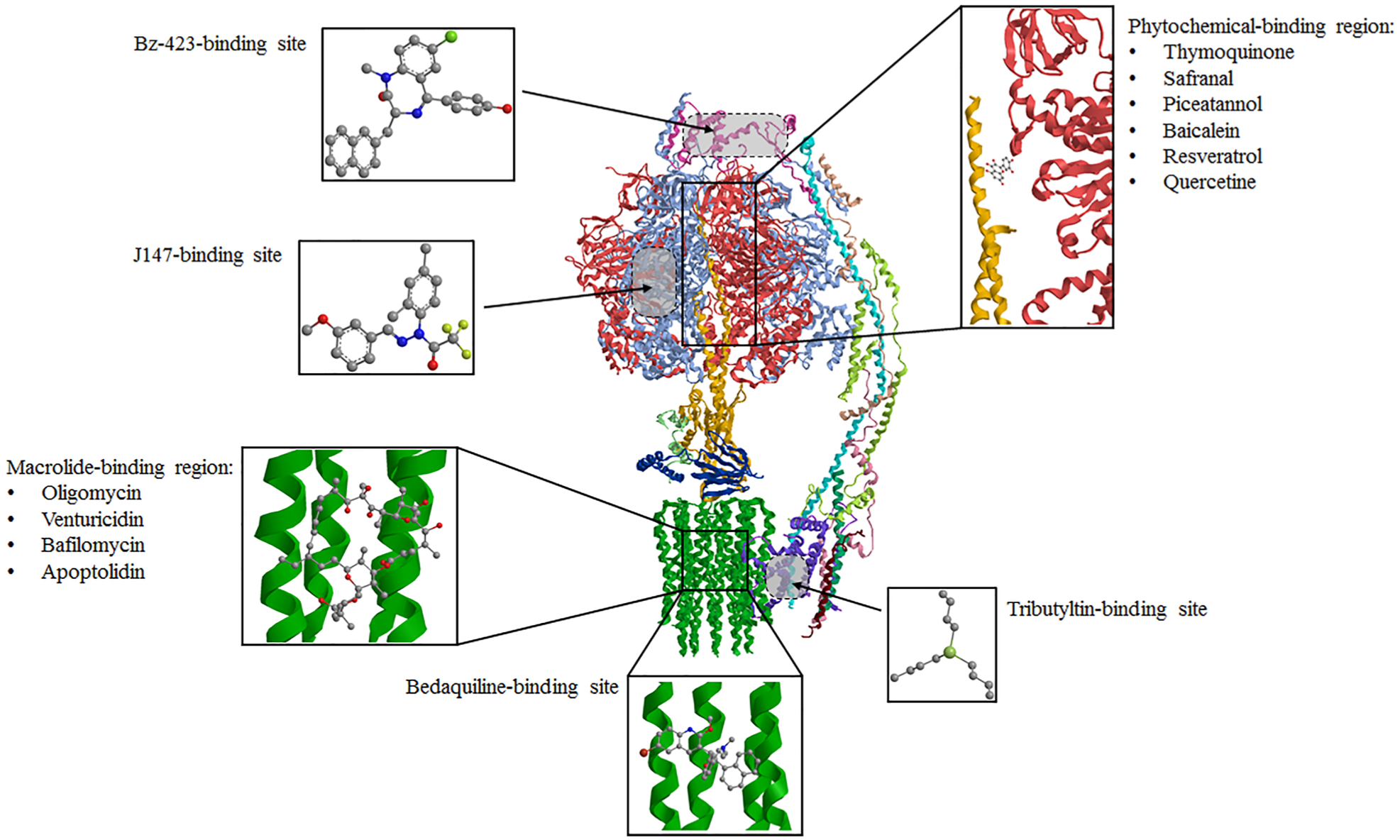
Binding sites of F1FO-ATP synthase inhibitors. The inhibitor structures are in ball and stick mode, while the enzyme is drawn as a ribbon (modified PDB ID codes: 6B8H).
