
Abstract
RNA-binding proteins (RBPs) are pleiotropic factors that control the processing and functional compartmentalization of transcripts by binding primarily to mRNA untranslated regions (UTRs). The competitive and/or cooperative interplay between RBPs and an array of coding and noncoding RNAs (ncRNAs) determines the posttranscriptional control of gene expression, influencing protein production. Recently, a variety of well-recognized and noncanonical RBP domains have been revealed by modern system-wide analyses, underlying an evolving classification of ribonucleoproteins (RNPs) and their importance in governing physiological RNA metabolism. The possibility of targeting selected RNA–protein interactions with small molecules is now expanding the concept of protein “druggability,” with new implications for medicinal chemistry and for a deeper characterization of the mechanism of action of bioactive compounds. Here, taking SF3B1, HuR, LIN28, and Musashi proteins as paradigmatic case studies, we review the strategies applied for targeting RBPs, with emphasis on the technological advancements to study protein–RNA interactions and on the requirements of appropriate validation strategies to parallel high-throughput screening (HTS) efforts.
Introduction
The posttranscriptional control of gene expression in eukaryotic cells defines all the steps related to mRNA metabolism, from its maturation/processing to its subcellular localization and decoding for protein production. The RNA-binding proteins (RBPs) participate in the formation of ribonucleoprotein (RNP) complexes by recognizing and dynamically binding to both coding and noncoding selective RNA targets, thus profoundly influencing gene expression. 1 Recently, genome-wide identification of RBPs and their RNA targets has been facilitated by unbiased and high-throughput approaches, such as next-generation sequencing and mass spectrometry. A census published in 2014 reports 1542 human RBPs interacting with all known classes of RNAs. 2 The majority of RBPs are found ubiquitously expressed and directly or indirectly implicated in the process of protein synthesis, while at least one-third of them have yet unrevealed biological functions. 2 RBPs bind to defined motifs in target RNA via RNA-binding domains. 3 These motifs are distinguished as RNA recognition motifs (RRMs), which are the most abundant protein domains in eukaryotes, 4 the heterogeneous nuclear RNP (hnRNP) K homology domains (KH), 5 the DEAD box helicase domains, 6 the double-stranded RNA-binding motifs (DSRMs), or the zinc-finger domains. 7 The analysis of the RNA cis elements among 205 genes from 24 diverse eukaryotes indicated a good correlation between sequence specificities and protein domain similarities. 8 Recent studies have uncovered hundreds of RBPs lacking conventional RNA-binding domains. This reveals the potential for novel types of RNA-binding modules mediated by new interaction modes. These could be favored by intrinsically disordered regions, protein–protein interaction (PPI) interfaces, and enzymatic cores. 9
Interestingly, RBP-focused pharmacological studies have highlighted the existence of a significant posttranscriptional impact mediated by small molecules exerting antitumor properties. For instance, the antitumor activity of resveratrol has been associated with the production of pro-inflammatory cytokines via modulation of the far upstream element-binding protein 2 (KHSRP) 10 and more recently via activation of the mRNA-decay factor tristetraprolin (TTP). 11 The small molecule PTC-299 12 or PTC-510 13 has been reported to target the 3′-untranslated region (UTR) of vascular endothelial growth factor (VEGF) mRNA in tumor cells. Furthermore, the antibacterial compound enoxacin has been proven to enhance the stability of selected miRNAs (premiR-125a, prelet-7, and premiR-30a), acting as tumor suppressors by binding to the TAR RNA-binding protein 2 (TRBP2). 14 This evidence suggests that the enhancement or inhibition of the activity of RNPs can be exploited to modulate the posttranscriptional control of gene expression with potential benefits in disease-related states.
In the Online Mendelian Inheritance in Man (OMIM) database, ~150 RBPs targeting both mRNAs and noncoding RNAs (ncRNAs) are linked to human diseases. 7 However, the majority of RBPs lack traditional enzymatic pockets or functional epitopes that could classify them as canonical “druggable targets.” Indeed, the explored drug targets are largely biased toward enzymes, easily assayable in high-throughput screening (HTS) format, and often use small-molecule cofactors inspiring the design of drug-like molecules. This bias has been reinforced by the hectic popularity of “hot” targets and by pharmaceutical companies often competing for the very same target families, that is, G-protein-coupled receptors, protein kinases, metallopeptidases, proteases, nuclear hormone receptors, and phosphodiesterases. 15 Proof of this biased approach is that 90% of the 823,179 drug-like compounds targeting human proteins are directed against 278 targets, which are classical enzymes or membrane protein targets. 16 These results are associated with the exploring of narrow chemical spaces corresponding to the “accessible doped space” so far considered. 17 This triggers a vicious circle where commercial libraries used in HTS fail to produce useful compounds for new targets, corroborating the notion of their undruggability.
The most striking example of the recent enlargement of the druggable space is the development of several compounds interfering with PPIs, especially targeting epigenetic reader domains such as bromodomains, chromodomains, PHD zinc finger, and Tudor domains. While the majority of PPI surfaces have undruggable features (shallow and polar), a number of them become pharmacologically attractive. The above-mentioned epigenetic reader domains have aromatic and hydrophobic binding sites for neutralized lysines (acetylated or methylated), which recognize their substrates with affinities in the low micromolar range. Therefore, competition through small molecules is a reasonably achievable task. 18 Indeed, various bromodomain inhibitors are currently in clinical trials for the treatment of solid and hematological malignancies. Another class of traditionally classified undruggable targets is represented by the transcription factors, whose activity has been proven to be successfully modulated by several strategies, including the blockage of protein–DNA binding. 19 Small compounds are probably not able to disrupt the transcription factor–DNA interaction, given the large interacting surface, but they can instead interfere with the dynamics of the DNA-binding domains (DBDs) and impede the subsequent molecular recognition of their target sequence. 20
In this context, RNPs represent a new, largely unexplored, druggable space. The challenge of targeting RNPs relies on the application of new strategies to identify and interfere with dynamic protein–RNA surfaces. Indeed, the notions uncovered with PPIs may be viewed as the archetype for the development of small molecules interfering with other types of molecular recognitions, in particular for RNP complexes and specifically for RNA reader/protective domain rather than RNA modifying enzymes ( Fig. 1 ). RNA reader/protective domains have aromatic and hydrophobic pockets or channels recognizing nucleobases whose polarity can be further reduced by epitranscriptomic modifications. For RNP disruption, the well-known purine-mimicking chemical space (i.e., fragment libraries and kinase inhibitors) is of possible advantage. The above considerations expand the concept of druggability, even implying the possibility of multiple downstream effects elicited by the RNP after interference by the small molecule, in a cell-specific context.
