
Abstract
Over the past century, a multitude of derivatives of structural scaffolds with established antimicrobial potential have been prepared and tested, and a variety of new scaffolds have emerged. The effectiveness of antibiotics, however, is in sharp decline because of the emergence of drug-resistant microorganisms. The prevalence of drug resistance, both in clinical and community settings, is a consequence of bacterial ingenuity in altering pathways and/or cell morphology, making it a persistent threat to human health. The fundamental ability of pathogens to survive in a multitude of habitats can be triggered by recognition of chemical signals that warn organisms of exposure to a potentially harmful environment. Host immune defenses, including reactive oxygen intermediates and antibacterial substances, are among the multitude of chemical signals that can subsequently trigger expression of phenotypes better adapted for survival in that hostile environment. Thus, resistance development appears to be unavoidable, which leads to the conclusion that developing an alternative perspective for treatment options is vital. This review will discuss emerging medicinal chemistry approaches for addressing the global multidrug resistance in the 21st century.
Introduction
In order to pursue chemotherapy successfully we must look for substances which possess a high affinity and high lethal potency in relation to the parasites, but have a low toxicity in relation to the body, so that it becomes possible to kill the parasites without damaging the body to any great extent. We want to hit the parasites as selectively as possible. In other words, we must learn to aim and to aim in a chemical sense. The way to do this is to synthesize by chemical means as many derivatives as possible of relevant substances.
—Paul Ehrlich, Über den jetzigen Stand der Chemotherapie. Berichte der Deutschen Chemischen Gesellschaft,
The effects of Penicillium on bacteria had been known prior to the now famous studies of Alexander Fleming. However, the reports describing the phenomenon received little attention before the clinical importance of penicillin was established. Investigating the possibility that one organism can interfere with another’s growth dates back to the early days of microbiology in the 19th century.1,2 By the end of the century, this phenomenon was widely accepted and has been given a name: antibiosis. Antibiotics are either bactericidal (kill bacteria) or bacteriostatic (stop bacteria from growing).
Antibiotic mechanisms of action are based on prevention of DNA/RNA, folate, and protein syntheses and destruction of the cell wall/membrane. Bacterial resistance to antibiotics has been known since the 1940s, when a colony of Staphylococci survived lethal action of the compound with a newly determined structure: penicillin. 3 Similarly, resistance to streptomycin was reported shortly after its discovery. 4 These findings led to the conclusion that a population of preexisting resistant organisms exists, even in the absence of drug-generated evolutionary pressure. However, it was not until the 1970s when the rate of bacterial resistance accelerated, eventually leading to the current global health problem.5,6 Looking back from our current understanding of how ancient bacterial resistance toward antibiotics is,7,8 we realize that resistance is unavoidable and accepting the necessity of developing new strategies for addressing it is pending. Developing resistance is a response of a pathogen to the selective pressure of antibiotics on the existing millennia elements of resistance (e.g., R-plasmids). 9 That response is triggered not only by natural antibiotics but also by antibiotics prepared via organic synthesis (e.g., fluoroquinolones). 10 Resistance mechanisms include expression of hydrolytic enzymes such as the β-lactamases; removal of the antibiotic by efflux pumps; modification of the antibiotic’s molecular target, rendering it unable to bind the drug; and circumvention of antibiotic toxicity.
In addition to bacterial ingenuity in altering pathways and/or cell morphology, human behavior (e.g., overutilization of miracle drugs, modest commercial opportunities of producing future infectious disease medicines) has contributed to reaching the magnitude of antimicrobial resistance (AMR) we face today with clinically relevant antibiotics. It is estimated that by 2050, the lives lost to AMR will exceed the 8.2 million a year currently lost to cancer, 11 and the economic burden caused by AMR worldwide might be as high as $100 trillion. 11 In 2017, the Centers for Disease Control and Prevention (CDC) detected more than 220 cases of a rare type of nightmare bacteria that are virtually untreatable, according to a report released on April 3, 2018. Although the CDC has warned of the danger of antibiotic-resistant bacteria for years, this new report helps illustrate the scope of the problem. The CDC’s principal deputy director, Anne Schuchat, said she was surprised by the extent resistance has spread. “As fast as we have run to slow (antibiotic) resistance, some germs have outpaced us,” Schuchat said. “We need to do more and we need to do it faster and earlier.” 12
Challenges in antimicrobial drug discovery are imposed by the vast microbial genetic diversity and the fact that it is technically difficult and takes a long time to make a novel antibiotic: “Converting an early chemical prospect into a medicine that can be used in people is a profound scientific challenge, the difficulties of which are not going to be mitigated by a change in the commercial landscape or public policy.” 13
In addition, the design of new antibiotics ought to make them less prone or even impervious to resistance development. This is a challenging goal, since the distribution of AMR is not always predictable. Illustrative to that is the finding that the gene encoding for a hydrolyzing enzyme (New Delhi metallo-β-lactamase, NDM-1) of β-lactam antibiotics is a chimeric gene. The latter resulted from the fusion of a progenitor type metallo-β-lactamase gene with a partial sequence of a gene conferring resistance to aminoglycosides, probably created de novo in Acinetobacter baumannii. 14
The chemical approaches used to create new lead molecules (aside from efforts in new natural product isolation) are (1) combinatorial chemistry generally resulting in primarily sp2-hybridized molecules, (2) methods that develop diverse libraries of complex scaffolds, and (3) modifications to previously identified antibiotic compounds. This review will focus on recent developments encompassing the two latter approaches attempting to address AMR through synthetic and medicinal chemistry means.
The Antibiotic Tree: Where Are We At?