Drugs Targeting FO
More recently, the membrane-embedded portion FO revealed its attractiveness as a target of lipophilic drugs.29,30,41 Some natural compounds structurally related to oligomycin 21 have been shown to target FO and to similarly inhibit the ATP synthase. Moreover, the susceptibility of c-subunits to posttranslational cysteine (Cys) thiol oxidation21,42 discloses a potential capability to modulate the enzyme’s susceptibility to inhibitors. The sulfur-containing Cys is especially susceptive to posttranslational modifications of its thiol group, which may act as a chemical switch according to the cell redox state. 43 Interestingly, bacterial F1FO-ATP synthase contains less Cys than the mitochondrial enzyme, and these Cys are apparently not essential for the enzyme functions. Accordingly, the enzyme function in E. coli is fully maintained when all 21 native Cys are replaced by alanines. 44 Posttranslational modifications are poorly documented in bacteria and probably confined to some proteins and species. 45 Conversely, the mitochondrial F1FO-ATP synthase is a hot spot for oxidative posttranslational modifications involving thiols. Interestingly, some thiols of FO are essential to bind the antibiotic oligomycin, which blocks the H+ flux within FO. In detail, oligomycin binds to the c-ring by interacting with the amino acid side chains of two adjacent c-subunits (in general identified as c1 and c2) and covers the H+ binding site of Glu residue on c1, 20 which is involved in ion translocation. 46 If these thiols are oxidized, the F1FO-ATP synthase becomes refractory to oligomycin and other macrolide antibiotics such as venturicidin, apoptolidin, and bafilomycin 21 ( Fig. 2 ). The proteolipid subunits of the c-ring frame a common drug-binding region and bind closely related macrolide antibiotics in distinct sites that share a common region.20,21,30 Therefore, an intriguing hypothesized antibacterial strategy may be based on the potential desensitization of the mammalian enzyme to these antibiotics by exploiting a chemical treatment that oxidizes these crucial thiols. In this case, the so-called drug-binding region of the F1FO-ATP synthase will become an effective target for antibiotics,20,21 which will leave the host enzyme unaffected. 30 Working in this direction, unfortunately up to now, the antibiotic-desensitizing properties were found only for the highly toxic organotin compounds,42,47,48 which can bind to the FO domain 27 ( Fig. 2 ) and whose biocidal effects prevent their use in therapy.
Tuberculosis is a major health problem and still a great threat for humans, because of the increasing resistance to traditional antimycobacterial drugs, which make the need for new therapeutic tools really pressing. 49 Mycobacterium tuberculosis is usually fought by mixtures of drugs that act on different targets. 50 The peculiar structure of the c-ring of mycobacterial F1FO-ATP synthase, which catalyzes only ATP synthesis and cannot operate in the reverse ATP hydrolysis mode, offers new therapeutic options to fight tuberculosis. The new diarylquinoline drug, also known as bedaquiline (BDQ) trade name Sirturo, and code names TMC207 and R207910, 14 binds to the c-ring 15 and also interacts with the mycobacterial ε subunit 51 by inhibiting the F1FO-ATP synthase, which is essential for Mycobacterium growth. The BDQ molecule approaches the c-ring and binds the region of the proton-binding sites ( Fig. 2 ) by resembling the DCCD and oligomycin mode of inhibition of F1FO. 15 BDQ would also act as an uncoupler by electroneutral H+/K+ antiporter. 52 According to Nath’s model, BDQ binds H+ from outside the mycobacterial cell and translocates H+ inside along the electrochemical gradient. The transport is electroneutral because, simultaneously, K+ is transported outside. 52 However, different from classical uncouplers, BDQ dissipates only ΔpH and does not interfere with the membrane potential (Δψ), which would be maintained by the negatively charged mono-anion succinate translocation from outside to inside through FO or by the electrically equivalent K+ transfer from inside to outside. The futile cycle created by BDQ accelerates respiration. The preserved Δψ would impair succinate homeostasis and could be the ultimate cause of mycobacterial death. Accordingly, succinate could be translocated outwards coupled with H+ by the redox reactions but could not reenter because of the Δψ. According to this model, the depletion of internal succinate required for the Krebs cycle and oxidative phosphorylation would kill mycobacteria. 53 Despite some aspects that remain to be fully understood, this recent model, sustained by Δψ measurements, provides new insights into the BDQ bactericidal mechanism. Even if, unfortunately, BDQ-resistant strains are increasing, 19 and BDQ cardiovascular side effects of this drug are still a matter of concern, 54 the assessment of BDQ properties opens the path to the design of new antimycobacterial drugs targeting the F1FO-ATP synthase. Accordingly, novel compounds named 5228485 and 5220832 selectively targeting the F1FO-ATP synthase of M. tuberculosis and showing excellent bactericidal activity in vitro have been identified. 13 These compounds, which show low toxicity to mammalian mitochondria, interact with mycobacterial c-subunits by binding to different amino acids with respect to BDQ.