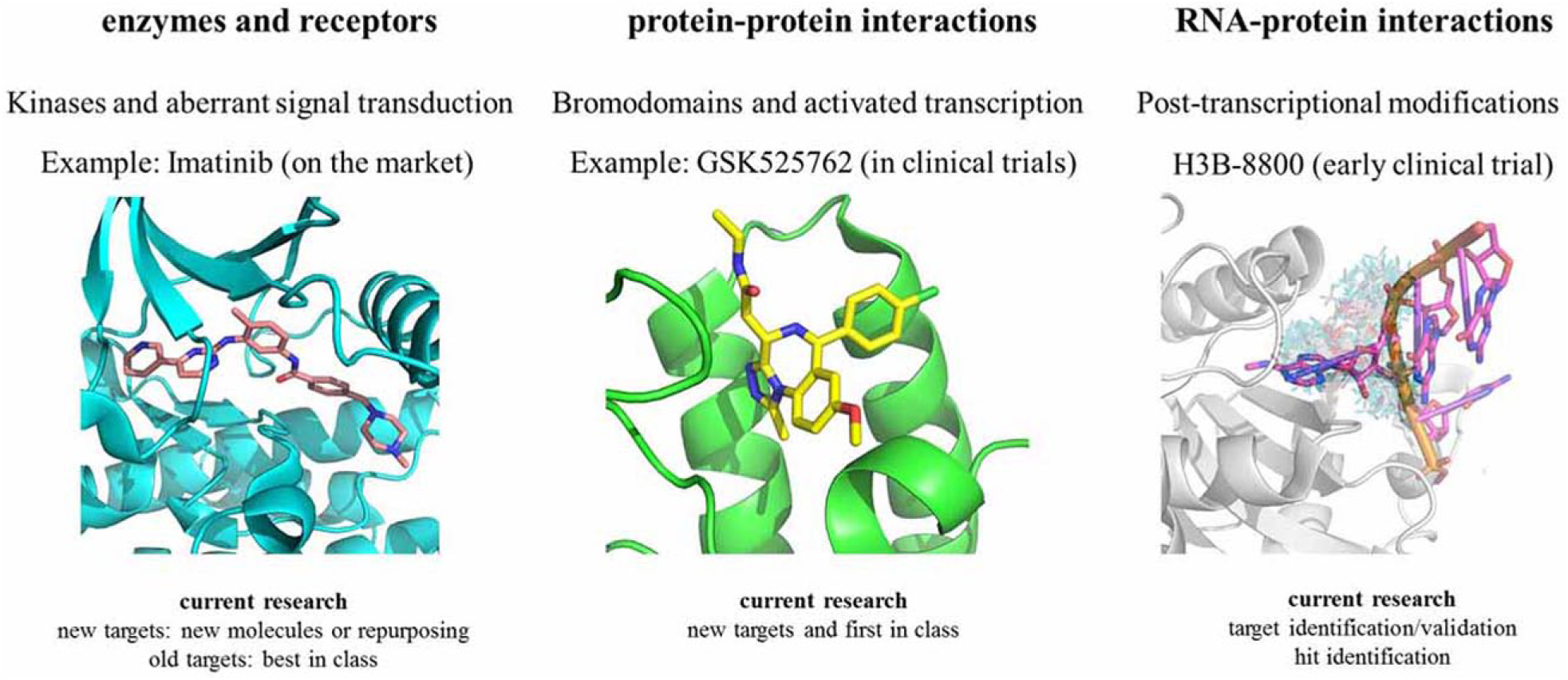
Expanding view of druggability: from the traditional concept of druggability, targeting enzymes or recognized epitopes, to the druggable interface responsible for RNA–protein interactions.
Here we review the current high-throughput experimental approaches to probe RNA–protein interaction in RNPs. An interesting proof of principle that interfering with the binding of RBPs to their target RNA is a promising strategy in drug discovery has been recently provided by the SF3b spliceosome complex and is outlined in the next section. We also describe the physiological roles and clinical relevance of other important RNP targets, with the most successful screening strategies employed so far to identify small molecules interfering with them.
In Vitro Approaches to Study and Challenge RNA–Protein Interactions
High-resolution structural insights of macromolecular complexes provide valuable biological information on the specific interaction between the ligands. However, techniques such as x-ray crystallography, nuclear magnetic resonance (NMR), and cryo-electron microscopy (cryo-EM) have to be complemented by studies probing the dynamics and flexibility of a RNA–protein complex formation. 21 A complete picture about the integrated biophysical and structural methods used to capture the structure and dynamics of RNPs is reviewed by Schlundt et al. 22 Briefly, the dynamic features require solution techniques such as NMR, small-angle scattering, or fluorescence spectroscopy to complement structural analysis based on crystallography, NMR, and cryo-EM.
Several biochemical and biophysical techniques have been successfully employed to assay RNA–protein interactions in vitro and challenge their inhibition by small molecules. A combination of different approaches is usually adopted to unravel the mechanism by which the inhibition is exerted. Nevertheless, a primary screening assay should be able to identify both classes of interactors, binding to either the RNA or the protein, the first being more unspecific and prone to off-target effects ( Fig. 2b ). The challenging approaches aiming at identifying small molecules specifically binding RNA are well described by Deigan Warner et al. in a recent review. 23 In addition, small molecules might act by altering the protein conformation and changing the RNA binding site ( Fig. 2c ).
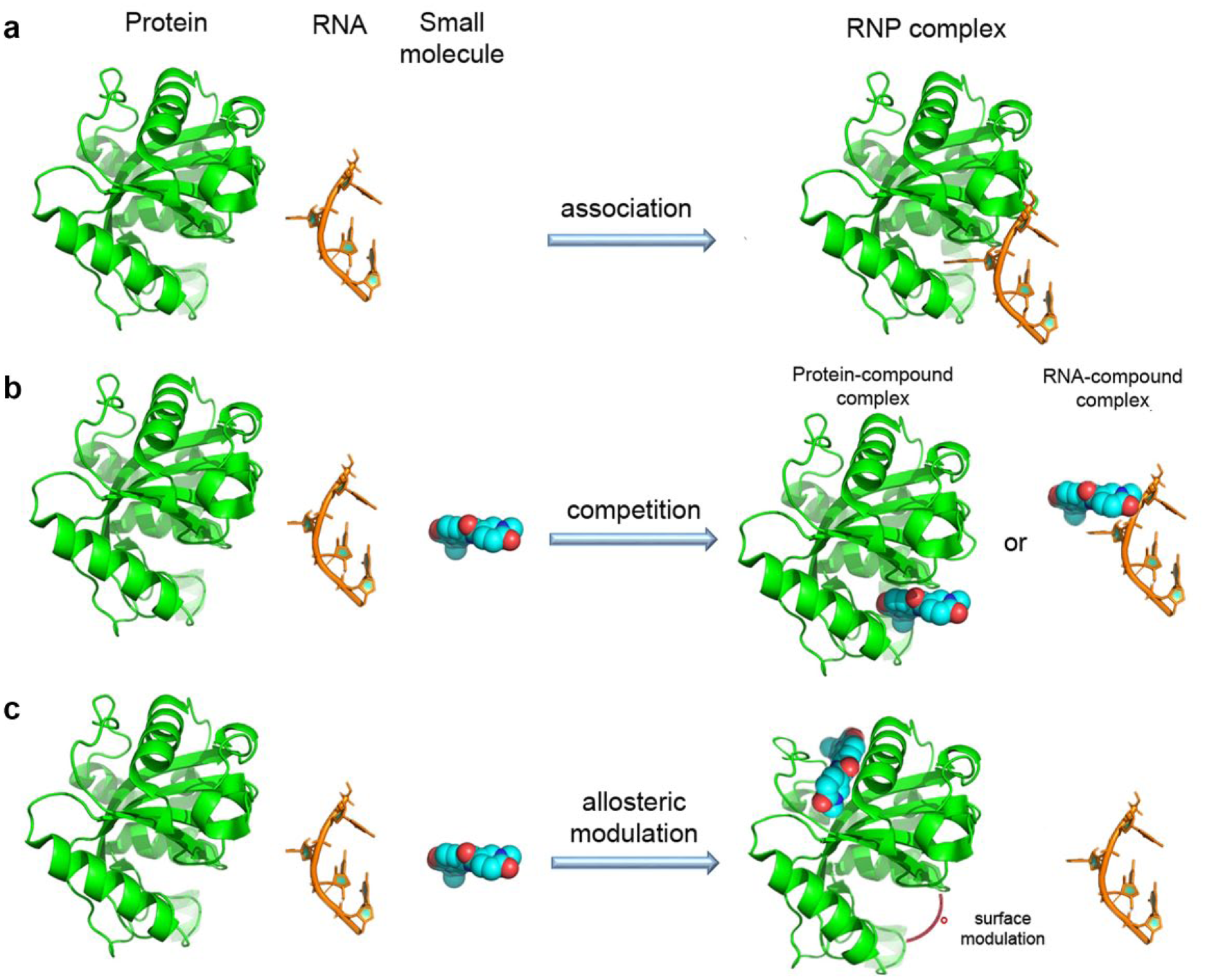
Multiple mechanisms by which a small molecule can interfere with the RNA–protein complex. (
The common assays employed in screening campaigns are biochemically based, involving the purification, labeling, and/or immobilization of one or both the binding partners. In Table 1 we describe the screening methods for small molecules interfering with RNA–protein interactions and those used for secondary assays and hit validation, including emerging techniques that could be adopted for screening purposes. Given the size difference of the two partners and the low complexity of the assay, fluorescence polarization (FP) has frequently been selected in HTS campaigns, while electrophoresis mobility shift assay (EMSA), even coupled with fluorescent or infrared probes, still represents one of the standard orthogonal tests for hit confirmation. Recently, the establishment of nonradioactive bead-based assays, such as AlphaScreen (Amplified Luminescent Proximity Homogeneous Assay), and of new high-throughput instruments has improved the screening power of technologies such as microfluidic capillary mobility shift assay (MMSA) or surface plasmon resonance (SPR).