The majority of today’s most effective antimicrobials are natural products isolated in the 1940s from soil-colonizing bacteria and fungi. Of the principle antibiotic scaffolds in current clinical use, fluoroquinolones, sulfonamides, trimethoprim, and more recently oxazolidinones are products of organic synthesis. Natural product scaffolds of clinical significance include β-lactams, aminoglycosides, tetracyclines, glycopeptides, and macrolides.15,16 Since their initial discoveries, the antimicrobial industry has focused on fine-tuning existing classes of antibiotics to improve their antimicrobial spectrum, efficacy, and safety as well as to combat clinical resistance in the later years of the 20th century. The semisynthetic approach for achieving the latter has been the most commonly used approach for most of the past century. 7 However, multiple variation of natural product scaffolds with a mode of action on conserved targets has contributed to the development of multidrug-resistant (MDR) bacterial strains. High frequencies of MDR bacteria have been grouped under the acronym ESKAPE: Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp. ESKAPE pathogens are responsible for the majority of nosocomial infections and are capable of escaping the biocidal action of antimicrobial agents. By the mid-1990s, the enthusiasm for making yet another incremental improvement to a β-lactam, macrolide, or quinolone had diminished (example of diversification of existing scaffold, of vancomycin, is shown in Fig. 1 ). Then, in 1995, determination of the complete DNA sequence of a bacterial genome from Haemophilus influenzae revived interest in the development of antibacterials. “The prospect of hundreds of new genes to explore as possible targets sparked new interest in antibacterial discovery and fired the imagination.” 13 From then on, attempts at finding a novel antibacterial agent in the pharmaceutical industry have been generally directed toward three discovery approaches: new molecular targets, found by comparative genomics; new structures for old molecular targets; and cell-based screening for novel structures. There are several excellent reviews on the subject, with lessons learned from the era of the great expectations of comparative genomics, nicely presented in the reflective papers of the GlaxoSmithKline (GSK) and AstraZeneca experiences.13,17
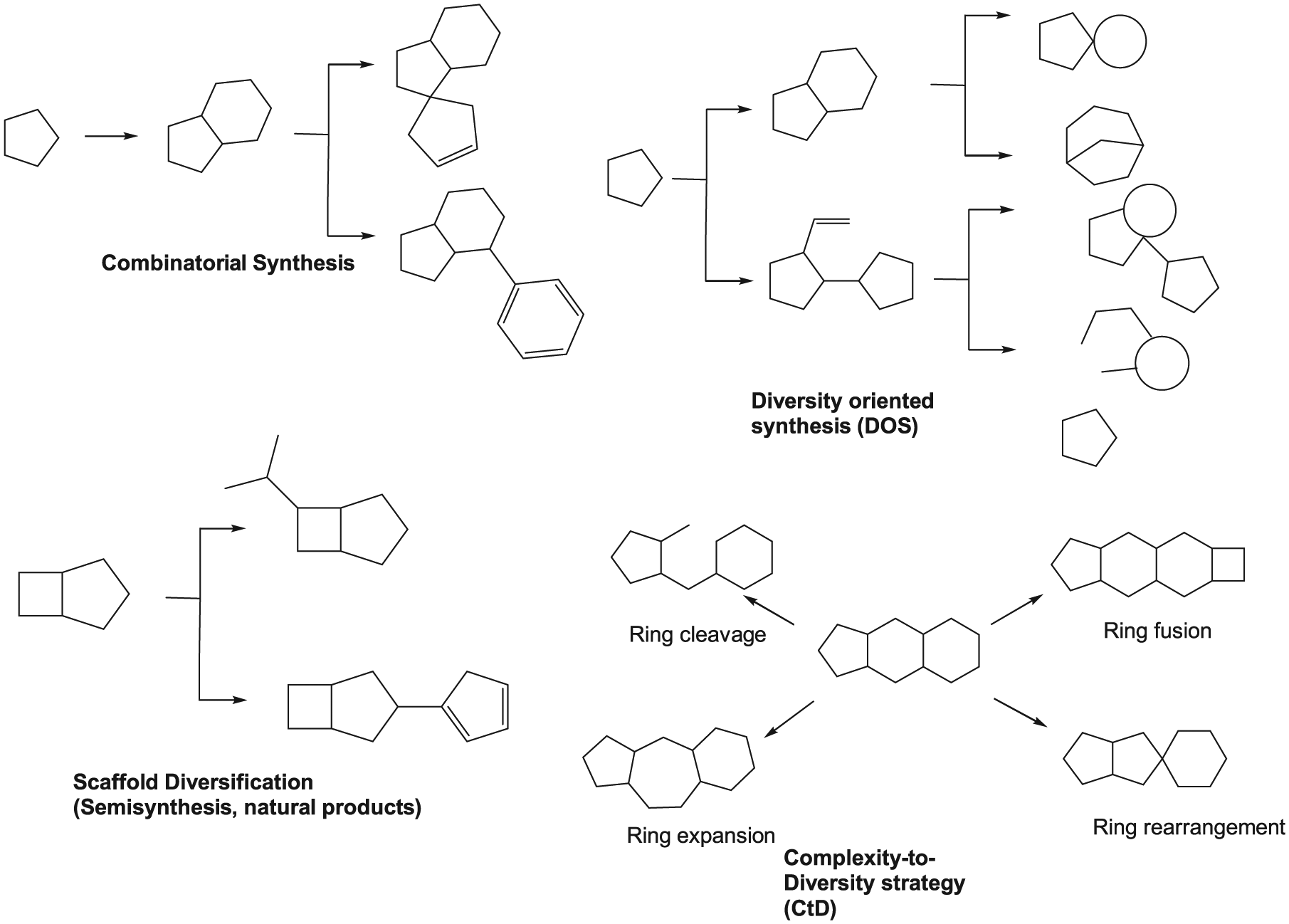
Traditional (semisynthesis and combinatorial) and innovative (diversity-oriented synthesis, complexity-to-diversity) synthetic approaches.