F1FO-ATP Synthase as a Target of Cardioprotectants
The F1FO-ATP synthase susceptibility to oxidative stress suggests that it can be adequately modulated by exogenous compounds that modify the cell redox state. Many cardiovascular diseases are featured by oxidative stress, 55 which, among diffused damage to biomolecules, causes posttranslational modifications on the enzyme. Accordingly, pharmacological manipulations of oxidative and nitrosative pathways are known to be beneficial in patients with heart failure.56,57 Moreover, oxidative stress in cardiovascular diseases often stems from mitochondrial dysfunctions. In recent years, pharmacological research has been focused on the development of compounds that specifically target mitochondrial components to treat cardiovascular pathologies. Unfortunately, until now, no drugs specifically conceived to modulate mitochondrial functions have been available. 58 However, many hints suggest that the beneficial effects of some currently used cardioprotectants are due to their chemical effects on the F1FO-ATP synthase. During heart failure, the α subunit of F1 forms disulfide bonds between Cys294 on neighboring α subunits as well as between Cys294 and Cys103 on the γ-subunit. The same Cys294 can also be S-glutathionylated and S-nitrosylated. The formation of disulfide bonds and the glutathionylation (RS-SG) at these regulatory sites inhibits the F-ATPase activity, thus suggesting that these bonds modify the enzyme conformation and prevent catalysis. 32 Among the effects of resynchronization therapy (RST) in heart failure, some chemical modifications are especially interesting. An adequate RST in heart failure patients affects the mitochondrial subproteome by modifying some proteins involved in cellular redox control and oxidative phosphorylation pathways. 59 The RST reverses the disulfide bond formation and replaces it with S-nitrosylation accompanied by a recovery of the F-ATPase activity. It seems likely that the cardiac RST therapy may also stimulate the mitochondrial antioxidant defense systems 60 or enhance the reducing status in the molecular environment. Reversible Cys oxidations may protect against permanent oxidative damage to the F1FO-ATP synthase, which would decrease ATP production. 60 On the other hand, during heart failure, F1FO-ATP synthase inhibition would have the physiological ability to limit ATP consumption, contributing to ATP homeostasis, reducing the Δψ and consequently the driving force for Ca2+ uptake. 61 It seems likely that Cys294 in the F1FO-ATP synthase α subunit, localized on the enzyme surface and surrounded by several basic amino acids residues, would act as a redox switch. Under physiological conditions, it is probably deprotonated and susceptive to oxidants. Most likely, at first Cys294 is oxidized to sulfenic acid, thus causing conformational changes, which in turn expose other side chains such as Cys103 of the γ subunit. When S-glutathionylated or involved in disulfide bonds between two Cys in adjacent α subunits as well as between Cys294 and Cys103 of the γ subunit, these cross-links block the enzyme rotation and consequently ATP production. The RST would stimulate the cellular antioxidant efficiency, break the disulfide bonds, and favor S-nitrosylation, a posttranslational modification that is compatible with the catalytic activity. 60 Thus, this single modifiable Cys in the F1FO complex would act as a redox modulator of cellular ATP concentration. 62 However, because an increase in S-nitrosylation of α subunits of F1 causes a dose-dependent decrease of the enzyme activity and S-nitrosylation inhibits the F-ATPase activity during ischemia/reperfusion, 57 most likely the effects on the catalytic activity of the F1FO complex may depend on the targeted Cys and still poorly defined variables. It is not clear if, at least under some conditions, S-nitrosylation can also lead to S-glutathionylation. 55