Biochemical, Biophysical, and Cell-Based Techniques Used to Assay the RNA–Protein Binding and Its Modulation by Small Molecules.
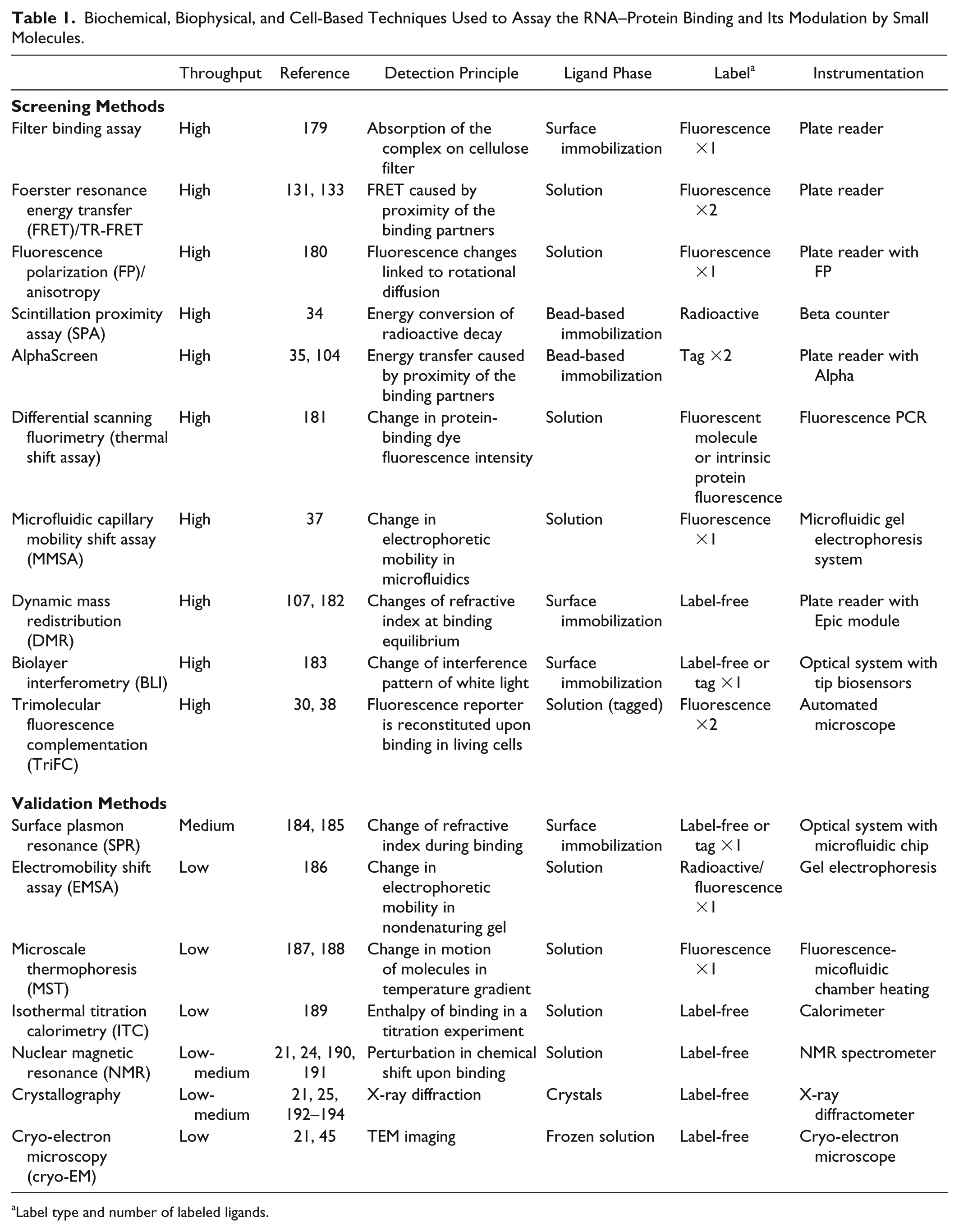
Label type and number of labeled ligands.
Additionally, fragment screening is nowadays possible with a low-medium throughput, also by NMR, and especially by x-ray crystallography. The latter allows obtaining the structural details required for the subsequent fragment-growing campaign, with the limitation of being applicable only to targets easily and reproducibly crystallizable.24,25 The throughput/molecular details and the success ratio are optimal when two or more strategies (molecular docking, HTS, NMR, and x-ray crystallography) are used in combination.26–28
The success of these biochemical and biophysical approaches also depends on the combination with cell-based methods addressing the activity of RBPs at the intracellular level. The analysis of subcellular localization of RBPs, participation in nuclear transcriptional and/or posttranscriptional events, cytoplasmic accumulation in functional compartments, and the exerted effect on cargo RNA decay and translation require multiple approaches, enabling the quantification of the intracellular effects of a small molecule interfering with an RBP.
