
Abstract
New cellular model systems for drug metabolism applications, such as advanced 2D liver co-cultures, spheroids, and microphysiological systems (MPSs), offer exciting opportunities to reproduce human biology more closely in vitro with the aim of improving predictions of pharmacokinetics, drug–drug interactions, and efficacy. These advanced cellular systems have quickly become established for human intrinsic clearance determination and have been validated for several other absorption, distribution, metabolism, and excretion (ADME) applications. Adoption will be driven through the demonstration of clear added value, for instance, by more accurate and precise clearance predictions and by more reliable extrapolation of drug interaction potential leading to faster progression to pivotal proof-of-concept studies. New experimental systems are attractive when they can (1) increase experimental capacity, removing optimization bottlenecks; (2) improve measurement quality of ADME properties that impact pharmacokinetics; and (3) enable measurements to be made that were not previously possible, reducing risk in ADME prediction and candidate selection. As new systems become established, they will find their place in the repository of tools used at different stages of the research and development process, depending on the balance of value, throughput, and cost. In this article, we give a perspective on the integration of these new methodologies into ADME optimization during drug discovery, and the likely applications and impacts on drug development.
Introduction
A number of cellular absorption, distribution, metabolism, and excretion (ADME) assays are used for lead optimization and to inform the selection of clinical candidates. Assays include metabolic stability, cellular permeability, transporter-mediated uptake, and efflux, as well as enzyme induction. The standard cellular model for metabolic drug clearance assessment is the short-term suspension culture incubation of multiple-donor pooled cryopreserved human hepatocytes.1,2 This system has the advantages of availability, acceptable cost, reproducibility (between experiments and between donor pools), and a full complement of the main metabolic enzyme systems (cytochrome P450 [CYP], UDP-glucuronosyltransferase [UGT], esterase), as well as more minor contributors (e.g., sulfotransferases, flavin monooxygenases, aldehyde oxidase). The system is deployed for a number of applications along the ADME value chain, such as intrinsic clearance determination, metabolite identification, and enzyme phenotyping. More specialized formats for cryopreserved human hepatocyte assays, for example, plated, sandwich-cultured, or co-cultured hepatocytes and hepatocytes suspended in human plasma, are used in drug uptake and efflux studies, 3 induction measurements, 4 and time-dependent inhibition risk assessments. 5 Recent developments have shown that advanced 2D hepatocyte co-culture systems (in which hepatocytes are cultivated together with supportive nonparenchymal cells) may be applied to many of these processes and, more interestingly, to the study of the combination of multiple processes to overall drug metabolism.6,7 Following on from interesting proof-of-concept studies,8–10 such advanced cellular model systems need now to be placed within the palette of tools used in routine drug development and show consistent added value in the optimization and selection of drug candidates, especially in consideration of limited-throughput capability and cost.
Early ADME screening is typically performed using one assay system per endpoint and with a focus on throughput, turnaround times, and simple data outputs. New and multiple endpoints are envisaged in future studies, not only to investigate drug–drug interactions (DDIs), but also to run more mechanistic studies for processes that last for a longer time, such as cellular infection 7 or disease-state cell culture conditions. Those applications are more focused toward the detailed characterization of drug candidates and the development of data packages for regulatory submission than in the initial phases of chemical optimization. Data from new cellular tools will be integrated in modeling and simulation approaches, such as physiologically based pharmacokinetic (PBPK) modeling, which is now routinely used to bring together knowledge of human physiology with drug-specific property information for the prediction of human pharmacokinetics (PK). PBPK modeling is increasingly recognized and accepted by health authorities in new drug applications. 11 However, the range of application with regulatory impact is currently restricted to areas of high prediction confidence, such as cytochrome P450-mediated DDIs. This limited scope is in part related to missing verification of quantitative in vitro–to–in vivo translation for other applications leading to reduced confidence.12,13 Expansion of the scope of PBPK approaches to optimize clinical development may be addressed by emerging advanced cellular or microphysiological systems (MPSs), if they can be demonstrated to improve quantitative translation for reference drugs. They may then become standardized and established within the industry so that sufficient regulator confidence is achieved to allow waiver of clinical studies through in vitro and PBPK predictions.
The main focus of this review is the application of newer hepatocyte systems to metabolism applications in ADME screening and clinical candidate selection. Not covered are tissue slices, hepatoma cell lines, and induced pluripotent stem cell-derived hepatocytes, or highly exploratory experimental systems without published ADME application using drug molecules.14,15 In vitro–to–in vivo correlations have frequently been performed for animal species to gain confidence in the predictions made from in vitro to man. Some of the new cellular tools are also available, for example, as rat, mouse, and cynomolgus monkey versions. However, with the exception of some cross-species metabolite identification studies, 16 reports of new cellular system validations have overwhelmingly focused on human cells, which is reflected in this review.
In addition to more predictive assays, ADME scientists have a high interest in the simplification of existing assays or their replacement with less resource-intensive methods. New in silico technologies have the potential to turn the information contained in large corporate ADME datasets into prospective predictions informing synthesis and testing strategies. One particular opportunity is the adoption of machine learning (ML) tools as presynthesis selection methodologies and as a way to focus experimental resources. An example of this application will also be described to illustrate the way in which the landscape of ADME screening, within which new cellular tools would be applied, is also changing.
Clearance Determination for High-Stability Compounds
Experimental Models
Metabolically stable molecules with a prolonged half-life and exposure in the human body are often designed to enable once-daily administration of drugs in moderate quantities. 17 The measurement of intrinsic clearance in vitro and the prediction of in vivo clearance form an important and well-established process to minimize the risk of unacceptable PK properties. Various in silico, in vitro, and in vivo methods have been developed for the prediction of human clearance.18–21 Suspension cultures of pooled cryopreserved hepatocytes are widely used as a moderate-throughput screen for metabolic stability in the lead optimization stages of drug discovery. 22 However, the activities of many drug-metabolizing enzymes degrade rapidly in suspended hepatocytes with loss of functionality observed within a few hours. 23 This limits effective incubation times to 3–4 h, acceptable for intrinsic clearance assessment of most compounds but insufficient for metabolically stable substances.17,24,25 One way to prolong hepatocyte activity and functionality is to incubate the cells as a 2D-plated, monolayer culture.23,25 Although extended cell lifetimes can be attained using plated hepatocytes, significant loss of enzyme activity over the first 24 h is still observed.22,23,26 One reason for the instability may be the flattened morphology of the cells in the 2D culture and the reduced polarization due to altered cell–cell and cell–matrix interactions. 8 New cultivation methods, such as co-cultivation with supportive nonparenchymal cells (e.g., fibroblasts), spheroid generation, or a combination of both, prolong the functionality and viability of hepatocytes to several weeks under media replacement conditions or to 4–5 days in the absence of new media.27–29 Two commercially available systems have been reported on most frequently: The HµREL system uses a mixed co-culture of hepatocytes and fibroblasts. 30 In contrast, the HepatoPac system features a highly controlled number of hepatocytes attached to micropatterned portions of a microplate surface. These are supported by surrounding mouse fibroblasts in a very highly optimized proportion and micropatterning geometry. 28 (For a detailed overview of this liver bioengineering, see recent reviews by Underhill and Khetani.31,32)
Intrinsic Clearance Measurability
The benefit that long-term co-culture models bring to clearance determinations of metabolically stable compounds is exemplified in Figure 1 . This shows data from an in-house study of the metabolism of verapamil and tolbutamide by suspended hepatocytes and HepatoPac co-cultures. Both hepatocyte systems could be used for the determination of verapamil intrinsic clearance with values (±95% confidence intervals) of 28.1 ± 1.3 µL/min/106 cells and 32.4 ± 2.0 µL/min/106 cells determined in suspended hepatocytes and HepatoPac, respectively. In contrast, much longer incubation times were required when studying the depletion of tolbutamide, a low-clearance drug. An in vitro intrinsic clearance of 0.72 ± 0.65 µL/min/106 cells was estimated for tolbutamide using suspension hepatocytes, whereas in the HepatoPac system, an intrinsic clearance of 0.76 ± 0.33 µL/min/106 cells was obtained. While the values are similar, there is a substantial difference in the magnitude of the confidence intervals. The percentage confidence interval in the HepatoPac system was ~40%, whereas with suspended hepatocytes it was ~90%. Importantly, confidence intervals ranging down to “nearly no metabolic clearance” would translate into high uncertainty in PK predictions.