In this molecular context, the beneficial effects of nitrite, abundant in many vegetables and now recommended to prevent cardiovascular diseases, most likely could depend on posttranslational modifications on the F1FO-ATP synthase. 56 During hypoxia/reoxygenation cycles in cardiac mitochondria, a sudden loss of membrane potential is accompanied by an increase in reactive oxygen species. 63 Upon oxidative stress, nitrite would generate the radical ∙NO2, which in turn would promote the formation of tyrosyl radicals from protein tyrosine residues. 22 By binding to the enzyme-ATP complex (ES), consistently with the uncompetitive inhibition mechanism, 56 nitrite would produce a two-step posttranslational modification: the first step generates tyrosyl radicals, whereas the subsequent step yields the ES complex and dityrosine. Dityrosine formation would be favored by the conformational change of the catalytic sites during ATP hydrolysis, which, by making closer two adjacent aromatic radicals, would make them bind together. Nitrite does not form nitrotyrosines, the most common posttranslational pathological modification of tyrosine, 64 but dityrosine. Consistently with this radical mechanism, dityrosine formation promoted by nitrite and the F1FO inhibition are enhanced by oxidative stress. 56 Consistently, the benefits of nitrite could be at least partially ascribed to the decreased ATP dissipation. 23
F1FO-ATP Synthase as a Target of Anticancer Drugs
The inhibition of the mitochondrial ATP production could be an efficient antiproliferative weapon, to kill malignant cells. Even if cancer cells mainly rely on glycolysis for ATP production (the Warburg effect), 65 many natural phytochemicals that target the F1FO complex were claimed as beneficial to fight cancer and prevent metastatic dissemination. Most studies addressed the catalytic portion F1 that binds various natural compounds. The F1FO-ATP synthase has about 12 discrete inhibitor binding sites, including peptides and other inhibitors located at the interface of the α/β subunits on the F1. Antitumor peptides bearing the α-helical structure interact with the β DEELSEED site of F1. 66 These peptides are derived from various animal taxa, including insects, yeasts, and amphibians. Some of them also display antimicrobial activity, due to a shared antiproliferative mechanism. The anticancer activity of some polyphenols may be at least partially ascribed to their binding to the F1FO-ATP synthase. 66 Among phytochemicals, only genistein binds to the FO sector, 25 because of its hydrophobicity and membrane penetration, but recent studies did not confirm this target. Other natural and synthetic compounds such as apoptolidin have been shown to inhibit the ATP synthase by specifically targeting FO and particularly the c-ring. The natural macrolide apoptolidin, which selectively leads transformed cells to apoptosis, revealed significant similarity between the apoptolidin aglycone and oligomycin. Accordingly, apoptolidin binds to the macrolide binding region of FO 35 ( Fig. 2 ). Other natural and synthetic polyketide macrolides (mandelalides) are highly cytotoxic, especially on cells with an oxidative phenotype. Some of these compounds bind to the F1FO-ATP synthase and stimulate caspase-dependent apoptosis in HeLa cells after inhibition of glycolysis by D-deoxyglucose. 67 However, the F1FO-ATP synthase inhibition by apoptolidin and analogues does not always correlate with the induction of cell death. 68 Most likely, the antiproliferative properties of these compounds are only partially referable to the F1FO-ATP synthase inhibition, and alterations in the signaling pathways are involved. However, the properties of these compounds suggest that relatively small molecules binding to the F1FO-ATP synthase can be exploited to modulate the apoptotic pathway. An alternative approach to kill unwanted cells by driving them to apoptosis may be to favor the lethal activity of the F1FO-ATP synthase, namely, to stimulate the formation of the mPTP, as considered below.