Current methods developed to track RNA–protein interactions in the context of the cells include Foerster resonance energy transfer (FRET) 29 and trimolecular fluorescence complementation (TriFC), a novel method based on the well-established bimolecular fluorescence complementation (BiFC) technique ( Fig. 3 ). 30 Both techniques allow for imaging of molecular interactions by using fluorescent probes: while FRET measures the intensity change of a dual fluorescence spectrum, fluorescence complementation only measures the specific fluorescence, with advantages in reducing the background noise. Despite the promising technical asset, no screenings have been so far reported using the TriFC assay, probably due to the technical challenges still associated with the fluorescent complementation: the low temperature required by many FC systems (below 37 °C), the self-assembly tendency of the split fragments that increases the background, and the irreversibility of fluorescent protein complementation. 31
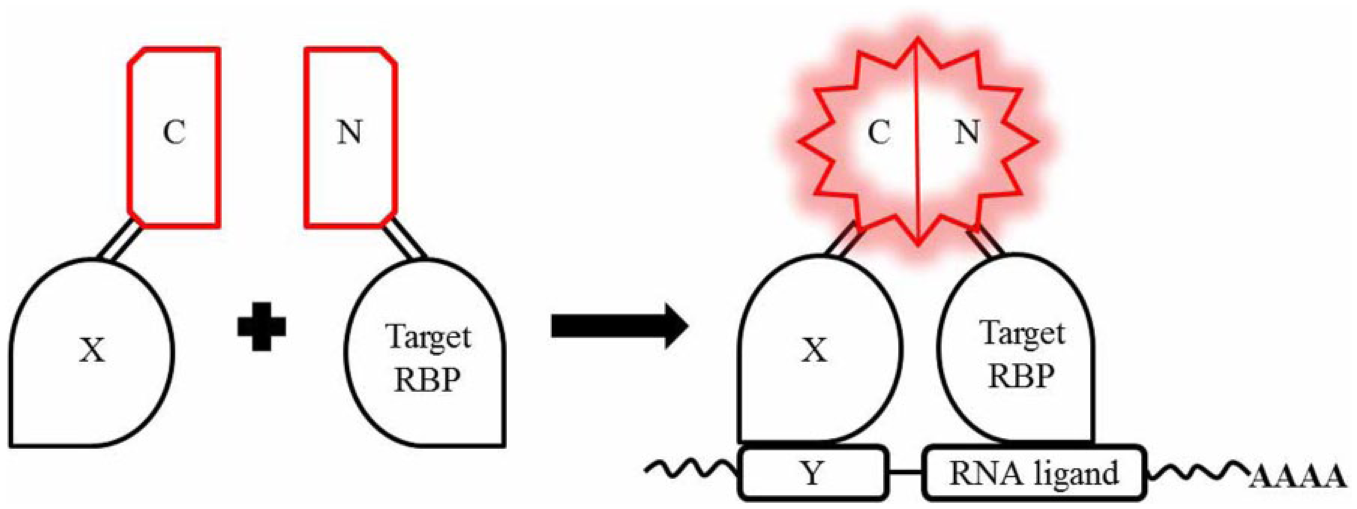
TriFC system for the detection of RNA–protein interaction. The C fragment of the fluorescent protein is attached to a protein X involved in a known RNA–protein interaction (X-Y). The complementing fragment N is fused to the target RBP. If the RBP interacts with a sequence of interest within the reporter mRNA close to the Y sequence, the two fragments N and C are brought into proximity to fold into a fluorescent product.
Initially, the idea of targeting RNA–protein interactions arose from the study of viruses like HIV. This represents a paradigmatic example of a complex system under strict control of a few “simple” RNA–protein interactions and extends the possibility to directly interfere with the surface of the molecules that evolved to bind to selected RNA motifs. 32 Tat and Rev are small viral proteins involved in the mechanisms of transcriptional activation and posttranscriptional regulation (splicing, transport, etc.) and exert their function through interaction with their respective RNA targets, trans-activation-responsive element (TAR) and Rev-responsive element (RRE). 33 Because the Tat-TAR axis is essential to HIV viral transcription, it has been the subject of intense efforts aimed at developing therapeutic interventions. Since 1985, several screening campaigns have been performed.34–36 The aminoglycoside class of antibiotics has been proven to inhibit the Tat-TAR complex, by binding TAR RNA, and neomycin derivatives have been proposed as leads for multitarget HIV inhibition. 33
Interestingly, the high-affinity binding between Tat and TAR has been exploited by several authors to validate novel methods for RNA–protein interactions.37,38
Other viral RNA–protein interactions, such as NS1 protein–influenza virus RNA, 39 HIV-1–Matrix protein, 40 HIV Rev–RRE,35,36 and HCV-IRES, 41 have been targeted by biochemical assays for screening small molecules. Table 2 reports these references, together with the other target RBPs described in the following paragraphs.
Screening and Screening Assays to Identify Small Molecules Interfering with RNA–Protein Interactions.
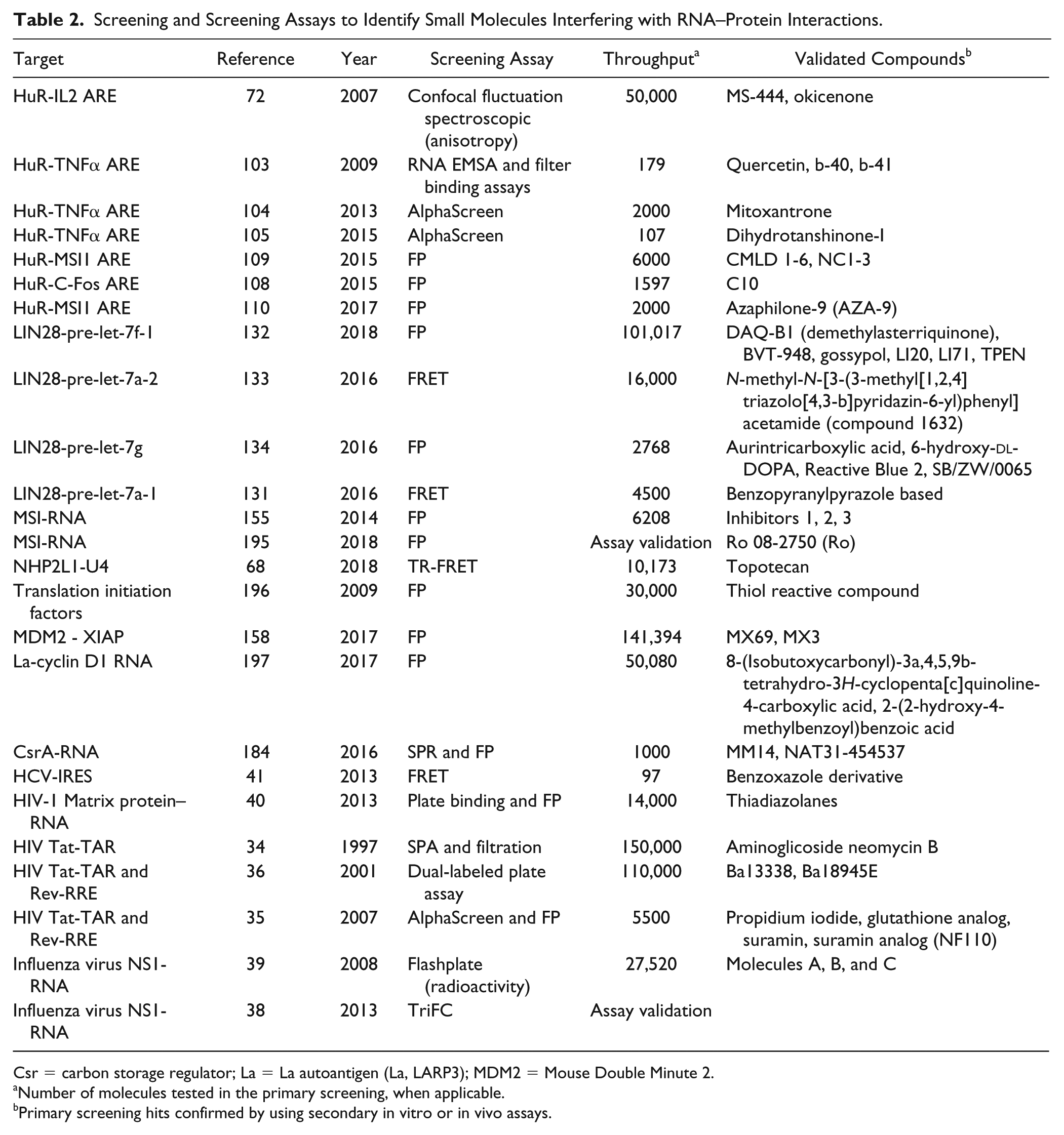
Csr = carbon storage regulator; La = La autoantigen (La, LARP3); MDM2 = Mouse Double Minute 2.
Number of molecules tested in the primary screening, when applicable.