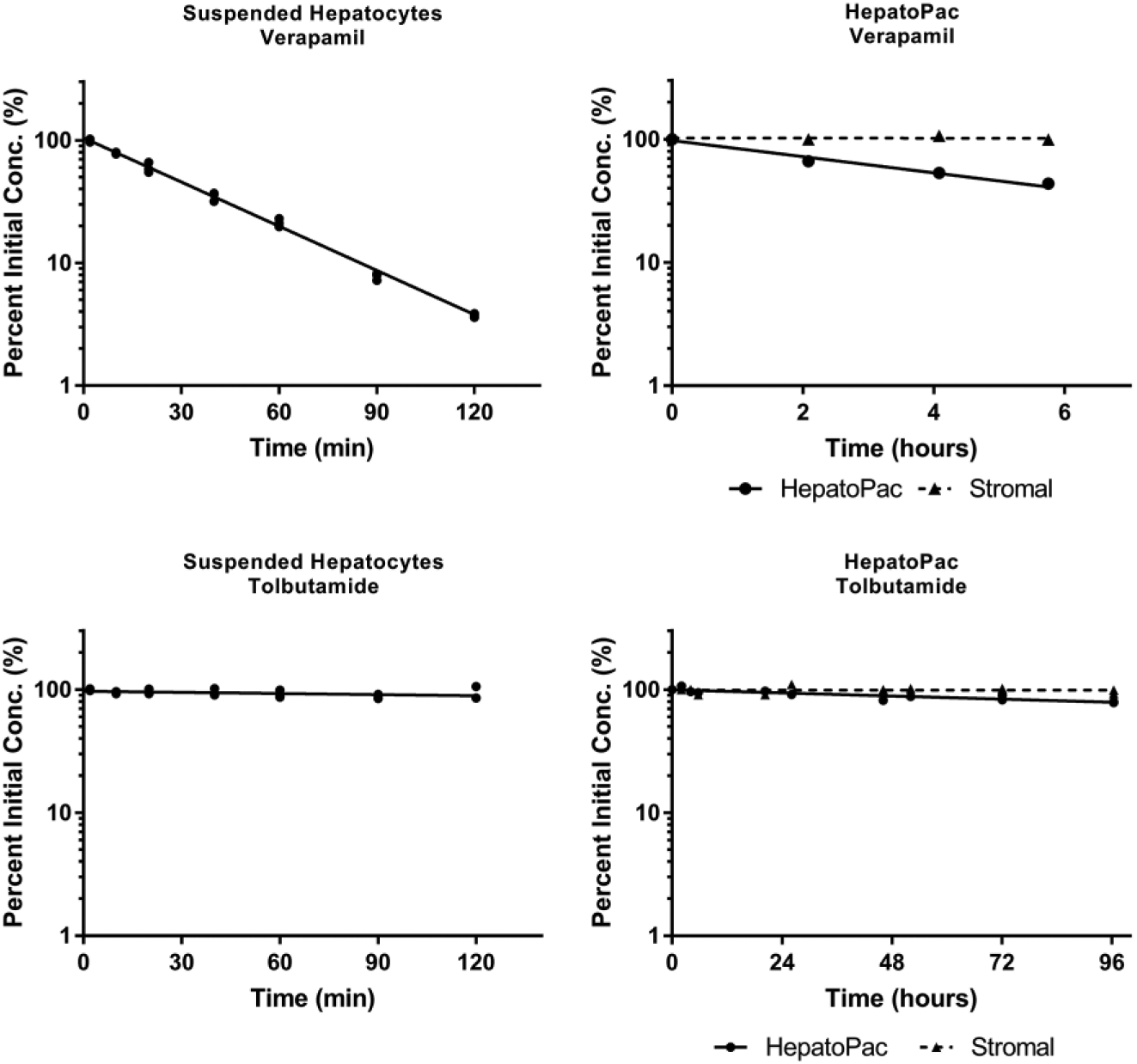
Compound depletion over time in two different hepatocyte models. Verapamil and tolbutamide were incubated in suspended hepatocytes and in HepatoPac co-cultured hepatocytes. The natural logarithm of the percent of the remaining compound with time data was fit to a linear regression curve using GraphPad Prism 6.07 software. Verapamil, a high-clearance compound, had an intrinsic clearance of 28.1 ± 1.3 µL/min/106 cells in suspended hepatocytes and 32.4 ± 2.0 µL/min/106 cells in HepatoPac cells. For tolbutamide, a low-clearance compound, an intrinsic clearance of 0.72 ± 0.65 µL/min/106 cells in suspended hepatocytes and 0.76 ± 0.33 µL/min/106 cells in HepatoPac was obtained. The contribution of stromal cells to the metabolism in HepatoPac cells was additionally determined and subtracted from the degradation observed in the HepatoPac cells.
Figure 2 explores the relationship between intrinsic clearance and confidence interval further. A wide range of compounds tested in different studies were used for this analysis, which shows an inverse correlation between percentage confidence interval and intrinsic clearance. Based on these data, a threshold of 3 µL/min/106 cells is proposed for suspension hepatocytes, above which the determination of the intrinsic clearance has an acceptable precision. While it is possible to shift this threshold to some extent by increasing the number of measurements or through further bioanalytical method optimization for individual compounds, such additional optimization is unlikely to be practical for medium-throughput screening applications. Accurate and precise determination of intrinsic clearance in long-term cultures depends on good maintenance of enzyme activities and highly reproducible cultures in each well, essential parameters for system usage.