Antimicrobials Are in a Drug Class of Their Own: Building the Right Antimicrobial Molecule Is the Hard Part
An extremely important factor when searching for new drugs is the chemical diversity of compounds available to screen. Comparative genomics give new targets; however, finding the suitable lead for a new target has been disappointing. Chemical collections of most large pharmaceutical companies have been exhausted for possible antibacterials mainly by using high-throughput screening (HTS). 13 The analysis of intensive HTS campaigns from that time by research groups at GSK 13 and AstraZeneca 17 offer some explanation for this lack of success. The reasons for the latter are complex. However, the limited diversity of chemical space of their libraries as sources of difficulty in identifying new antibacterials has been recognized in both reports.13,17 The initial excitement for the opportunity to find chemically diverse compound libraries has been spurred by the development of combinatorial chemistry ( Fig. 2 ). The latter has been extensively used in industry to generate lead compounds. From 1999 to 2008, 45 of the 50 Food and Drug Administration (FDA) approvals for first-in-class small-molecule new molecular entities originated from a screen. 18 However, this approach has not paid off for the class of antimicrobials as expected, since it generally results in the sp2-hybridized molecules.19,20 Essential gene screens and HTS technologies allow for testing of millions of compounds in a short period of time, but they have not resulted in new antimicrobial drugs. 21 The reason is the limited makeup of screening libraries.22,23 In addition, Lipinski’s rules, which are set for combinatorial chemistry, do not include the drug class of antibiotics. 24 The lack of success to fit antibacterials in the combinatorial approach is because they (marketed drugs and natural products) tend to be larger and more complex than the average screening compound.13,25 In addition, gram-positive agents have larger molecular weight, larger polar surface area (PSA) of 243 Å, and lower ClogD7.4 (–0.2) than comprehensive medicinal chemistry or normal drugs, which have a PSA of 70 Å and clogD7.4 of 1.6.26–28 The gram-negative agents are generally smaller, are more polar, have a PSA of 165 Å, and have a ClogD7.4 of (–2.8).26,28
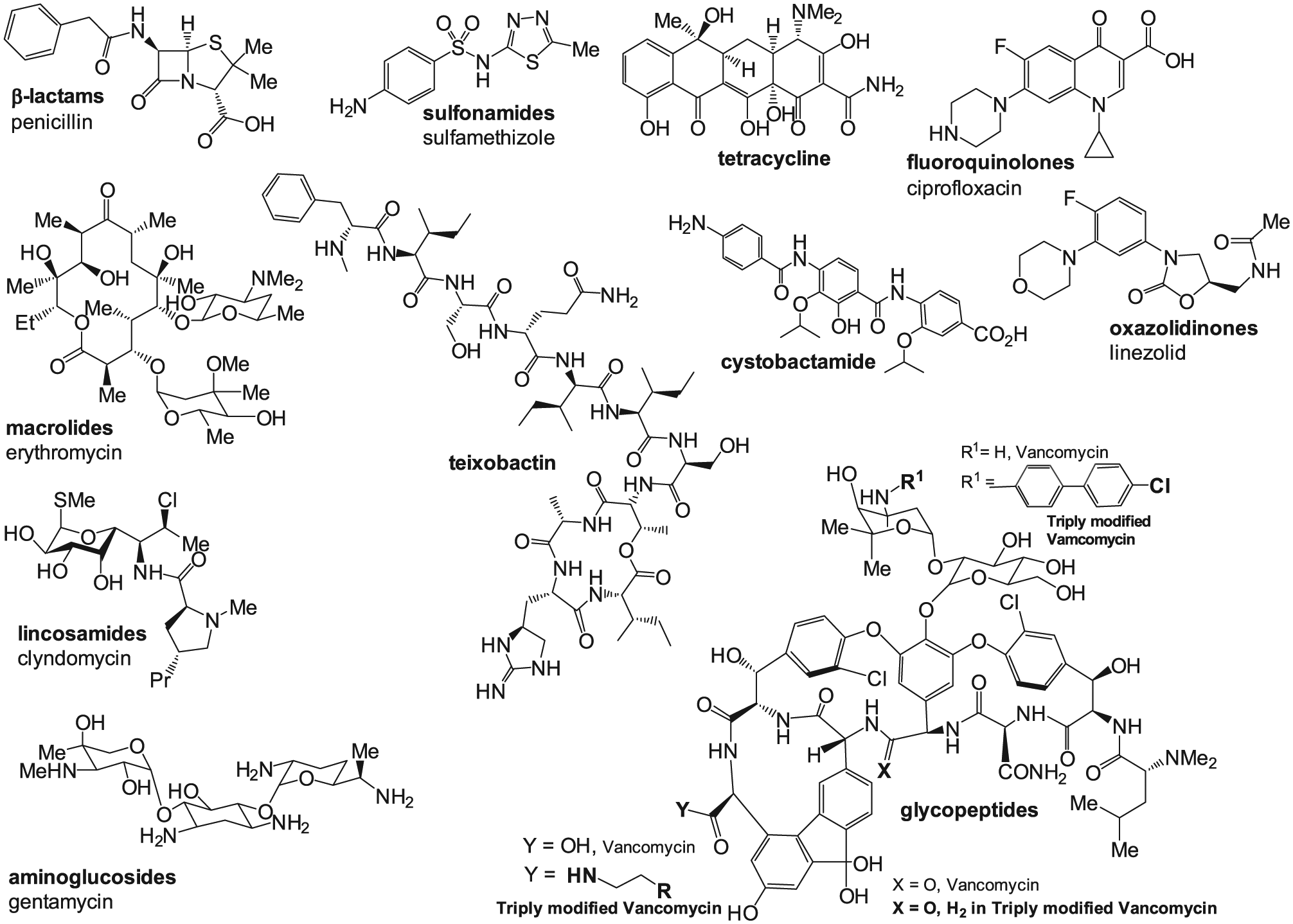
Representative classes of clinically relevant and newly discovered antibiotics (teixobactin and cystobactamides). The fragments shown in bold represent the peripheral structural changes of vancomycin that led to improved antimicrobial potency and provided additional synergistic mechanisms of action, including those that are associated with membrane-binding properties.