Primary screening hits confirmed by using secondary in vitro or in vivo assays.
SF3b Complex and the First-in-Class Small Molecule in Clinical Trial
Splicing and alternative splicing of mRNA precursors is necessary for the removal of introns and the production of mature mRNAs. These events involve almost the entire human transcriptome and strongly contribute to the regulation of cellular gene expression programming. In the spliceosome machinery, the splicing factor 3B subunit 1 (SF3B1) protein is the largest component of the heptameric SF3b subcomplex in the U2 small nuclear RNP (snRNP) and supports the splicing via the recognition and selection of the branch point adenosine (BPA).42,43 SF3B1 is functionally associated with PHD finger protein 5A (PHF5A), SF3B3, and SF3B5 and forms a scaffold complex of ∼250 kDa that allows the subsequent recruitment of additional splicing factors. 44 SF3B1 is composed of 20 HEAT repeats that form a superhelical spiral. This spiral wraps around PHF5A and establishes contact regions with the polypyrimidine tract of the pre-mRNA and the BPA. In the apo form, the SF3B1 protein assumes an open conformation. Binding to the RNA induces SF3B1 to close up on the RNA itself. 45
Aberrations in RNA splicing, leading, for example, to intron retention, are common across cancers in comparison to the normal counterpart.46,47 Somatic mutations in genes encoding core spliceosomal proteins and associated RNA splicing factors, such as SF3B1, U2 small nuclear RNA auxiliary factor 1 (U2AF1), and SRF2, are commonly present in several cancers, including myelodysplastic syndromes (MDSs), 48 chronic myelomonocytic leukemia (CMML), 48 and chronic lymphocytic leukemia (CLL), 49 and in a number of solid tumors, including breast, 50 lung, 51 uveal melanoma, 52 and pancreatic carcinomas. 53 Mutated splicing factors confer a strong vulnerability to the associated cancer type as shown by genetic and pharmacological perturbation of the splicing process.54,55 These mutations represent a genetic link between dysfunction of the splicing machinery and cancer, thus providing a strong rationale for targeting the spliceosome as a new anticancer therapy. 56
The development of spliceosome-targeting small molecules accelerated when potent anticancer natural compounds isolated from bacteria as FR901464, herboxiedenes, and pladienolides57–59 turned out to target the SF3b subcomplex of U2 snRNP and to interrupt early stages of spliceosome assembly. Using biotinylated chemical probes, spliceostatin, a methylated pladienolide derivative of FR901464 and E7107, was found to exert antitumor activity by modulating the splicing process via the SF3B complex.60,61 Later efforts have led to the chemical synthesis of herboxiedenes 62 and of FR901464 analogs, a new class of small molecules called sudemycins, that showed similar splicing inhibition as spliceostatin. 63
Based on important preclinical indications,54,64 E7107 entered clinical trials for locally advanced or metastatic solid tumors (Study E7107-A001-101; trial registration ID: NCT00499499).65,66 Unfortunately, the trials were suspended due to toxic bilateral optic neuritis.
The ability of E7107 to modulate the spliceosome activity has been readily explained by structural studies using cryo-EM: E7107 binds to the branch point of the adenosine-binding pocket, at the interface of SF3B1, namely, the HR domains 15–17, and PHF5A, in a hydrophobic region. Comparison of the apo form of the tetrameric (SF3B1, PHF5A, SF3B3, SF3B5) complex with the bound ligands showed that E7107 binds to the pre-mRNA-unbound form or the so-called open conformation of the complex. The cryo-EM data indicate that E7107 directly competes with pre-mRNA for binding to the apo form of the complex, suggesting that E7107 would more easily inhibit weaker pre-mRNA substrates. 45
Starting from E7107, an orally available modulator of the SF3b complex, called H3B-8800, was generated by medicinal chemistry. The competitive mechanism of action of H3B-8800 is similar to that of pladienolide, and H3B-8800 potently binds and inhibits splicing catalysis by both WT SF3B1 and its mutant forms in vitro. However, differently from pladienolide, H3B-8800 preferentially kills spliceosome-mutant epithelial and hematologic tumor cells. Mutations of either SF3B1, SRSF2, or U2AF1 confer to cancer cells a dependency on the remaining WT spliceosome. H3B-8800 causes the enrichment of short, GC-rich intron retention in mRNAs encoding RNA splicing factors, providing an explanation for H3B-8800’s preferential killing of spliceosome-mutant tumor cells. 67
H3B-8800 has been proposed for the treatment of genetically defined subsets of cancer with RNA splicing factor mutations. H3B-8800 is currently being tested in a phase 1 clinical trial (NCT02841540) for MDSs, acute myeloid leukemia, and CMML. Notably, H3B-8800 and E7107 well represent the first-in-class small molecules that modulate the activity of RNP complexes with a therapeutic outcome and a precise indication of use.
Interestingly, other well-known drugs, such as topotecan, have been recently shown to inhibit RNA splicing. 68 In this work Diouf et al. report a time-resolved (TR)-FRET screening to find small molecules disrupting the interaction between NHP2L1 protein and U4, revealing an additional posttranscriptional activity of this drug.
HuR
Human antigen R (HuR; also known as ELAVL1) is among the most widely studied RBP. It regulates the splicing, stability, and translation of thousands of coding and noncoding RNAs.69–71 The posttranscriptional function has been described for a wide number of transcripts bearing AU-rich elements (AREs), whose turnover is critical for tissue differentiation and cell physiology.
HuR protein comprises three RNA-recognition motifs (RRMs): RRM1, RRM2, and RRM3. The N-terminal ones are arranged in tandem (RRM1–RRM2), whereas RRM2 and RRM3 are joined by a hinge region. While the first two regions are involved in the binding to target RNAs, 70 RRM3 has been reported to bind to poly-A tail and to be involved in protein oligomerization.72,73 The HuR crystal structure of RRM1–RRM2 was solved in both the RNA-free and RNA-bound form of the protein and revealed a conformational change undergone by HuR after RNA binding. 74
HuR is ubiquitously expressed, is normally located in the nucleus, and shuttles to cytoplasm upon cell stresses as DNA damage or hypoxia. 75 In the nucleus, it exerts posttranscriptional functions such as splicing76,77 and alternative polyadenylation. 78
HuR is fundamental during the development of the embryo, as its deletion leads to embryonic lethality, and affects the adult tissue homeostasis.79,80 It plays a role in the maturation of lymphocytes (B and T) and macrophages and regulates the expression of specific chemokines and cytokines.81–83 By binding to AREs in target mRNAs, it modulates the expression of specific transcripts coding for proteins involved in inflammation,83,84 cell division, 85 angiogenesis,86,87 senescence, 88 apoptosis,89,90 immune,91,92 and hypoxia response. 93 Therefore, HuR is involved in the cellular response to different stimuli and impairment of its related gene expression can impact different disease processes. HuR was reported to be implicated in cancer and chronic inflammation and linked to cardiovascular, neurological, and muscular pathologies. 94
HuR posttranscriptional regulation can sustain cancer traits, such as increased cell proliferation and survival, elevated local angiogenesis, immune recognition evasion, tumor cell invasion, and metastasis, due to its stabilizing function of many key mRNA encoding proteins implicated in carcinogenesis. 95 Furthermore, overexpression of HuR or its cytoplasmic localization is associated with tumor progression and poor prognosis in various cancer types, including breast,96,97 colon,98,99 ovarian, prostate, pancreatic, and oral cancer. 95 HuR nucleo-cytoplasmic translocation in malignant cells has been reported for many tumors and correlated with advanced clinicopathological parameters and decreased patient survival rate. 100 Moreover, observations from animal models confirm a critical role for HuR in sustaining colon cancer formation and progression. 101 Therefore, HuR has been proposed as a valuable drug target based on all the reported posttranscriptional regulations involved in different pathologies and its ubiquitous expression in malignant samples. 102
The pivotal role of HuR in several diseases, such as inflammation and cancer, led to searching for inhibitors/modulators able to impact HuR activity. 102 Different compounds have been identified as able to interfere at the level of RNA-HuR complex formation.