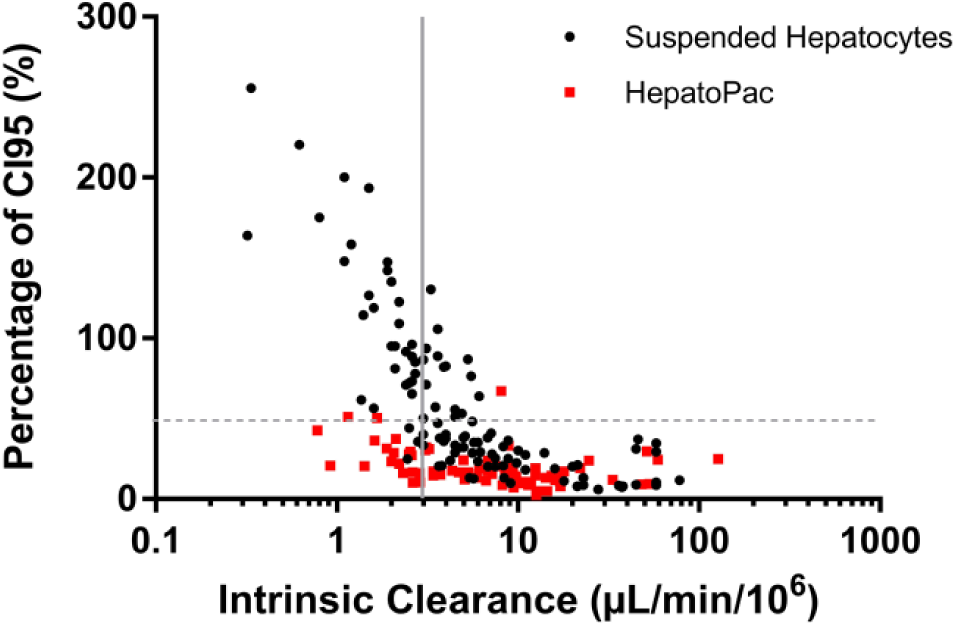
Ninety-five percent confidence interval plotted as percentage of intrinsic clearance. In-house clearance determination studies conducted in two different hepatocyte cell systems are compared. Red squares represent data from the long-term hepatocyte culture (HepatoPac) and black dots represent data from suspended hepatocytes. Studies in long-term, co-cultured hepatocytes were more precise compared with suspended hepatocytes, particularly for low-clearance compounds. Experiments with suspended hepatocytes were typically run in singlicates (2, 10, 20, 30, 60, 90, 120 min) and experiments with HepatoPac in singlicates (120, 1200, 1560, 2880, 4320, 5760 min) and in duplicates at 120 and 5760 min time points. Solid grey line indicates the threshold value of 3 µL/min/106 cells. Dotted grey line shows 50% of the 95% confidence interval. Apparent intrinsic clearance values are shown, without correction for protein binding. All compounds were tested at concentrations below Km.
Extrapolation
The use of extended incubation time systems for more precise in vitro intrinsic clearance determinations is now well established and can be expected to result in more precise in vivo clearance predictions for low-clearance compounds.32,33 Chan et al. 34 and Lin et al. 35 established the utility of micropatterned hepatocyte co-cultures for the prediction of human metabolic clearance. In these studies, in vivo clearance could be predicted for ~60%–70% of compounds within twofold and for 70%–90% of compounds within threefold of actual values. Bonn et al. 36 and Hultman et al. 30 reported almost as good predictivity for hepatic clearance using a nonmicropatterned co-culture system. These levels of predictivity compare well with those reported for the short-term suspension culture systems for higher-clearance drugs. 37 The long-term hepatocyte co-cultures also have the advantage that a broader range of intrinsic clearance values can be determined, with accurate measurement down to ~0.2 µL/min/106 cells, approximately 10-fold lower than attained in classical suspension culture experiments. 14 A comprehensive study by Da-Silva et al. showed the benefit of long-term co-cultures in human clearance prediction: 38 Using the same batch of primary human hepatocyte cells, they determined the intrinsic clearances for 15 drugs in suspension, monolayer, and HepatoPac cultures. Here the HepatoPac system clearly outperformed the other culture systems in prediction accuracy. The quality of predictions was further improved when taking into account the potential for albumin-assisted drug uptake, with 14 of 15 predictions within twofold of the in vivo clearance.
In this way, advanced cellular systems have become established for the intrinsic clearance determination of highly metabolically stable compounds and the accuracy for predictions of human in vivo clearances could be improved compared with conventional cell cultures. This new market for premium intrinsic clearance products is a likely entry point for other systems currently in development. In a challenge to long-term co-culture models, Lancett et al. recently showed data indicating that new culture supplements and Matrigel overlay may be employed to sufficiently slow the decay of enzyme activities in monoculture-plated cells to enable multiple-day usage. 39 They reported the Matrigel overlay to enable robust intrinsic clearance predictions over a duration of 30 h, with high gene expression levels for CYP, UGT, and nuclear receptors maintained over this time course. The uncomplicated nature and high cell density (350,000 hepatocytes per well in a 24-well plate) of this system could enable measurement of in vitro intrinsic clearance for highly metabolically stable compounds at lower cost than using the sophisticated co-cultures. 39 Alternative approaches, such as spheroids or more MPSs, are discussed later.
Drug–Drug Interactions
DDIs can significantly change the exposure of a co-medicated drug, potentially resulting in toxicity or loss of efficacy. 40 Avoidance of DDIs forms an important part of lead optimization. In contrast, the qualification of potential DDI issues contributes to extended clinical pharmacology programs, adding costs and slowing progression of candidate drugs to the market.
Time-Dependent Inhibition
For simple determinations of inhibition potency, reversible enzyme inhibition is easily measured using acellular systems, such as liver microsomes, and the clinical effects can be predicted using PBPK modeling approaches. 41 In contrast, time-dependent inhibition (in which the net effect on enzyme activity in vivo at steady state is a competition between enzyme inactivation and de novo synthesis 42 ) is more complex and can benefit from the use of cellular systems. 43 While rates of enzyme inactivation are readily measured using acellular systems, these have the tendency to overpredict the in vivo effect.42,44 Short-term cultures of hepatocytes suspended in human plasma have been reported to show improved prediction of clinical DDIs due to time-dependent inhibition.5,45,46 An example from an in-house study, in which a strong CYP3A4 time-dependent inhibitor (troleandomycin) was applied to the HepatoPac culture, is shown in Figure 3 . The compound caused rapid and concentration-dependent loss of enzyme activity, sustained after removal of the drug from the medium. After treatment with 0.1 and 1 µM troleandomycin, full culture activity was nevertheless regained after 48 h. With 10 µM treatment, only a partial return to baseline was attained, potentially due to irreversible cellular damage. Takahashi et al. used stable-labeled amino acid incorporation into newly synthesized proteins to examine the half-life of cytochrome P450 3A4 resynthesis in HepatoPac cultured cells. 10 This study took advantage of the equilibrium between enzyme degradation and resynthesis in long-term hepatocyte culture systems with stable enzyme expression levels. These studies indicate how long-term cultures have the potential to allow the concentration–time profiles of interacting drugs and their metabolites to be combined with effect measurements to enable more in vivo relevant DDI assessments to be made.