F1FO-ATP Synthase as a Target of Pore Modulators
Compounds able to prevent the mitochondrial permeability transition, the master player in apoptosis and necrosis, are increasingly considered as beneficial tools in cytoprotection. Because the mitochondrial permeability transition is ascribed to the mPTP, these compounds should be pore shutters. Assuming that, as strongly suggested by recent advances, the mPTP formation involves the F1FO-ATP synthase,11,69 compounds acting on the enzyme complex may modulate the mPTP. The 1,4-benzodiazepine (Bz-423), inhibitor of F1FO-ATP synthase, binds to the OSCP subunit at the same site as cyclophilin D (CyPD), the only known protein modulator of mPTP. 70 Bz-423 induced apoptosis by sensitizing the mPTP to Ca2+ ( Fig. 2 ). 11 CyPD interactions with the OSCP subunit decrease the hydrolysis of MgATP, whereas cyclosporin A (CsA) increases the Mg-ATPase activity by displacing CyPD from its binding site. 71 Moreover, CsA has long been known to inhibit the mPTP. 72 The CsA-dependent mPTP inhibition is responsible for the immunosuppressive action by calcineurin. The mPTP opening as an end-stage event of mitochondrial demise contributes to cellular damage in spinal cord injury. 73 The nonimmunosuppressive CsA analog, NIM811, improves function recovery in spinal cord injury. Therefore, in the central nervous system, the mitochondrial function can be potentially exploited as a drug target. 73
Recent evidence indicates that both Ca2+ and Mg2+ elicit F1FO-ATP synthase activities are referable to the same F1FO-complex, 74 and when Ca2+ rises in mitochondria, the enzyme complex switches to the Ca2+-activated mode, the inner mitochondrial membrane is depolarized, 63 and the F1FO-ATP synthase works “in reverse,” namely, it hydrolyzes ATP. According to an intriguing model, when the enzyme complex is activated by Ca2+ instead of by the natural cofactor Mg2+, the higher steric hindrance of Ca2+, which would insert in the catalytic sites of F1 in replacement of Mg2+, would trigger conformational changes. 75 These changes, once transmitted to FO, would detach the two joined monomers of the dimer, thus opening the mPTP. 39 In this case, the F1FO-ATP synthase would act as a “death enzyme,” leading to cascade events that ultimately cause cell death. 76 Thus, any compound that preferentially inhibits the Ca2+-activated F1FO-ATP synthase with respect to the Mg2+-activated F1FO-ATP synthase can be potentially thought of as a p -shutter and be used to limit or prevent cell death. This property can be exploited as an efficient molecular tool to treat mPTP-related mitochondrial dysfunctions, which are a common feature not only of mitochondrial disorders but also of a variety of human pathologies. However, to prevent mPTP formation and be used in therapy, the pore shutter should discriminate between normal and mutated or modified (pathological) enzyme complexes, such as those found in some neurodegenerative human diseases, namely, maternally inherited Leigh’s syndrome and neuropathy, ataxia, retinitis pigmentosa. 77 Pathogenic mutations often involve changes in the Atp6p (or a) subunit of the F1FO-ATP synthase, which contains most of the residues involved in proton translocation through FO. 46 Other pathologies are featured by increased mPTP formation. These diseases are often associated with oxidative stress, which in turn favors mPTP opening. Drug design could be addressed to different directions. A first approach could aim at obtaining molecules that selectively bind to the mutated amino acid residues in genetic diseases so as to modify their properties chemically and allow/improve the mitochondrial F1FO-ATP synthase function.