In 2007, Meisner et al. identified the first low-molecular-weight HuR inhibitors. The authors screened 50,000 compounds using a confocal fluctuation spectroscopic assay with a shortened variant of recombinant HuR. The most potent hits that have been found were dehydromutactin, MS-444, and okicenone. The mechanism of action of these compounds has been proposed through mathematical and experimental analysis and resulted in the prevention of HuR homo-dimerization by binding the first two RRMs. This is reflected by inhibiting HuR activity as nucleus–cytoplasm shuttling, cytokine expression, and primary T-cell line activation. 72
In 2009, Chae et al. identified chemical inhibitors of the interaction between HuR and the ARE of tumor necrosis factor alpha (TNFα) mRNA using RNA-EMSA and a filter binding assay. 103 In particular, they generated a recombinant glutathione S-transferase (GST) fusion protein with HuR and TTP, and used these recombinant proteins and radiolabeled RNA containing the ARE sequence from TNFα to perform RNA-EMSA. From the screening of a total of 179 chemicals, three compounds, quercetin, b-40, and b-41, respectively, showed a half-maximal inhibitory concentration (IC50) below 10 μM. The IC50 values of the three compounds were 1.4, 0.38, and 6.21 μM, respectively, for the binding between HuR protein and TNFα mRNA. Further experiments were performed in RAW264.7 cells treated with lipopolysaccharide (LPS), quercetin, and b-40, and a decrease in the stability of TNFα mRNA and the secreted protein was observed. 103
A novel biochemical assay to study in vitro HuR protein–RNA complex formation, feasible for HTS, has been proposed in 2013. This assay is based on AlphaScreen technology, in which the binding efficacy is evaluated between a purified human HuR protein and a probe with the sequence of TNFα ARE. This method allowed to calculate HuR binding parameters under saturation binding and kinetic conditions, quantifying HuR-RNA Kd in the low nanomolar range. The results were further validated by fluorescent probe-based RNA-EMSA. In this work, 2000 small molecules were screened, and after a secondary validation with RNA-EMSA, mitoxantrone was identified as a modulator of the RNA-binding activity. 104
Furthermore, in 2015 the same group identified the natural compound dihydrotanshinone-I (DHTS), belonging to the family of tanshinones, as interfering with HuR binding activity. DHTS was further validated with complementary assays such as the EMSA, NMR titration, and molecular dynamics simulation, demonstrating that DHTS interacts with HuR at the level of the mRNA binding site, thus preventing the complex formation.105,106 The in cellulo confirmation has been performed with different cell lines toward the control of TNFα, CTNNB1, and ERBB2 mRNA targets. 105
Recently, DHTS has been demonstrated to inhibit in vivo xenograft tumor growth in a HuR-dependent model without systemic toxicity. 106 As a consequence, DHTS has been considered the leading compound for the synthesis of analogs, called tanshinone mimics, in order to enhance the efficacy in inhibiting HuR activity. 107
In 2015, Wang et al. presented an integrated approach to select inhibitors of HuR-RNA interaction using fluorescence-based HTS. This method was used to screen a library of 1597 compounds and hit validation was performed through the NMR method with saturation transfer difference (STD) detection. In this case, the authors identified the mechanism of action of these compounds, based on the disruption of HuR oligomerization, thus blocking the RNA binding. 108
A further screening was performed in 2015 by FP assay, using HuR protein and an ARE oligo from Musashi RNA-binding protein 1 (MSI1) mRNA, a HuR target. Wu et al. performed an HTS of about 6000 compounds; the potential disruptors were then validated by AlphaLISA, SPR, RNP immunoprecipitation (IP) assay, and luciferase reporter studies. These compounds inhibit HuR-ARE interactions at the nanomolar range and prevent HuR function by competitive binding with HuR. 109
Lastly, in 2017, through another FP competition HTS assay, Kaur et al. isolated the compound azaphilone-9 (AZA-9) that derives from the fungal natural product asperbenzaldehyde. AZA-9 is able to bind to HuR and inhibit HuR-ARE interaction with an IC50 of 1.2 μM. Data were validated with SPR, in which the authors were able to verify the direct binding of AZA-9 to HuR. NMR methods identified the involvement of critical RNA-binding residues in binding with AZA-9. Computational docking was then applied to propose a predictive binding site for AZA-9 in the RNA-binding cleft of HuR. 110 Similar to others, this work was able to identify HuR-RNA disruptors in vitro.
Considering the structural diversity of the compounds discovered so far and their mechanisms of interfering with ELAV protein−mRNA complexes, the structure–activity relationship is still a challenge.
LIN28
LIN28 protein was first described in Caenorhabditis elegans in association with its important role in development and in developmental timing. 111 LIN28 is highly expressed in the early developmental stages and in stem cells, decreases upon differentiation, and is normally absent in most differentiated cells in the adult.112,113
The mammalian genome encodes for two LIN28 paralogs: LIN28A and LIN28B. The term LIN28 will be used to refer collectively to LIN28A and LIN28B in this section. The human LIN28A protein, which is composed of 209 amino acids, and the human LIN28B protein, which consists of 250 amino acids, share a high degree of homology in their structure and function. 114 Both LIN28A and LIN28B proteins present a cold-shock domain (CSD) and a zinc knuckle domain (ZKD), composed of two CysCysHisCys (CCHC) zinc finger domains.115–118 These highly conserved regions are responsible for interacting and binding with target miRNAs and mRNAs.