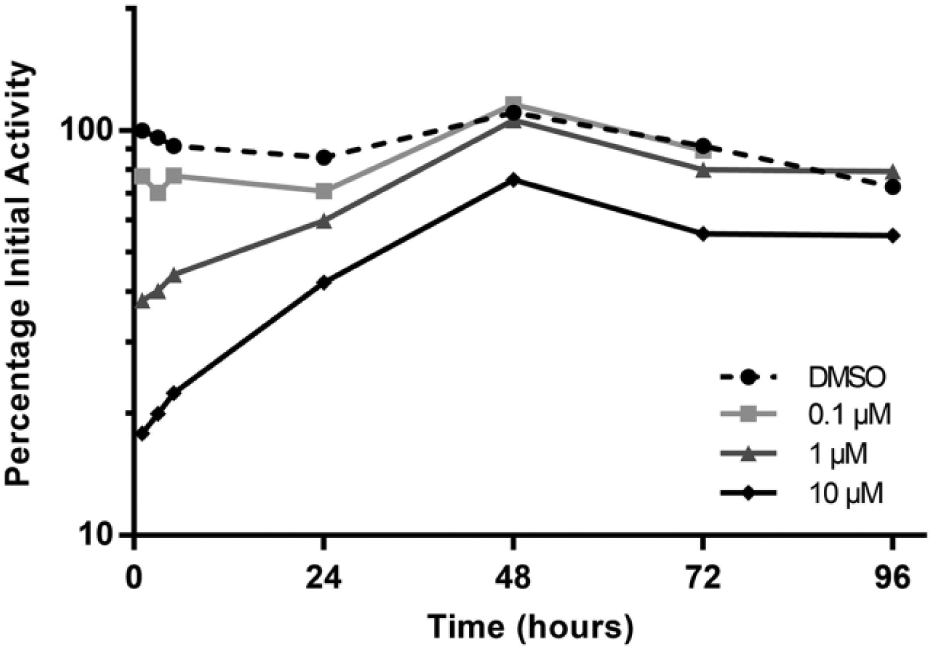
Cell activity recovery following time-dependent inhibition of CYP3A4 by troleandomycin. Troleandomycin was incubated for 1 h in HepatoPac cells at three different concentrations and then washed away using two times media exchange. Formation of 1′-hydroxymidazolam was investigated as a CYP3A4 enzyme marker in 1 h incubations at several time points up to 5760 min (96 h). Enzyme inactivation was concentration dependent and recovery to baseline activity was reached 48 h after removal of troleandomycin in the hepatocytes incubated with 0.1 and 1 µM. A recovery of the enzyme activity of about 60%–70% to DMSO control activity was observed in the cells treated with 10 µM troleandomycin, whereby the activity parallels baseline activity after 48 h.
CYP Induction
During drug development, compounds are routinely screened to identify whether they cause induction of multiple drug-metabolizing enzymes and transporters via the aryl hydrocarbon, constitutive androstane, or pregnane X receptor pathways. Well-characterized markers for these different induction pathways are mRNA levels and enzyme activities of CYP1A2, CYP2B6, and CYP3A4. Reliable testing of those markers in vitro is crucial because the induction of drug-metabolizing enzymes can have marked effects on drug bioavailability. As an example, rifampicin has been reported to reduce the exposure of eliglustat, flibanserin, or isavuconazonium sulfate by more than 95%. 47 Such exposure changes carry risks of underexposure and therapeutic failure and may require dose adjustment or contraindication. The inductive effect can last for several days before return of enzyme activities to baseline levels. 48 The typical in vitro test system for induction studies is plated primary human hepatocytes, using multiple individual donor batches known to be responsive to induction stimuli. 49 Cells are exposed to the inducing agent over a period of 2–3 days and induction is assessed via mRNA and enzyme activity measurements. 14 Similar to the determination of metabolic clearance, a drawback is the loss of activity during the incubation. 8
Figure 4 illustrates the effect of the strong CYP3A4 inducer rifampicin on monocultured plated hepatocytes and micropatterned co-cultured hepatocytes. The inductive effect could clearly be observed in both hepatocyte models, but the baseline from which it was detected was quite different. Using monoculture plated hepatocytes, the inductive effect is measured against a background of a declining enzyme activity. In contrast, the HepatoPac system shows an induction effect above a baseline that does not decline with time, potentially a better reflection of the human situation. Moore et al. showed induction validation data for the HepatoPac system, including extensive gene expression profiling. 9 Ramsden et al. used the HepatoPac system to investigate the suppression of enzyme activity by interleukin-6 (IL-6), a cytokine that indirectly downregulates CYP3A4.50,51 This long-term (11-day) study allowed observation of both enzyme suppression and enzyme resynthesis, leading to the recovery of activity after removal of IL-6. Such experiments are only possible using long-term hepatocyte cultures. Dixit et al. showed that rifampicin had a 10-fold lower apparent EC50 in the micropatterned co-culture compared with monocultured hepatocytes. 8 It was postulated that a higher organic anion transporter (OATP1B1) mediated active uptake in the HepatoPac system, resulting in an equivalent cellular rifampicin concentration and inductive effect at lower media rifampicin concentrations, better reflecting the in vivo potency of rifampicin.8,9
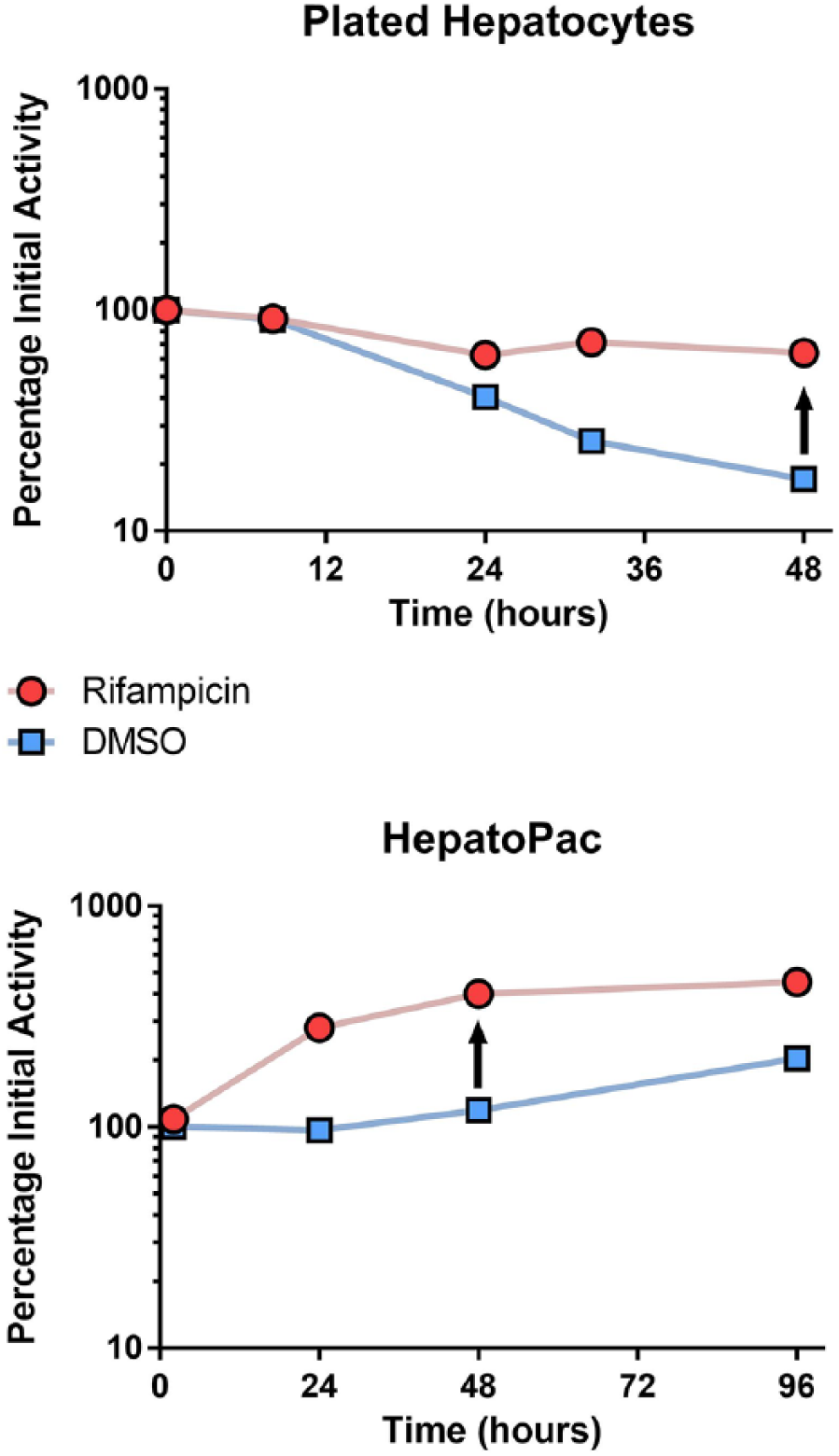
Induction of CYP3A4 enzyme activity in monocultured and micropatterned co-cultured hepatocytes. Rifampicin (10 µM) was applied with media exchange every 24 h to plated, monocultured hepatocytes or HepatoPac cells. The enzyme activity (1′-hydroxymidazolam formation) was measured at several time points and plotted as percent of initial CYP3A4 activity against time. DMSO-treated cells without inducer were used as a control. The inductive effect of rifampicin is seen in both cell models. In the monocultured hepatocytes, the enzyme activity of the DMSO-treated cells decreased to 15%–20% of the initial activity, while the baseline activity in the HepatoPac culture did not decrease with incubation time.