Some suggestions are provided by known antiapoptotic proteins. Bcl-XL is an anti-apoptotic protein that acts at the outer mitochondrial membrane 78 but can also target the mitochondrial inner membrane. When Bcl-XL is co-localized with the β subunit of the F1FO-ATP synthase, 79 it acts as an anti-apoptotic regulator by blocking the mPTP opening. The chemotherapeutic agent ABT-737, which mimics BH3, the only protein that binds to Bcl-XL, reverses the Bcl-XL binding to the β subunit and increases the membrane leak conductance. 80
Another approach that can be applied to treat all (genetic and nongenetic) mPTP-related pathologies is to find appropriate pore shutters. Other than in mitochondrial dysfunctions of variegated etiology, selective inhibitors of the Ca2+-activated F1FO-ATP synthase, such as nitrite, could shut the mPTP, prevent cell death, and play a main role in cytoprotection in different body districts. 23 When Ca2+ rises in mitochondria, the enzyme complex activated by Ca2+ hydrolyzes ATP. In this case, the enzyme inhibition by nitrite would lessen ATP dissipation. 56 Accordingly, high nitrite concentrations may efficiently block the F-ATPase activity sustained by Ca2+ without affecting that activated by Mg2+. The enhanced enzyme inhibition by nitrite under oxidative stress, which features many diseases, spanning from diabetes to cancer and neurodegenerative diseases, may be helpful to limit cellular injury under pathological conditions. Nitrite constitutes a good representative of small molecules that directly acts on the F1FO-ATP synthase. The list of mPTP-related pathologies on one hand and, on the other, of mPTP blockers is fated to increase with the knowledge in this field. The advantage of small molecules over complex compounds as drugs targeting mitochondria relies in their easier incorporation in mitochondria without requiring to be vehiculated. 81 Because the mPTP opening plays a key role in the progression of myocardial cell death secondary to reperfusion in myocardial infarction, a new pharmacological approach using a small molecule based on a 1,3,8-triazaspiro[4.5]decane targets the c subunit of the F1FO-ATP synthase and inhibits the mPTP activity by decreasing the apoptotic rate during reperfusion. 82
F1FO-ATP Synthase, Aging and Neurodegenerative Disorders
Many diseases share with aging some basic mechanisms of cell death. In turn, the incidence of some severe pathologies is tightly linked to an increased life span and increases with age. Because mPTP formation is one of these age-related mechanisms, it is clear that the pore shutters may have multiple therapeutic roles and also display anti-aging properties. However, if pore modulators targeting the F1FO-ATP synthase can be effective in the treatment of neurodegenerative disorders and other mPTP-related diseases, alternative therapeutic approaches, also based on the F1FO-ATP synthase modulation, have been considered. Recently, the synthetic compound J147 emerged as a promising therapeutic molecule because it rescues severe cognitive deficits in aged, transgenic mice. J147 targets the mitochondrial α-F1-ATP synthase (ATP5A; Fig. 2 ) and causes a modest enzyme inhibition that drives intracellular Ca2+ to activate AMP-activated protein kinase to inhibit mammalian rapamycin target, a known mechanism that extends the life span from worms to mammals. 19 Developing treatments for amyotrophic lateral sclerosis aim at maintaining the mitochondrial function 83 and often reveal a close parallelism between mPTP formation and the activity of the F1FO-ATP synthase. The increase in ion conductance due to the mPTP formation and recorded within the c-subunit of the F1FO-ATP synthase 12 is inhibited by the neuroprotective drug dexpramipexole, which induces conformational changes in the F1FO complex. 84 Therefore, the mutually linked modulation of the F1FO-ATP synthase and of the mPTP strongly supports the ongoing development of drugs targeting the ATP synthase to treat neurodegenerative disorders.
Ectopic F1FO-ATP Synthase: A New Target for Drugs?