The let-7 family of miRNAs is the most studied LIN28 interactor and consists of different members, including let-7a–i, mir-98, and mir-202. LIN28 binds to precursor forms of let-7 (pri- and pre-let-7) using both the CSD, which binds to let-7 terminal loops and contributes to most of the LIN28-let-7 binding affinity, and the ZKD, which recognizes a highly conserved GGAG sequence motif. 117 Upon binding, LIN28 prevents let-7 maturation and leads to decreased levels of mature let-7, which in turn cannot exert its tumor suppressor activity on its multiple target genes, such as RAS, MYC, and high-mobility group-A2 (HMGA2). 114 LIN28A initiates pre-let-7 degradation via recruitment of the terminal uridylyltransferases (TUTases) TUT4 and TUT7, which oligouridylate the pre-let-7 RNA in the cytoplasm.119–121 The mechanism of LIN28B-mediated let-7 inhibition, as well as its cellular localization, remains controversial. LIN28B has been shown to directly bind to precursor let-7 transcripts in the nucleus and to prevent the pri-let-7 cleavage mediated by Microprocessor,122,123 but also to bind to pre-let-7 in the cytoplasm and prevent its processing mediated by Dicer. 124 Furthermore, LIN28B has also been demonstrated to promote pre-let-7 uridylation by TUT7. 121 Compelling evidence shows that LIN28 also acts by directly binding to target mRNAs presenting GGAGA sequences enriched within their loop structures. 125
LIN28 is a recognized oncogenic driver, which is abnormally expressed in ~15% of human cancer cell lines and has been associated with a dismal prognosis. 126 High levels of LIN28 are found in different tumor types, such as glioblastoma, prostate, gastric, ovarian, and breast cancers. 114 Its overexpression has been shown to induce different tumors, such as Wilms tumor, 127 neuroblastoma, 128 and hepatocellular cancer, 129 in mouse models. LIN28 has also been demonstrated to have an important role in cancer stem cell formation and tumor metastasis. 130 LIN28A and/or LIN28B are useful cancer stem cell biomarkers in several cancer types. 130 Moreover, their expression has been associated with resistance to chemotherapy in several cancers. 114
In recent years, four HTSs aimed at identifying molecules able to disrupt LIN28-let-7 interaction have been reported.131–134
Using a FRET assay based on a GFP-LIN28B donor and a black-hole quencher-labeled let-7 acceptor, Roos et al. screened 16,000 small drug-like molecules and selected 203 molecules, which were reevaluated in triplicate in a new screen. Different secondary assays, including a luciferase reporter gene assay; RT-qPCR on let-7a, let-7f, and let-7g; and enzyme-linked immunosorbent assay (ELISA), were performed to validate the 14 confirmed molecules. N-Methyl-N-[3-(3-methyl[1,2,4]triazolo[4,3-b]pyridazin-6-yl)phenyl]acetamide, which displayed an IC50 value of 8 μM, was selected. The molecule was proven to bind to LIN28A and not to the let-7 RNA in a pull-down experiment. The selected molecule was able to induce differentiation in murine embryonic stem cells (ESCs) and to decrease clonogenic growth of four different cancer cell lines. 133
Lim and colleagues also developed a FRET-based assay to identify small molecules able to inhibit the LIN28-let7 interaction. Forty-five hundred drug-like molecules were screened and the primary hits were validated using EMSA. A benzopyranylpyrazole-based molecule, which presented IC50 values in the low micromolar range in FRET and EMSA, was selected as a hit. Structure–activity relationship studies revealed that the carboxylic group in the para position in the phenyl ring attached to the pyrazole was essential for the inhibitory activity. The selected molecule was proven to bind to the CSD of LIN28A by SPR and differential scanning fluorimetry. Finally, the cellular effects of the molecule were demonstrated by miRNA level quantification through qRT-PCR, a reporter gene assay, and Western blot analysis. 131
Lightfoot et al. developed an FP assay, through which they screened 2768 small molecules. The 64 primary hits were retested using the same assay to assess for reproducibility, and the 21 confirmed hits were then validated in a radioactivity-based EMSA. Next, the four validated hits were tested for their ability to prevent LIN28 blockage of pre-let-7 cleavage by Dicer. 6-Hydroxy-
Very recently, Wang and colleagues also used an FP assay to screen 101,017 compounds belonging to 17 different libraries in order to identify small-molecule inhibitors of both the CSD and ZKD of LIN28A. Since CSD has a higher affinity for the target RNA compared with the ZKD, compounds able to selectively interfere with ZKD-RNA binding are difficult to detect with assays using native full-length LIN28A. Therefore, in order to increase the sensitivity in detecting ZKD-RNA inhibitors, a point mutation able to weaken CSD-RNA binding was introduced in the LIN28A protein structure. The HTS identified 350 molecules, which were then prioritized based on their IC50 values calculated by performing a dose–response FP titration. Evaluation of the ability of the most potent compounds to prevent TUT4-mediated uridylation allowed to select six promising compounds. Structural studies performed by NMR and/or STD spectroscopy experiments elucidated the mechanism of action and the binding site of two compounds, with TPEN binding to the ZKD and LI71 binding to the CSD. 132
Musashi
Musashi (MSI) protein was first identified in Drosophila as an essential factor for correct asymmetrical division of cell precursors generating external sensory organs. 135 In mammals, two orthologs have been identified, both described as critical regulators of stem cell differentiation.136,137 The MSI1 protein has been found selectively expressed in neural stem cells, while MSI2 has been found in a variety of other tissues, including hematopoietic stem cells (HSCs). MSI2 has also been demonstrated as an independent factor able to enhance the regenerative potential of HSCs through a posttranscriptional mechanism that negatively regulates the aryl hydrocarbon receptor (AHR) signaling and induces a pro-self-renewal phenotype. 138
Musashi proteins belong to the A/B hnRNP class. They are characterized by two N-terminal RRMs that are responsible for binding to the UAG-context sequence in the 3′-UTR of target RNAs. 139 Both proteins interact with RNA by RRM1, in a molar ratio of 1:1, as demonstrated in complex with Numb5 RNA, 140 and the RRM2 adds affinity. 141 Other structural insights relate to the minimal recognition sequence, UAGGUAG, required to interact with RNA. 142
The global reprogramming capacity has prompted the investigation of Musashi proteins in cancer. Accumulating evidence from many aggressive forms of solid tumors, such as colorectal adenocarcinomas; breast, lung, pancreatic, glioblastoma, or endometrial cancer; 143 and hematologic malignancies, 144 suggest that Musashi proteins are potential markers of cancer stem cells (CSCs). 145 On the basis of maintaining cancer stem cell populations, Musashi proteins have been proposed to regulate cancer invasion, metastasis, and the development of more aggressive cancer phenotypes, with modulation of drug resistance. 145 Elevated levels of MSI2 were found in non-small cell lung cancer (NSCLC) tumor specimens, and its higher expression was associated with disease progression, positively correlating with the metastatic potential of tumor cells through the regulation of TGF-bR1/SMAD3 signaling. 146 Different reports have indicated that MSI2 overexpression can be detected in 70% of AML patients and correlates with poor prognosis.147,148 The same trend of clinical outcome has been demonstrated in gastric cancer for MSI1, 149 or in cervical cancer, 150 colon cancer, 151 and breast cancer 152 for MSI2. In addition, the overexpression of MSI2 strongly influenced the chemoresistance of ovarian cancer cells, NSCLC cells, and pancreatic cancer cells; 145 consistently, the downregulation of MSI2 sensitized ovarian cancer and acute myeloid leukemia cells to the pharmacological treatment.153,154
These findings suggest that Musashi proteins can be a promising therapeutic target for cancer, and their modulation can also represent a potential strategy to optimize conventional therapeutics.
Recombinant MSI1 and MSI2 proteins were used for an FP-based drug screening of more than 6000 compounds. 155 The small molecule Ro 08-2750 (Ro) was identified as an inhibitor of the RNA–protein interaction by FP and EMSA, with IC50 in the low micromolar range. 156 By microscale thermophoresis (MST), the Kd of Ro was evaluated around 12 µM, with appreciated selectivity in comparison with an evolutionarily related RBP, SYNCRIP. Structural data also demonstrated that the amino acids F66 and R100 are crucially involved in Ro binding. The validation of biochemical data has been performed using leukemia cell lines, where Ro induced differentiation and apoptosis together with a transcriptional program resembling MSI2 depletion. In addition, Ro inhibited leukemogenesis in an MLL-AF9 mouse model.
MSI1 targets include the mRNA encoding for the tumor suppressor protein APC, and one strategy to interfere with Wnt and Notch pathways was to identify compounds that could inhibit MSI1 RNA-binding activity. Also in this case, the screening was performed by FP using GST recombinant MSI1 protein and the natural product gossypol was identified with an inhibitory equilibrium dissociation constant in the high nanomolar range. 157 Complementary approaches, including SPR, confirmed a direct interaction of gossypol with the protein. The inhibition of MSI1-RNA interaction was effectively observed in vivo with inhibition of colon cancer growth upon oral administration of gossypol.