Despite these clear system advantages, long-term culture systems are not in widespread use for induction work due to (1) the lower-cost incumbent (2D monoculture) methodology and (2) a higher level of experimental difficulty due to the low cell number (3200 hepatocytes per well in a 96-well plate), which impacts mRNA yields and the amount of metabolism products formed. Clear demonstration of the value of the more complex cellular system is therefore needed for more widespread system adoption. Furthermore, prediction of enzyme induction via translation of in vitro data in PBPK models is an area where the scope of regulatory impact is currently limited. For example, although the Food and Drug Administration (FDA) and Pharmaceuticals and Medical Devices Agency (PMDA) have accepted induction modeling based on data from monocultured hepatocytes, 52 the European Medicines Agency (EMA) has stated that they do not have confidence in this approach yet. 53 Difficulties include the limited physiological relevance of the currently used systems, that effects on extrahepatic tissues (such as intestine) are not well addressed, and that the short incubation times limit the in vitro characterization. Therefore, the use of more advanced, long-term co-culture systems shows considerable promise in this area. High-quality validation of predictivity, especially in cases where compounds exhibit both time-dependent inhibition and induction effects, could enable long-term hepatocyte cultures to become the recommended system for induction studies.
Multiple-Endpoint Studies
Consistent Multiple-Parameter Measurement
Advanced cellular systems allowing long-term culture are clearly needed when long-term effects, such as steady-state DDIs due to mechanism-based inhibition and induction on enzymes or modification of disease phenotype, are to be assessed. In addition, there is a role for long-term ADME systems in improving PK prediction through enhanced data consistency. Ultimately, multiple in vitro ADME data (e.g., determination of transporter activities, DDIs, metabolic stability) are brought together in a PBPK model for human PK prediction and DDI simulations 14 ( Fig. 5 ). Essential for confidence in the simulations are model inputs from high-quality in vitro assays. Although the various assays may individually have been performed under optimized conditions, those conditions may vary considerably. The capability of a long-term system to be used for a combination of both short- and long-duration experiments also enables multiple parameters to be measured in a single consistent system involving the same media, solvent content, and free drug concentration. A study by Kratochwil et al. that combined data from active uptake, active efflux, and metabolic clearance, in the clearance assessment for a hepatitis B virus (HBV) candidate drug, illustrated how such data packages could be put together. 7 The simultaneous generation of transport and metabolism clearance data using the same system removed the possibility for gross mismatch in the data due to different test system usage. Both in vitro efflux transport clearance and metabolic clearance were combined for the prediction of human clearance.
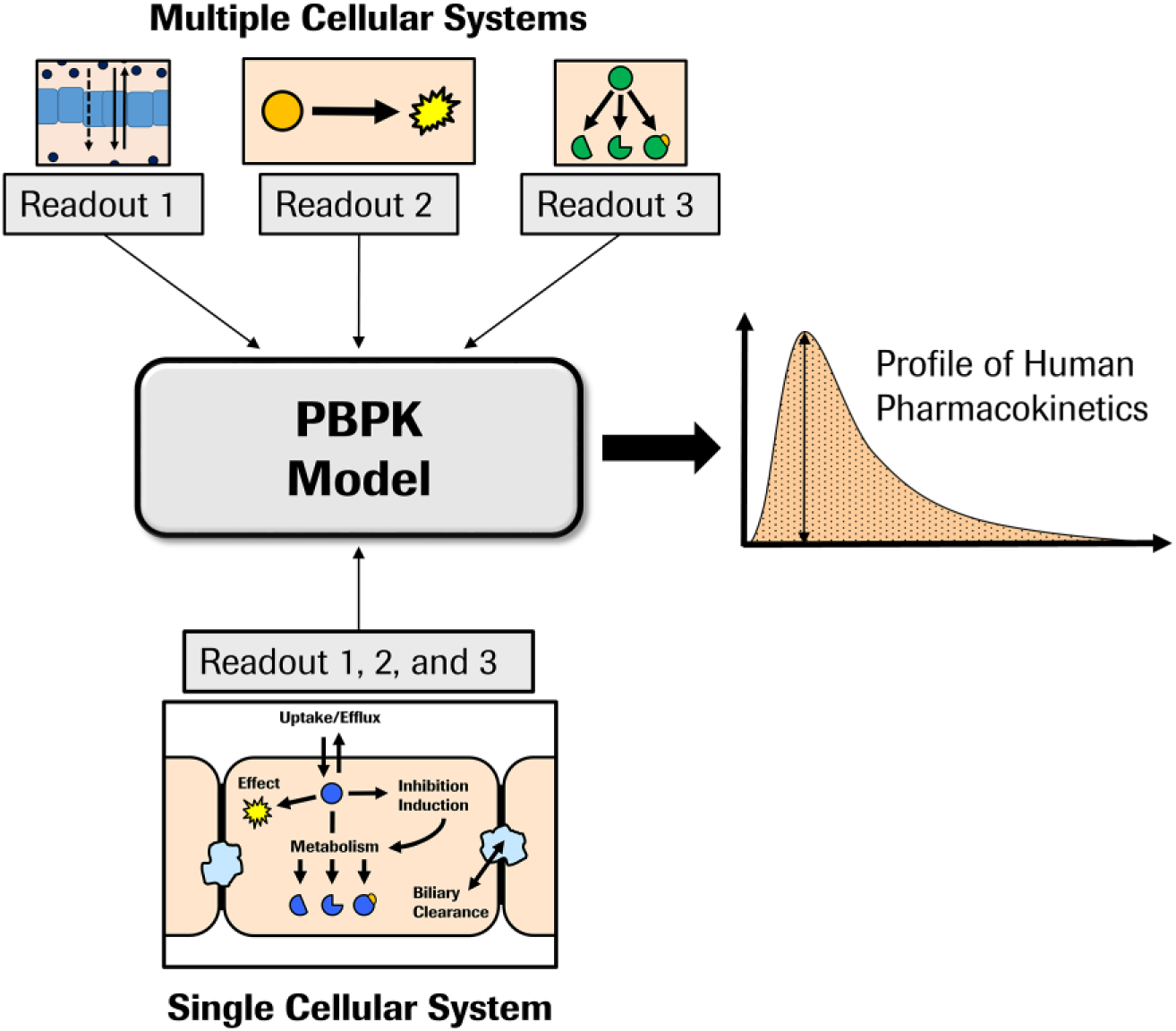
Schematic representation of human PK simulation using in vitro data from multiple individual cellular assays or from a single cellular (e.g., long-term culture) system. Currently, specialized cellular systems are used to investigate different parameters that are brought together in a computational model for human PK profile simulations. One quality of a long-term cell culture system is the capability to implement multiple endpoints, enabling consistent incubation conditions for multiple readouts and enabling interacting processes to be probed. As a result, the generation of multiple readouts from a single cellular system could enhance the quality of input parameters into the computational model and give additional information about the PK and pharmacodynamics of a drug.