For a long time in eukaryotes, the F1FO-ATP synthase has been confined to the mitochondrial inner membrane. However, the occurrence of F1Fo-ATP synthase or of its subunits, both nuclear and mitochondrially encoded, 26 in extramitochondrial membranes such as the plasma membrane of tumor and normal cells, namely, endothelial cells, hepatocytes, and adipocytes, and also in the endoplasmic reticulum, cannot be neglected. This peculiar F1FO-ATP synthase was defined as “ectopic,” because it is found in extramitochondrial membranes. Interestingly, some ectopic F1FO-ATP synthase subunits act as cell-surface receptors for various ligands, a feature not always shared with the mitochondrial enzyme, which extends its potentiality as putative drug target. The origin and biological function of the ectopic F1FO-ATP synthase, which shows oligomycin sensitivity and both ATP synthase and ATP hydrolase activities, 85 are still a matter of debate. The overexpression of ectopic F1FO-ATP synthase under peculiar physiopathological conditions and its receptor-like behavior shoulder its involvement in cell signaling for the regulation of vascular tone, cholesterol metabolism, carcinogenesis, and metastatic progression.36,86 The ectopic F1FO-ATP synthase is a typical “moonlighting protein,” whose function apparently depends on many variables such as localization, expression, ligand concentration, and so on. 86 Thus, the ectopic enzyme may be a potential target for a wide variety of drugs. However, before setting out therapies targeting the ectopic F1FO-ATP synthase, its role must be clarified. Moreover, on considering drugs targeting the F1FO-ATP synthase, their possible dual effect on the ectopic and on the mitochondrial enzyme complex should be evaluated. Recently, the interaction of some phytophenols and natural peptides with the ectopic F1 was considered as a putative mechanism that contributes significantly to the anticancer properties of these compounds. 66
Because of its unique properties of a crucial enzyme complex able to drive the opposite conditions of life and death in cells, the F1FO offers great opportunities as a drug target in therapy. Accordingly, compounds able to bind to the F1FO and to affect the enzyme function may be used as pharmacological tools to selectively address unwanted cells to death or, on the contrary, to prevent the cell’s lethal fate, not only due to mitochondrial impairment or dysfunctions but also to mPTP opening, in which the Ca2+-activated enzyme is most likely involved. 77 The main challenge in exploiting new molecules as antibiotics to overcome antibiotic resistance is blocking the enzyme’s vital role as an ATP maker in only unwanted (pathogen) cells without affecting the host enzyme. To this aim, slight structural differences between the prokaryotic and eukaryotic F1FO complex can be exploited. Moreover, as recent advances suggest, the mammalian enzyme could be made refractory to known antibiotics by posttranslational modifications, induced by chemicals and unable to affect the prokaryotic F1FO-ATP synthase.
Assuming that the Ca2+-ATPase is involved in the mitochondrial pathway of apoptosis, some compounds targeting the enzyme complex could constitute antiproliferative drugs or conversely delay or lessen cell death under severe pathological conditions. mPTP modulators that target the Ca2+-activated F1FO-ATP synthase and favor or block mPTP formation and opening may expand the range of therapeutic options. Accordingly, the symptoms of some severe neurodegenerative and cardiovascular disorders, as well as some age-related diseases—all conditions featuring increased mPTP formation—could be attenuated by the so-called pore shutters, such as nitrite, which would block the conformational transmission mechanism that opens the mPTP. On the other hand, mPTP activation by compounds that stimulate the Ca2+-ATPase activity could be exploited to kill malignant cells. From this fascinating perspective, novel drugs could be found among natural or synthesized compounds that selectively target the Ca2+-activated F1FO, namely, the putative deathly engine of mitochondria.
Molecular therapies based on F1FO modulation may offer new strategies, also in combination with existing therapies, to counteract insufficiently treated human pathologies. Accordingly, drugs targeting the F1FO may help to attenuate structural and/or functional mitochondrial defects, to overcome antimicrobial resistance, and also to improve the prognosis of mPTP-related diseases.
The attractiveness of the F1FO complex as a promising drug target is strongly substantiated by recent knowledge. Further studies may also expand these perspectives and define the pros and cons of the potential use of this enzyme in pharmacology, toxicology, clinics, and personalized medicine.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.