Intriguingly, Gu et al. found gossypol to be among the top hits of an HTS done with the aim of identifying inhibitors of the interaction between mouse double minute 2 (MDM2) RING and XIAP IRES. They later demonstrated gossypol’s inhibitory activity on the MDM2-VEGF 3′-UTR interaction.158,159 Through isothermal titration calorimetry (ITC) and fluorescence titration techniques, the investigators were able to determine whether the selected screening hits bind to the protein or to the RNA and found that gossypol binds to MDM2 RING, with a Kd of 5.21 µM. 159 Indeed, the biological effects of gossypol and some analogs have been shown in different cell lines and models, 160 demonstrating anti-inflammatory effects, 161 antiproliferative capacity through direct interference with the kinase domain of EGFR, 162 pro-apoptotic effects by enhancing the levels of Bcl-2, 163 and autophagic effects through activation of LC3. 164 This compound also induces an accelerated hemolytic toxicity when administered in vivo. 165 The interference with RBPs represents an additional perspective that contributes to defining the broad mechanism of action of gossypol and its preferential intracellular targets at the tissue-specific level.
Other RNPs Suggested as Potential Therapeutic Targets
TDP-43 is a nuclear protein of 414 amino acids, with two highly conserved RRM1 and RRM2 domains resembling the architecture of the members of the hnRNP family. TDP-43 participates in different processes, including DNA replication, repair, mRNA splicing, and translation. Mechanistically, TDP-43 recognizes both DNA and RNA cis elements. This protein is crucial for correct embryonic development and its role has been extensively investigated in neurodegeneration, starting from the identification of rare TDP-43 mutations in cases of sporadic and familial amyotrophic lateral sclerosis (ALS) and cases of frontotemporal lobar degeneration (FTLD). 166 TDP-43 mutations are the cause of disease for 6.5% of familial ALS cases, while in the rest of ALS and FTD patients no mutations have been found but the presence of TDP-43 aggregates. Other functional studies have demonstrated that TDP-43 is associated with aberrant exon 9 skipping, as the case of the cystic fibrosis transmembrane conductance regulator (CFTR) gene in cystic fibrosis. 167
An increased accumulation of this protein is surprisingly found in the cytoplasm of affected neurons, where TDP-43 aggregates have been demonstrated to be hyperphosphorylated, ubiquitinated, and cleaved at the C-terminus. 168 However, the pathogenetic role of this protein, in terms of specific neurotoxic effects, remains to be clarified. 169 The mislocalization in the cytoplasm suggests a loss of all the functions concerning transcriptional regulation, splicing, and mRNA stability.
Among RBPs, TDP-43 is certainly considered an interesting target to address neurodegeneration. However, the elucidation of the basic molecular functions of the protein and of the effects of their alteration after mutation and aggregation is required for a targeted approach. Technically, the interaction of TDP-43 with nucleic acids has already been explored with high-throughput assays. 170 Interestingly, the inhibition of TDP-43 aggregate formation has been explored by suppressing ataxin-2 expression (necessary for TDP-43 aggregate formation), by reversing translational suppression, by stimulating autophagic processing, or by decreasing TDP-43 mitochondrial localization. 171
Tristretaprolin (TTP) is an RBP containing tandem CCCH zinc finger (TZF) domains. It strongly binds to ARE sequences, mainly enriched in the 3′-UTR of mRNAs, regulating their stability and decay. TTP interacts with the CCR4-CNOT1 deadenylase complex and favors the decay of ARE containing mRNAs, including the ones encoding pro-inflammatory cytokines. TNFα is one of the main targets of TTP, which causes the specific degradation of this transcript, as clearly observed in macrophages. 172 Interestingly, the downregulation of TTP in mice leads to the development of autoimmune diseases due to the increased release of TNFα. 173 For these reasons, TTP has been proposed as a therapeutic target in inflammatory diseases, with the goal of enhancing its expression using small molecules. 174 In a more complex paradigm, TTP loss has been reported in several tumors and associated with poor prognosis for enhanced cancer cell proliferation, epithelial–mesenchymal transition, and tumor aggressiveness, as summarized by Guo et al. 175
Discussion/Conclusions
In conclusion, in recent years a number of reports have highlighted the role of alterations of the posttranscriptional control of gene expression in disease, and the multiple reasons why specific RBPs could be attractive drug targets. The advantages of targeting RBPs reside in their intrinsic pleiotropy and in their ability to simultaneously control entire subsets of mRNAs, which could in principle allow the small molecules interfering with them to affect complex cellular programs deranged in disease. Another attractive feature of RBPs is that several of them act as repressors of gene expression, or as activators that compete with ncRNAs. In this case, the inhibition of their activity by small molecules would in principle produce enhancement of the expression of the RBP targets. This would open up potential therapeutic solutions for single-gene haploinsufficiency diseases, to reconstitute physiological levels of expression of pathologically repressed genes.
Despite these exciting perspectives, the strategies to develop therapeutic approaches against RNPs face several challenges. The first directly relies on the RBP pleiotropy, which exposes any pharmacological interventions to the risk of unwanted effects, involving a fine tuning in the medicinal chemistry phase to reach the required specificity. Medicinal chemistry campaigns are also likely necessary for the development of lead compounds requiring large hydrophobic cores, as several of the RBP hits are predicted to contain. Again, in terms of selectivity, one of the main biological challenges of RNP complexes is first related to the strong avidity (often detected in the nanomolar range) that limits the competition kinetics of small molecules at the levels of the association between the ligands with a weak or no effect on the dissociation. Second, the presence of homologous protein domains shared by RBPs can be responsible, by competition, for a limited intracellular efficacy of the small molecules. It is worth noting that the efficient activity as well as the subcellular localization of an RBP is a result of the global protein conformation that assists the RNA recognition and is often posttranslationally modified to confer target preferences.
Future screening strategies should take care to use chemical libraries as diverse as possible and include noncanonical chemotypes (i.e., violating Lipinski’s rule of five), such as various natural compounds or macrocycles, as well as using fragment libraries to identify useful chemical scaffolds and hot spots in protein pockets. 176
Furthermore, computational predictions can be successfully applied to RNPs, analyzing targets with known 3D structure and scanning their shape in search of pockets with favorable physicochemical and geometrical properties (hydrophobicity, size, compactness, hydrogen bond donor and acceptor surface areas, and amino acid composition in pockets) or running fragment-based virtual screening.177,178 These methods are cost-saving and timely with respect to the experimental approach, with the drawbacks of being limited to targets with known structures and hardly applicable when significant flexibility and conformational adaptivity are present at the binding site.
These challenges can be successfully addressed when deep structural insights about the ligands are available and the dynamics of the biological process involving that specific RNA-RBP interaction is elucidated. For these reasons, compounds identified through primary and counter HTSs need to be validated by at least two rounds of complementary approaches: (1) a technical validation only successfully achieved by the smart combination of different (in principle) assays, and (2) a biological validation of the small molecule in cellular and in in vivo models where the pathophysiology of the RBP is known to generate specific readouts. These integrated approaches will be necessary to understand the pathological relevance of specific RBPs and their effect in regulating constitutive pathways, whose inhibition might result in potential toxicities when administered at the systemic level, as showed by the case of the drug E7107 targeting SF3B1.
In order to deal effectively with these obstacles, a number of tailored strategies can be suggested. The attempts so far conducted to explore the RNP as targets have generated important advancements in understanding their biochemical activity and additional efforts should be undertaken to implement novel technologies, suitable for HTS, to study the interactions and their dynamics at the intracellular level, with the final goal of discovering novel drugs or developing chemical probes as powerful research tools.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: A.P. gratefully acknowledges funding from Associazione Italiana per la Ricerca sul Cancro (AIRC) (17153), CARITRO Riposizionamento Farmaci (40102838), and CARIPLO, ricerca biomedica sulle malattie legate all’invecchiamento (40102636). A.Q. acknowledges AIRC (17026). V.G.D. acknowledges MIUR-FFABR-17.