Parameter Crosstalk
Most ADME assay systems have been optimized to limit the capacity for crosstalk between different processes, enabling clean and unequivocal assay outputs to be generated. However, this sometimes means that important processes may be missed. Examples include the time-dependent inhibition of CYP2C8 due to the formation of glucuronide metabolites of gemfibrozil or clopidogrel.54,55 Cellular systems with a full complement of uptake transporter and metabolism processes are suited to the study of complex interactions. Such combined process assessments have been reported using the HepatoPac system and would be a target area for the implementation of more complex cellular systems in the future. Dixit et al. and Moore et al. evaluated and confirmed the interplay between transporter activity and enzyme induction in the HepatoPac model.8,9,56 The aforementioned study of Kratochwil et al. reported on measurements of metabolism, metabolite identification, and induction under high uptake conditions in a single test system to obtain consistent model inputs. 7 Furthermore, cellular activities in healthy and HBV-infected HepatoPac cultures were assessed, which raised the interesting possibility of simultaneously investigating in vitro PK and pharmacodynamics in a cellular model of hepatic disease and added to literature precedents for using co-cultured hepatocytes in this way.7,57,58
Emerging Cellular Models
Spheroids
Hepatocyte spheroids have been successfully applied to the prediction of hepatotoxicity and for biotransformation studies, but have not to our knowledge been extensively applied in ADME screening.59–62 Spheroids can more fully replicate the in vivo architecture of the liver with 3D cell–cell and cell–extracellular matrix interactions, as well as a gradient of oxygen tension to encourage hepatocyte zonation.63,64 The 3D morphology enables the cells to retain their phenotypic function over several weeks. 27 Arakawa et al. evaluated 3D primary hepatocyte spheroids for the investigation of enzyme induction. 65 The expression of transcription factors and inducibility of CYP enzymes could be maintained for 14 days after spheroid formation. Bell et al. described multiple functional aspects in primary human hepatocyte spheroids. 27 They found that stable CYP enzyme activities and albumin secretion were maintained during a long-term culture (4–5 weeks) and showed that the liver spheroids can be used for long-term toxicity studies and for studies under disease-state conditions. Spheroids have also been used as a basis for rudimentary multiorgan systems. 66 Linked hanging drops containing spheroids were used to demonstrate that connection of hepatocyte and tumor spheroids enabled tumor growth suppression by hepatically bioactivated cyclophosphamide. However, individual spheroid incubations still suffer the limitations of low cell number per spheroid (e.g., low total metabolism capacity, low mRNA yield, and very small cellular volume for uptake measurements). Culture systems that enable arrays of spheroids to be incubated in a single microtiter plate well will therefore open up many more ADME applications. 64
Microphysiological Systems
Fluid flow and associated shear stress are important for hepatocyte activity.64,67,68 MPSs aim to include the flow of incubation media past one or more microtissues using microchannels and microstructures implemented into the devices (“chips”).63,69 By recreating physiological conditions in the form of flow rates and volumes, it is hoped that the in vivo perfusion conditions for oxygen/nutrient delivery and physical stimulation can be created.64,69 Sarkar et al. also used such a system to assess hydrocortisone metabolism, obtaining intrinsic clearance estimations very close to human in vivo clearance. 70 Tsamandouras et al. addressed the in vitro PK of six different drug compounds in a microphysiological liver system, using cryopreserved hepatocytes from five individual donors. This study made a significant step toward demonstrating MPS activity in a way relevant for drug metabolism scientists. 71 Further developments to include additional cell types have now also been reported. Sarkar et al. more recently described an immunocompetent MPS with hepatocyte and Kupffer cell co-culture and used it to study diclofenac metabolism. 72 More complex MPSs that combine multiple tissues with interconnected flow are also emerging. A major challenge for these systems is finding adequate culture conditions (e.g., media) for very different cell types (e.g., hepatocytes, enterocytes, neurons). For example, Tsamandouras et al. established an MPS that combines liver and gut organoids. 73 The liver organoid was composed of primary human hepatocytes and Kupffer cells, while the gut organoid was made by combining the intestinal epithelial cell lines Caco2-BBe and HT-29-MTX and primary monocyte-derived dendritic cells. Although the two-tissue MPS had no particular impact on the stability of either organ, the model successfully achieved an intestinal epithelial barrier, which allowed PK investigations of orally administered drugs (diclofenac and hydrocortisone) under experimental perturbations. Using the same MPS, Chen et al. reported a physiological crosstalk between gut and liver microtissues, namely, increased secretion of fibroblast growth factor 19 and concomitant downregulation of CYP7A1 in the liver organoid. 74 Adding lipopolysaccharide (LPS) to the circulation media (to emulate sepsis) revealed a crosstalk between cytokines released from the gut MPS that synergized with cytokines released from the liver organoid. 74 Multitissue MPSs have also been used to investigate drug efficacy in combination with more physiologically relevant conditions. For example, Imura et al. developed a microtissue system with intestine, liver, and tumor target cells combined. 75 The target cells were human breast carcinoma cells and the whole microsystem was used to evaluate intestinal absorption, hepatic metabolism, and drug effect on the target cells in one in vitro system. Clearly this is an area of rapid development with some labs bringing their MPSs to a state of validation, which may enable application to new drug development. Validation along the lines shown for some of the leading examples of co-cultured hepatocyte systems could potentially be followed to demonstrate equivalence or superiority of MPS. These include applications such as intrinsic clearance, 38 enzyme activities, 14 metabolite generation, 16 uptake, 7 and induction. 8
Other Organ Models for ADME Studies
Although metabolism of xenobiotic molecules happens mainly in the liver, other organs may also make relevant contributions to PK. This is especially the case for compounds whose (CYP-mediated) hepatic clearance is very low. New models for skin, lung, kidney, and intestinal metabolism have been reported, but the difficulty in obtaining active and reproducible tissue/cells still limits their application. 76 Of the extrahepatic metabolism organs, the intestine is perhaps the most important, due to its contribution to first-pass clearance and DDIs.77,78 Two interesting commercially available cellular models for intestinal metabolism have recently appeared. Human enterocytes have been isolated from individual donors and can be supplied as cryopreserved cells. The cells show activity across many different enzyme classes and are suited to qualitative exploration of intestinal metabolism.79–81 To date, primary enterocytes cannot be plated and cultured for long-term in vitro investigations as is possible with hepatocytes. This limits their application to short-term studies such as clearance and metabolite identification. However, when donors or culture conditions are identified, which enable these cells to be seeded into MPSs and retain their native enzyme and transporter activities, a significant step forward will be taken toward the generation of ADME-relevant multiple-organ systems. Another intestinal system of interest, EpiIntestine, boasts a multiple-cell-type microtissue layer available in a trans-well format. In this way, an effective barrier function is developed and the system can be used for the investigation of intestinal permeability and effect of drug transporters (P-gp, MRP-1, MRP-2, and BCRP). 82 The extension of this system to metabolic endpoints would potentially enable better assessment of first-pass metabolism in which permeability, transport, and metabolism elements are included.