The physicochemical properties of antimicrobial drugs26,29 have not been met by the majority of libraries of compounds from most commercial and pharmaceutical sources.26,27 The aforementioned approaches do not allow for coevolution of a high affinity for a biological target and the ability to reach the cellular site of action. These are two distinct advantages of starting from a natural product scaffold during lead optimization.27,30 Therefore, it is only conceivable that nature will continue to serve as a prolific source of new antibacterial leads. 31
New Structures for Old Molecular Targets: “A Good Target Is Better than a New Target” 13
There are several recent reports confirming the revived interest in collection of new29,32 and utilization of previously rejected (due to either toxicity or poor pharmacokinetics/pharmacodynamics, narrow antimicrobial spectrum) natural products. 29 Kibdelomycin, a novel natural product inhibitor of bacterial type II topoisomerase 32 that is unrelated to fluoroquinolone antibiotics, is such an example. This confirms the notion that the search for naturally produced yet unsuspected scaffolds for established drug targets expands our antimicrobial arsenal. A novel class of natural products from Cystobacter spp. has been recently identified 33 as part of a search for new compounds with activity against gram-negative bacteria. Cystobactamides ( Fig. 2 ) have structures of arylamides, and in a sense, they are peptides with unusual aromatic moieties, such as p-aminobenzoic acid. Cystobactamides are inhibitors of topoisomerases and thus can be regarded as natural quinolones. 33 They strongly inhibit the growth of various ESKAPE pathogens, 34 including the gram-negative Acinetobacter baumanii and gram-positive Enterococcus faecalis and Streptococcus pneumoniae at concentrations comparable to and even exceeding the activity of ciprofloxacin, a clinically relevant second-generation fluoroquinolone antibiotic.
Another representative of naturally occurring peptides, the cyclic depsipeptide teixobactin, has been discovered in a screen of uncultured bacteria (from the previously unknown β-proteobacterium Eleftheria terrae). 35 Notably, no teixobactin-resistant bacterial strain has been detected so far, regardless of directed efforts. The molecular target of teixobactin apparently is the same as that of vancomycin, lipid II, the precursor of peptidoglycan, but the binding site differs from the one of vancomycin. 36 Because teixobactin has shown excellent antimicrobial activity against several ESKAPE pathogens, including vancomycin-resistant strains, (0.5 to 0.03 µg/mL), it is considered to be one of the very promising lead compounds for antimicrobial drug development. 36
Synthetic approaches directed toward reengineering of natural products into antibiotics continue to rejuvenate the established scaffolds of the golden age of antibiotics. These approaches include diversity-oriented synthesis (DOS) and complexity-to-diversity (CtD). They allow for preparation of libraries of compounds through relatively few chemical transformations in a highly divergent manner.22,37–51 The DOS38–47 and CtD22,49–51 are directed toward addressing the specific requirements for a successful antibacterial structure. As mentioned earlier, average log D values (log P or log D values are the parameters for measuring hydrophobicity) for drugs targeting these organisms are −0.2 and −2.8, respectively, and 1.6 for other drugs. Additional data from approximately 3200 compounds emerging from AstraZeneca antibacterial discovery campaigns has expanded the aforementioned findings. 28 The hydrophobicity has been found to be highly distinct between active antibacterials and average log D in the screening compound library. The trend to improve in vitro activity of the lead scaffolds frequently led to increased log D, which in turn resulted in lowering in vivo activity. The balance between cell penetration and efflux 28 is especially important for gram-negative organisms such as Pseudomonas, which has numerous efflux pumps. Understanding the role of the cell envelope in compound entry and retention in the cell is needed, 52 if efforts in building tailored chemical libraries with improved whole-cell bioactivity are to be supported. 21 However, a high number of pharmaceutical companies are still synthesizing compounds with a mean ClogP >4. 53
DOS
The DOS strategy (in which simple starting materials are coupled to make diverse structures;
Fig. 1
), originally introduced by Schreiber,
41
has led to the discovery of complex bioactive scaffolds.
42
Compounds produced by DOS synthesis are comparable to natural products by their higher ratio of sp3-hybridized atoms and stereocenters as compared with compounds found in conventional (produced by combinatorial chemistry) screening collections. The descriptor Fsp3 is the number of sp3-hybridized carbon atoms in a compound divided by the sum of the sp3- and sp2-hybridized carbon atoms.
48
The benefits of a higher Fsp3, which include lower melting points and enhanced aqueous solubility, has been demonstrated.
19
Three antimicrobial compounds (out of 242 screened) with inhibition activity against S. aureus, including two UK epidemic methicillin-resistant strains (EMRSA 15 and EMRSA 16), have been discovered through the DOS approach.