The Right Tool for the Right Job
The increasing complexity of the cellular metabolism systems allows for a wider range of applications and ADME parameter determinations. However, while it is important that tools of sufficient complexity are applied to answer complex questions, cellular systems must also be simple enough to support medium-throughput, short-turnaround times for data generation during lead optimization. At early stages, simple cellular or subcellular systems are sufficient for ranking compounds within a series or for estimation of ADME parameters. When nearing candidate selection, more advanced cellular models are appropriate. Long-term processes, such as induction, time-dependent inhibition, or the metabolism of metabolically stable compounds, demand cellular systems that allow for long-term incubations and which can replicate in vivo hepatocyte functionalities. Figure 6 shows how increasingly complex cellular in vitro tools need to be matched with increasing complexity of the endpoints in an investigation. For instance, the lower-complexity corner of the chart contains cellular assays currently deployed for ADME screening, using systems such as suspension or plated hepatocytes, with individual parameter measurements such as intrinsic clearance, metabolite identification, active uptake, and enzyme induction. In the center of the chart, applications can be found where published examples exist, but the assays are not in widespread use. At the top part of the chart are the more highly complex systems, largely aspirational in nature at the current time, but with the potential to allow very complex ADME and safety questions to be addressed. Apparent in this representation is the steady shift toward additional validation of more complex cellular systems and their more routine application in areas where they can add value to lead optimization programs.
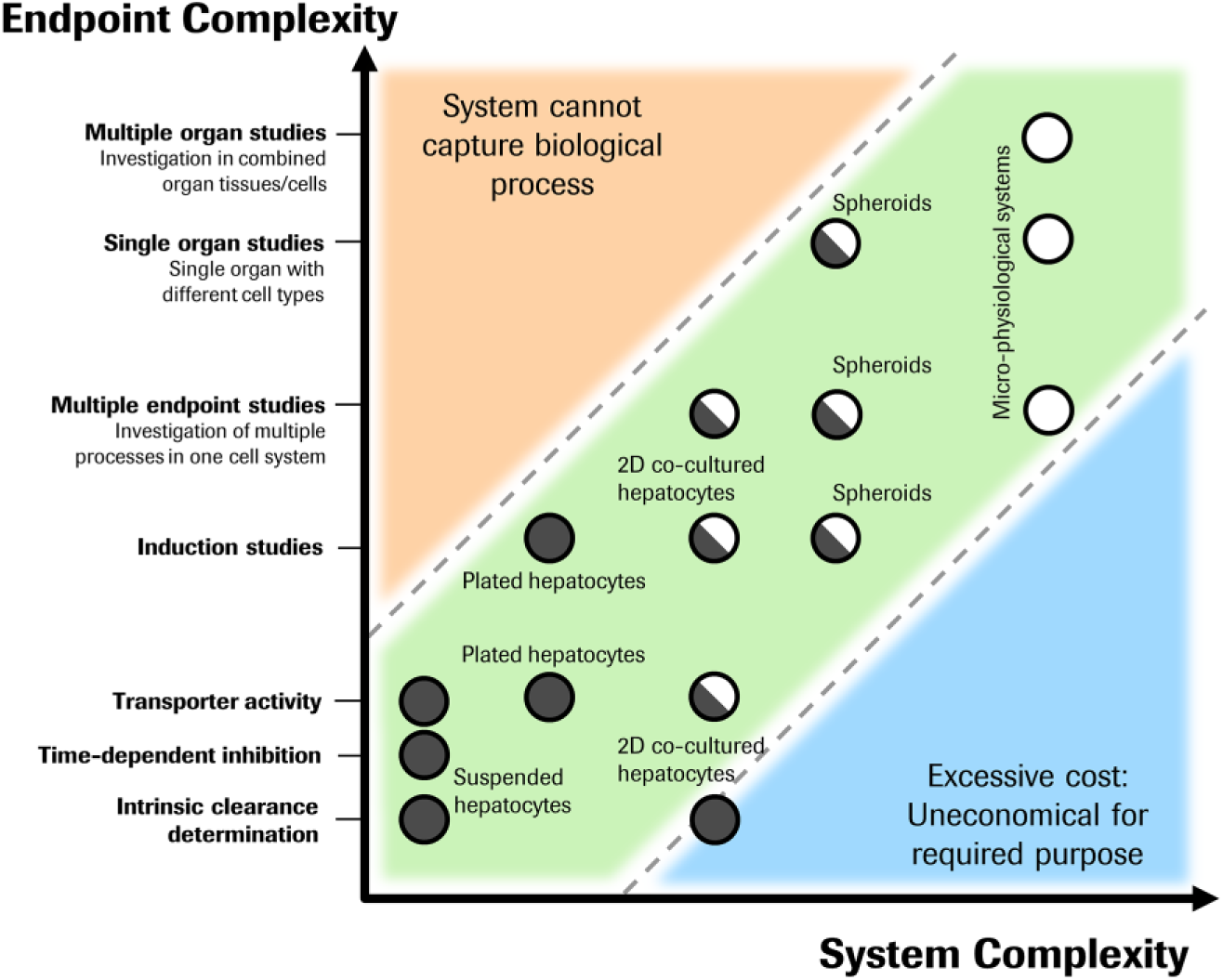
Application of cellular tools to different study types. Dots represent the intersections between the in vitro model and the applicable study type, with an increasing biological complexity of the study readouts on the y axis and an increasing complexity of the cellular tool described with the x axis. Filled circles, currently used and validated ADME assays; half-filled circles, application published but not in widespread use; open circles, potential new ADME assays. Lower right area: Indicates too great expense for the required purpose. Upper left area: Cellular model is not capable of capturing the biological readout.
Future Perspectives
Impact of Machine Learning on ADME Screening
ML and other in silico technologies have advanced in recent years, allowing predictions of ADME properties such as microsomal stability, CYP inhibition, solubility, and lipophilicity.18,83–85 ML methods differ from structure–activity relationship (SAR) and quantitative structure–activity relationship (QSAR) models of the past in at least two important aspects: (1) the self-building nature of the models, not requiring ADME scientist direction, and (2) the ability of the model to give a self-assessment of individual prediction quality. 86 The prediction quality self-scoring is of particular interest as it allows a more nuanced approach to be taken to tool implementation. ML technologies offer an exciting opportunity for ADME scientists to focus experimental resources on the most interesting of discovery molecules, which either have some chance of becoming candidate drugs or are highly informative about novel chemistry space that is not yet well predicted. 87 An active partnership between ADME and in silico scientists and medicinal chemists is needed for effective implementation of in silico tools. One element of this is the development of clear guidelines about when to use the prediction tools and when to generate new experimental data. For instance, a ML tool developed at Roche for the prediction of CYP inhibition has a very good capability to predict noninhibitors, due to the very large number of examples in the database. However, the confidence in prediction of inhibitory compounds is relatively low due to their underrepresentation in the dataset. A clear recommendation could therefore be formulated on the basis of the category and quality of prediction. If predicted inhibition effect is low and prediction confidence is “good” or “best,” then use ML tool prediction; otherwise, measure. An approach like this enables chemistry teams to make use of qualified prediction tools while also retaining access to experimental measurement. At the same time, ADME resources are focused on testing molecules of highest importance/greatest challenge and in silico scientists are able to propose the testing of additional molecules with the purpose of further model improvement. Although predictive ML tools are increasingly available for ADME applications, they have so far achieved only limited uptake. This is potentially due to a number of factors, including (1) the perceived quality gap between measurement and prediction, perhaps reinforced by the preponderance of categorical models (in contrast to numerical prediction models); (2) the incomplete shift from a “design–make–test” type of approach to more of a “predict–make–confirm” approach; and (3) the availability of medium- to high-throughput ADME screening methodologies largely preventing ADME data delivery from being a bottleneck in discovery project progression, reducing the urgency to evolve new methods of working. Nevertheless, the application of ML tools has the opportunity to impact early discovery screening and synthesis strategies significantly.18,88 Here, one could anticipate that the number of molecules tested in routine screens can be more than halved and have an even greater impact on presynthesis decision-making, where the generation of close analogues with poor physchem/ADME properties within established series could be substantially reduced.
New Cellular ADME Screening Systems
The scientific literature contains a clear bias toward the reporting of positive findings and an optimistic outlook for the different systems in which the publication authors are highly invested. It is sometimes suggested that new cellular systems will soon enhance ADME optimization in the early stages of drug discovery. To date, this optimism has not been substantiated by widespread application of new cellular systems for drug metabolism screening. Implementation of new cellular systems has been focused on low-volume, high-value-added applications. In addition to the obvious cost, throughput, and technical handling limitations that currently deter earlier deployment, the complexity and quality of data, generated by the advanced systems, exceed the project needs during the earliest phases of drug discovery and mainly find application when considering potential candidate drugs for use in humans. In this arena, however, there is great potential for additional ADME and toxicological de-risking of drug candidates and enhanced understanding of human PK and disease modulation. 89
What then are the likely next steps? In vitro toxicology systems are likely to develop via the stepwise availability of better individual tissue models in which the effects of the parent drugs and metabolites can be studied. In addition, drug metabolism systems are likely to develop with an initial focus on liver (metabolism), intestine (barrier function and metabolism), and brain (sensitive organ barrier function). The increasing availability of different tissue spheroids and the capability to generate mixed-cell-type spheroids, which can easily be added into simple flow systems, will quickly enable crosstalk experiments to be performed using different tissue types. Such systems could offer the advantages of very rapid and simple reconfiguration for each new experiment and putting the selection of tissues in the hands of the end users. At the same time, the active pursuit of MPSs that mimic human physiology and the translation of data from these systems directly into PBPK models with advanced mathematical modeling of the in vitro systems should enhance PK and DDI prediction. However, before more advanced cellular models can have regulatory impact, more validation is needed. 90 This should include a characterization of activities of the major enzymes and transporters as well as demonstration of in vitro–to–in vivo translation for a set of reference drugs, as was demonstrated by Kratochwil et al. 14 using different long-term co-culture models. Initially, recapitulation of a population average phenotype is needed for in vitro–in vivo extrapolations. Generation of disease model MPSs (e.g., hepatitis infection58,91) will broaden the use of MPSs and enhance their applicability to the clinical situation.
Conclusions
The generation of novel cellular systems for advanced ADME applications is progressing rapidly and holds promise to improve the quality and physiological relevance of in vitro data. The opportunity to assess multiple and more complex endpoints, encompassing PK, pharmacodynamic, and toxicological processes, may enable the new in vitro systems to become a viable alternative to animal testing in many areas. Moreover, consistent in vitro data can be integrated into a PBPK model that allows simulation of multiple endpoints in humans under variable conditions, such as different populations, pediatrics, or organ impairment. We foresee the incremental implementation of different systems where the balance between value, throughput, and cost is favorable and where the technical challenge of implementation is low. In addition to potential cost reduction, as economies of scale are realized and more competitors enter the market, the value of a given experiment can be increased through better experimental efficiency and an increase in the amount of information generated per well. Experimental efficiency improves with good establishment of the cellular tools, while the number of endpoints per well depends on the combination between biological complexity of the in vitro system and advanced study designs. Successful implementation of ML and advanced cellular models can lower the number of molecules tested in routine screens and enable more informed selection of candidate drugs. However, new cellular models require extensive, high-quality validation from multiple laboratories and use of relevant drug molecules in order to demonstrate their in vivo relevance, and therefore can be implemented in the pharmaceutical industry and accepted by regulatory authorities.
Supplemental Material
Supplemental_Material_for_Review_Cellular_Tools_by_Docci – Supplemental material for Application of New Cellular and Microphysiological Systems to Drug Metabolism Optimization and Their Positioning Respective to In Silico Tools
Supplemental material, Supplemental_Material_for_Review_Cellular_Tools_by_Docci for Application of New Cellular and Microphysiological Systems to Drug Metabolism Optimization and Their Positioning Respective to In Silico Tools by Luca Docci, Neil Parrott, Stephan Krähenbühl and Stephen Fowler in SLAS Discovery
Footnotes
Acknowledgements
The authors gratefully acknowledge the contributions of Birgit Molitor, Pascal Schenk, Florian Klammers, Aynur Ekiciler, Isabelle Walter, Andreas Goetschi, Vincent Monin, and Charles Tournillac, who generated much of the data used in the CYP inhibition, time-dependent CYP inhibition, and intrinsic clearance examples shown, and Dr. Michael Reutlinger, who generated the CYP inhibition ML tool. We thank Drs. Patricio Godoy and Kenichi Umehara for their helpful comments on the manuscript.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Stephen Fowler and Neil Parrott are employees of F. Hoffmann-La Roche Ltd. The studentship of Luca Docci is financially supported by F. Hoffmann-La Roche Ltd.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.