44
The most potent compound, (±)-gemmacin (
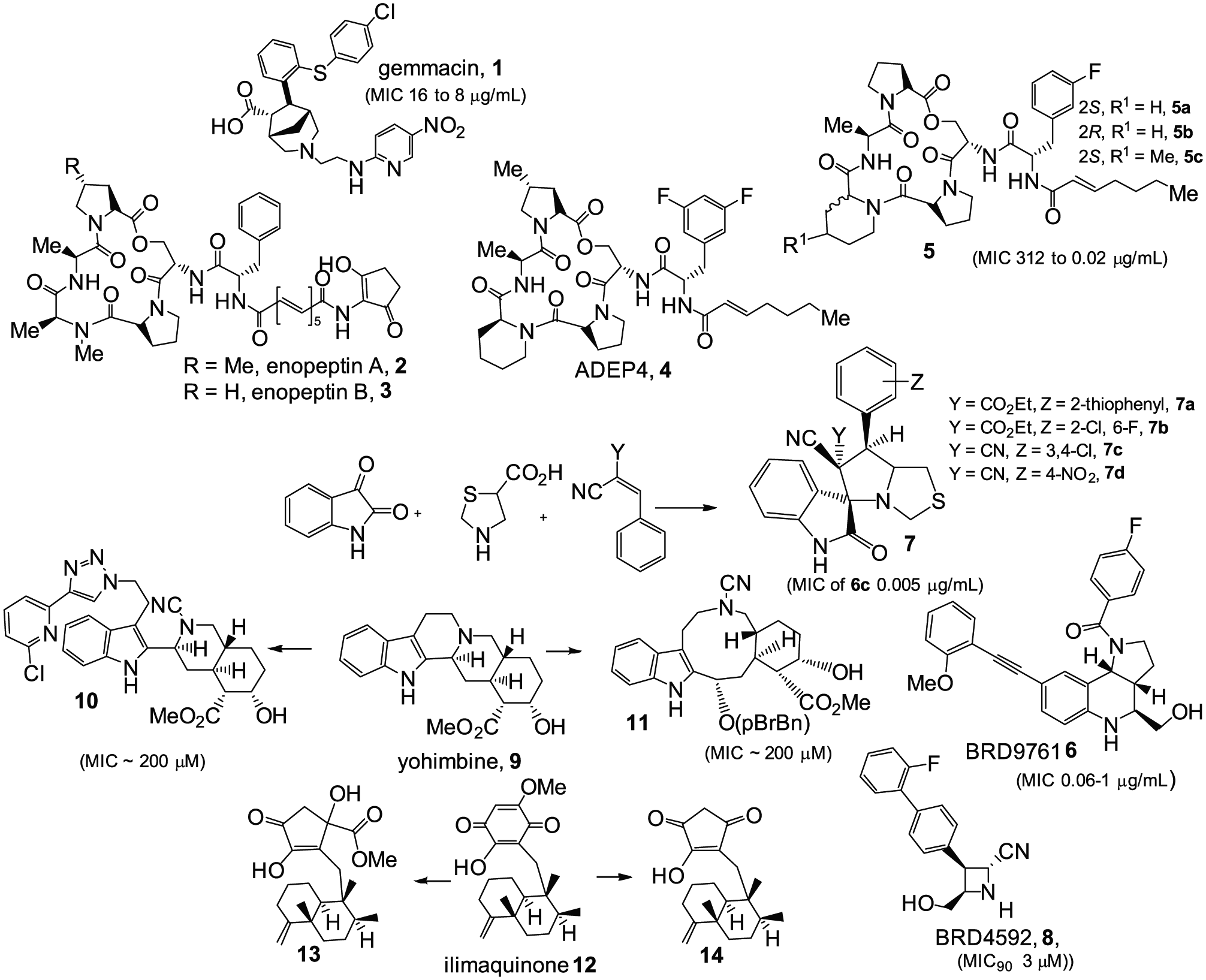
Structures of antibacterials from diversity-oriented synthesis (DOS) and complexity-to-diversity (CtD) approaches. Compounds
CTD Approach
The CtD approach introduced by Hergenrother and coworkers 22 uses chemoselective reactions to systematically alter core structures of natural products via reactions that distort the ring system. In contrast to traditional optimization campaigns, the goals of the CtD method are to enhance the inherent biological activity/improve druglike properties of a natural product.22,49–51 Several natural products from different structural classes such as gibberellic acid, adrenosterone, and quinine have been used as core structures to demonstrate the potential of the CtD approach. 22 Because there are many readily available natural products, the possibilities to achieve diverse and complex molecules using this approach are plenty. The CtD strategy has been inspired by how nature creates complex molecules using a common intermediate to generate a multitude of compounds that are very different from one another. 22 The resulting compounds from the CtD approach appear to meet the aforementioned goals: they have an average Fsp3 of 0.59, which is higher than that in the commercial collection (0.23), and the average ClogP is 1.1 log units lower than that in the commercial screening set (2.90 versus 3.99), which corresponds to a 12-fold reduction in hydrophobicity. 22
Using this strategy, small-molecule compounds with antimicrobial properties have been constructed from readily available natural products.
58
The indole alkaloid yohimbine is an example of such a starting natural product. Yohimbine, (
Fragment-Based Drug Discovery
Fragment-based lead generation (FBLG) is an alternative to traditional HTS. FBLG allows the discovery of novel inhibitors of known drug targets through a unique binding site, chemistry, or mechanism of inhibition, which allows for the evasion of existing resistance mechanisms. Most major pharmaceutical and biotech companies use fragment-based approaches as part of their lead generation strategies.
60
The underlying concepts of FBLG were implemented in silico in the 1980s and early 1990s.61,62 However, lead generation driven by experimental screening of fragments has been receiving widespread interest only since the introduction of SAR by nuclear magnetic resonance in the mid-1990s.63,64 This approach initially focused on the use of two-dimensional nuclear magnetic resonance techniques to screen for small compounds binding to the target protein, preferably to different adjacent subpockets in an active site. It appears, however, that most proteins have only a single energetic focal point, dominating the binding energy available in the active site. This so-called hot-spot65–67 is comparatively small and easily exploited with fragment-sized compounds. Pyrrolamides, prototypes of a novel class of DNA gyrase inhibitors, with activity against predominantly gram-positive bacterial pathogens, including resistant strains, have been identified using the FBLG approach by an AstraZeneca research team.
68
A representative compound from the pyrrolamide series (compound
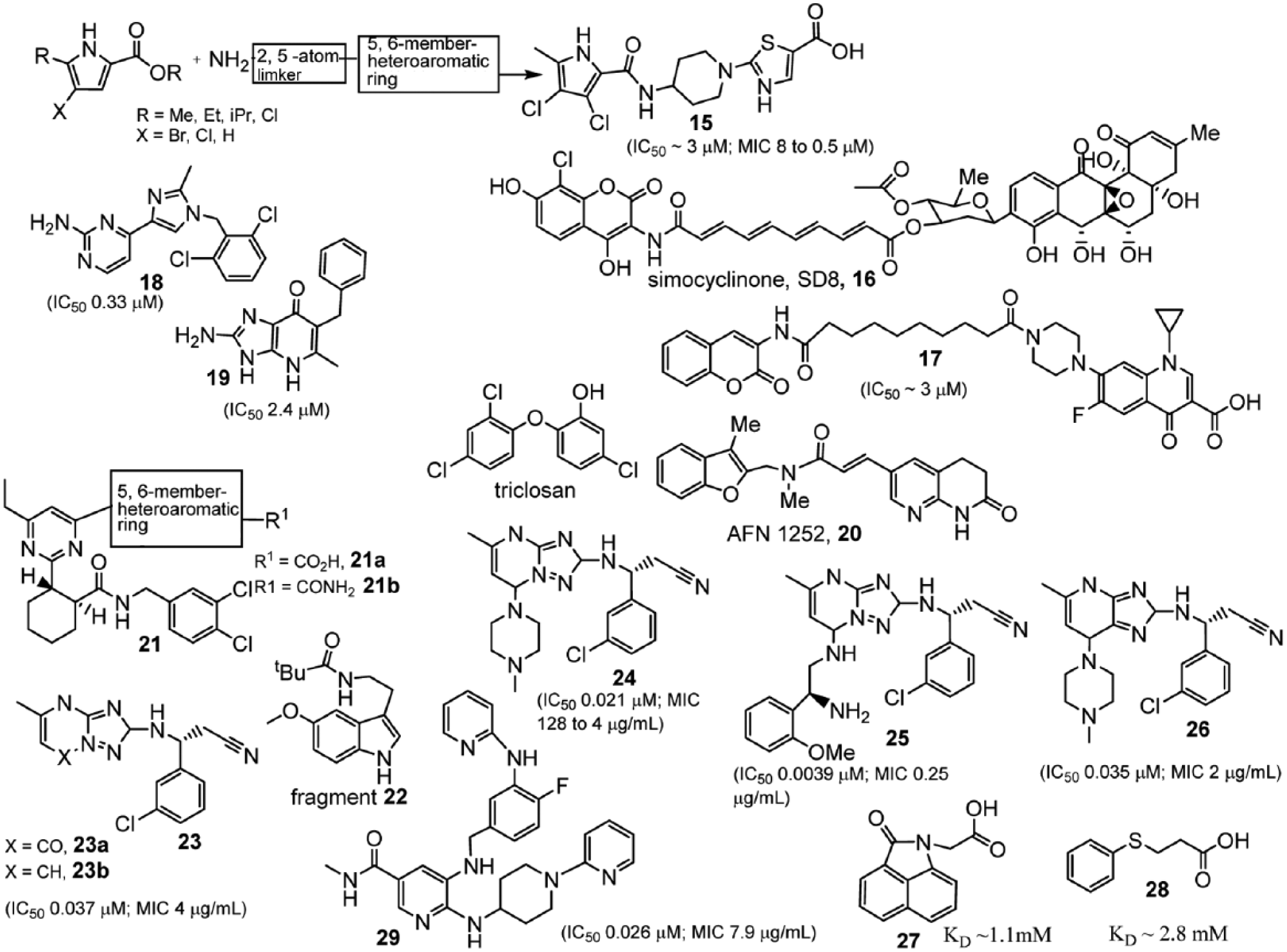
Antibacterials identified by the fragment-based lead generation (FBLG; compounds
Simocyclinone D8 (SD8,
The type II fatty acid biosynthetic pathway is an excellent target for antibacterial drug discovery.
70
A Pfizer research group described pyridopyrimidine-based potent biotin carboxylase inhibitors using the FBLG approach.
71
This strategy has led to identification of a structurally diverse collection of weak-binding but ligand-efficient fragments for biotin carboxylase adenosine triphosphate–competitive inhibitors (compound
Recently, the FBLG approach has been directed at another enzyme of the fatty acid synthesis pathway: FabI in the gram-positive bacterium, S. aureus. Type II fatty acid biosynthesis, as has already been mentioned, is a drug target that has been validated by several studies75,76 with clinically relevant molecules such as AFN-1252 (compound
A molecular drug target associated with coenzyme A is phosphopantetheine adenylyltransferase (PPAT; also known as CoaD). PPAT catalyzes the penultimate step in coenzyme A biosynthesis. PPAT is essential for bacterial growth. It shares relatively little sequence homology with the human ortholog, which makes is a good target for antibacterial drug development.86–88,89 Two compounds from AstraZeneca (compounds
Identification of compounds using the FBLG approach for the DNA-G–SSB interaction has been reported.
93
SSB protects single-stranded DNA during replication.94,95 A fragment-based screen has led to the identification of inhibitors (compound
Additional examples of FBLG in the antibiotic area, as well as advances that have supported the evolution of FBLG methods, have been nicely summarized in a recent review. 96
DNA-Encoded Chemical Libraries
A recent addition to lead-generation technologies is DNA-encoded chemical libraries or encoded library technology (ELT) to discover small-molecule binders to targets of interest.
97
Use of ELT has led to identification of a large number of molecules obtained via chemical synthesis that bind to proteins from diverse chemotypes, as well as to an understanding of SAR directly from the screening output.
97
A variety of methods for creating large DNA-encoded combinatorial libraries have been developed in the past two decades. Regardless of the individual synthesis techniques, ELT involves screening very large collections (typically >108) of small-molecule compounds using a technology that involves the conjugation of chemical compounds to DNA fragments.
98
This technology, however, has led to fewer clinical candidates as compared with other lead-generation approaches. This is mainly due to the current limited access to ELT by only several large pharmaceutical companies and contract research organizations
98
and by limitations posed by the types of chemistry that can be used to construct libraries on DNA. The latter, coupled with the challenges associated with building of large libraries with low molecular weight and low lipophilicity, has been recently highlighted.
99
However, successes in the area of infectious diseases100,101 using this technique might be an indicator of its future impact on drug discovery. A pool of 11 DNA-encoded libraries comprising more than 66 billion on-DNA compounds has been used for selections against various forms of InhA (enoyl-acyl-carrier protein reductase, the primary target of isoniazid). Cofactor-specific inhibitors of InhA that do not require activation by KatG (unlike isoniazid), many of which had bactericidal activity in cell-based assays (e.g.,
Old Structures: Delaying the AMR
Modifications to Previously Identified Antibiotic Compounds
The idea of taking a deeper look at some previously undeveloped leads is supported by the emerging view of the mechanism of action of daptomycin, a natural product (lipopeptide antibiotic), which, even though currently a therapeutic, initially was given little interest. 104 Other examples of a revived interest toward several natural products include hygrobafinomycin 105 (a member of a novel hygrolide [macrolide]) and tetrodecamycin 106 (a member of the tetronate family, whose antimicrobial activities and biosyntheses have been recently reported).
The structural diversity and complexity of natural product antibiotics have inspired many successful total synthesis campaigns for their preparation. 107 These syntheses are challenging, and aside from prompting the development of a synthetic methodology for overcoming a host of common obstacles, are for the most part impractical from the point of view of antibacterial development. However, the impressive achievements in synthetic methodology over the decades allow current efforts in the development of total synthesis of antibacterials to change in synthetic strategy. A shift from the original goal of reaching solely the molecule of interest (the synthetic target) to preparation of target analogs in order to establish SAR has started to occur. The newest developments in total synthesis of the main classes of antibiotics have been thoroughly reviewed, 107 and a special issue of the Journal of Organic Chemistry has been recently published that focused on both synthetic and biosynthetic chemistry of natural products, with an emphasis on antibiotics. 108
One recent success in the deliberate design of an antibacterial compound (vancomycin) based on the total synthesis may serve as a confirmation for applicability of the modification of existing natural product scaffolds in making the latter less prone to resistance development. 109 Certain features of glycopeptides have been identified that enable this class of antibiotics to avoid many mechanisms of resistance.110,111 Based on this knowledge, incorporation of peripheral structural modification in vancomycin led to enabling multiple synergistic mechanisms of action. Bacteria sense the presence of the antibiotic, 112 which triggers a change in the peptidoglycan termini from D-Ala-D-Ala to D-Ala-D-Lac. 113 This remodeling of the peptidoglycan allows for a 1000-fold reduction of the binding affinity of vancomycin for the altered target,114,115 resulting in a 1000-fold loss in antimicrobial activity. The three modifications made in the structure of vancomycin (triply modified vancomycin; Fig. 1 ), two peripheral ones (with synergistic mechanisms of action) and one pocket modification, have led to a vancomycin analog with three independent mechanisms of action, only one of which is dependent on D-Ala-D-Ala/D-Ala-D-Lac binding. This triply modified vancomycin demonstrated increased antimicrobial potency against VanA VRE (>6000-fold) as well as reduced susceptibility to resistance. 109 This accomplishment is a result of the total synthesis of the starting pocket modified aglycon(s) (26 steps), 116 enzymatic installation of the disaccharide (2 steps), 117 and subsequent addition of the two peripheral modifications (2 steps), demonstrating an impressive synthetic methodology. Peripheral structural changes lead to improved membrane binding. Traditionally, drug development has been focused on optimizing target affinity, selectivity, and protein binding. 118 A more recently explored approach is adding structural motifs, especially to drugs acting on membrane-associated targets (e.g., vancomycin). This could lead to increased drug concentration at the target site, in addition to improved membrane binding.119,120
New Life for Old Friends: β-Lactam Drug Combinations
Triply modified vancomycin is an example of a single molecule having three independent mechanisms of action, which allow for retention of its antibacterial activity in both susceptible and resistant bacteria. Identifying a single molecule able to block several resistance activities is challenging. That challenge could be attenuated by identifying prominent modes of resistance in order to develop specific inhibitors for them.
121
Illustrative to that approach is the success of the serine β-lactamase inhibitors. The first compound of this class to find clinical use in combination with amoxicillin (Augmentin) or ticarcillin (Timentin), clavulanic acid,
122
is produced by Streptomyces clavuligerus.
123
Clavulanic acid was discovered in 1976 by Beecham. Since then, different types of β-lactamase inhibitors have been explored, which led to the clinical implementation of penicillanic acid sulfones, sulbactam and tazobactam, in combination with various penicillins. Continued interest in β-lactams as antimicrobials is due to their low toxicity in humans and the good pharmacokinetics/pharmacodynamics profiles they enjoy. That is confirmed by the FDA approval of three new combinations of β-lactam (two representatives of the cephalosporins and one meropenem with intrinsic antibacterial activity)/β-lactamase inhibitor for the past 5 y, as opposed to one cephalosporin in the past 8 y. β-lactams with intrinsic activity of the β-lactam/β-lactamase combinations consist of a cephalosposin, Zerbaxa (2014), Avycaz (2015), and one of meropenem (2017). The non–β-lactam diazabicyclooctane (DBO) avibactam (
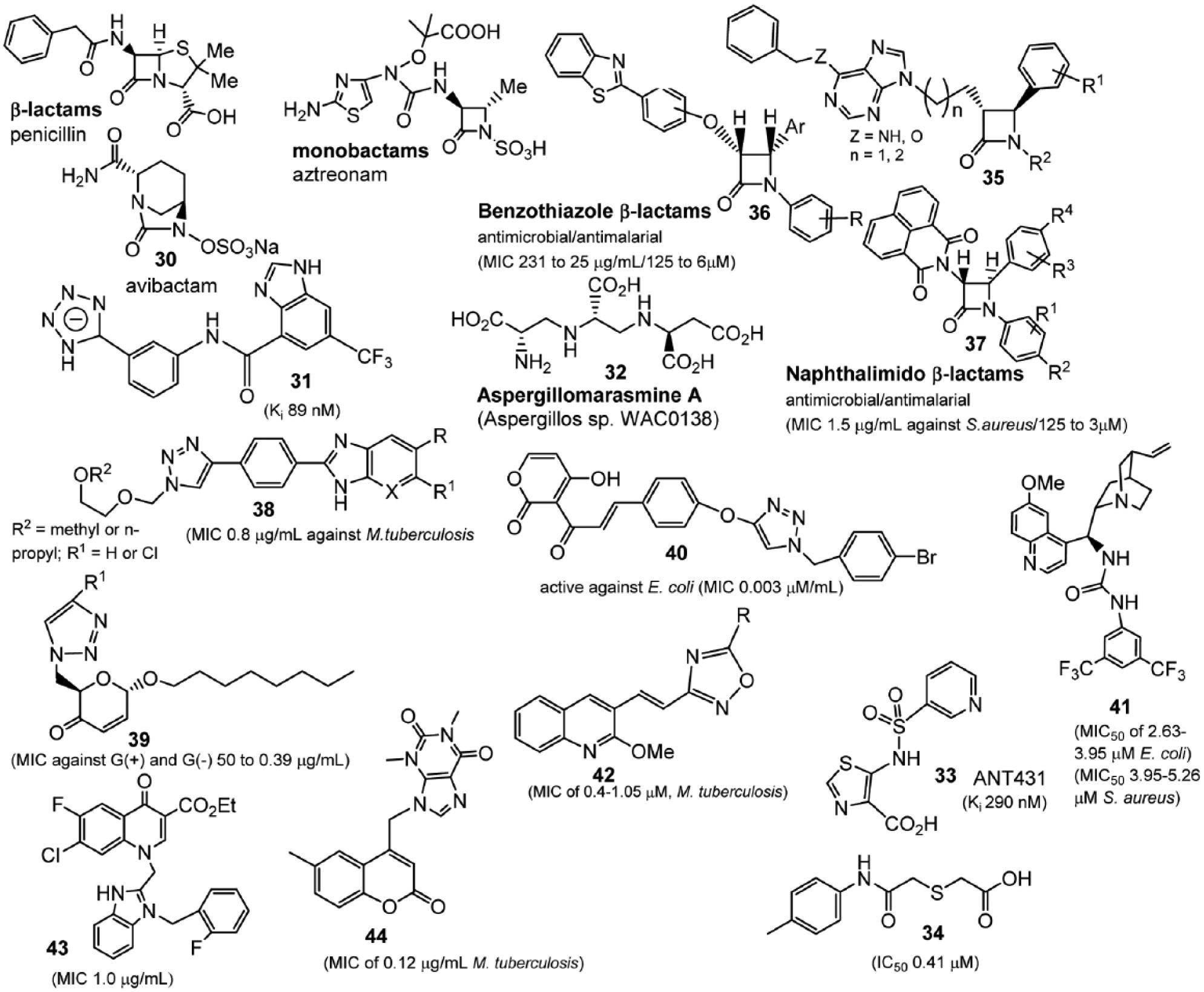
Examples of β-lactamase inhibitors (compounds
Although the attenuation of resistance to serine β-lactamases has continued for more than 30 y of clinical implementation, identification of a clinical candidate to the metallo-β-lactamases (MBLs) has been challenging. MBL inhibitors with good in vitro activity have been reported125–131 but have not demonstrated efficacy in animal infection models. An exception to this is the natural product aspergillomarasmine A (WAC0138,
Molecular Hybridization
Molecular hybridization is the chemical fusion of multiple pharmacophores via covalent bonds into a single molecule. The main reason for implementing chemical hybridization, as opposed to combining biologically active molecules, is associated with efforts to achieve a benefit in terms of therapeutic potential, potency, mode of action, pharmacokinetics, and so forth.141,142
An important incentive for applying the molecular hybridization approach is the delay of antibiotic resistance. Undoubtedly, there will always be an element of uncertainty as to whether combinations of different entities may alter the effectiveness of individual antibiotics. However, this strategy holds the promise of achieving structural novelty, which is a great challenge for medicinal chemistry and an even greater challenge for the design of antimicrobial compounds. 142 Several examples of hybrids141–148 as antibacterials are discussed briefly below.
The aziridine-based hybrid molecules (
β-lactams/benzothiazole hybrids
146
such as
Earlier examples using phenothiazine and 1,3,4-thiadiazole hybrids have demonstrated anti-Mtb activity with an MIC of 0.8 µg/mL of
Triazole hybrids have demonstrated promising antibacterial activity. Sugar-triazole hybrids show a broad-spectrum antibacterial (in addition to antifungal) activity. Compound
Dual anti-Mtb activity (growth inhibition and efflux pump inhibition) and a synergistic effect with antitubercular agents of hybrid triazoles has been reported, 149 with an MIC of 4 µg/mL and 8 µg/ mL, respectively.
The dehydroacetic acid-chalcone-1,2,3-triazole hybrids have demonstrated an enhanced antimicrobial activity as compared with the dehydroacetic acid alone.
150
Several compounds (
As a continuation of earlier studies on quinolone-containing hybrids with activity against both gram-positive and gram-negative organisms,
151
quinolone-thiourea,
152
quinoline-oxadiazole,
153
and quinolone-benzimidazole
154
hybrids have been prepared. In the former series, MICs of
The quinolone-oxadiazole hybrids (e.g., compound
Benzimidazole-quinoline hybrids (e.g.,
Anti-Mtb activity (MIC of 0.12 µg/mL) has been also reported for coumarin-theophylline hybrids (
New Targets Inspired by Natural Product Leads
All of the clinically relevant β-lactam antibiotics, including those recently approved by the FDA, contain an ionizable group either in the proximity (carboxylic acid, bicyclic, penicillin-like structures) or on (sulfonic acid, monobactams) the lactam nitrogen of the β-lactam ring (
Fig. 6
).
156
Until recently, it was generally accepted that for β-lactams to exert bactericidal activity, they must contain a scaffold, which specifically has an ionizable group at the lactam nitrogen within 3.6 Å of the β-lactam carbonyl carbon. The aforementioned ionizable group allows for β-lactams to be recognized by transpeptidases. However, there now appear to be exceptions to this scaffold requirement, since N-alkylthiolated β-lactams
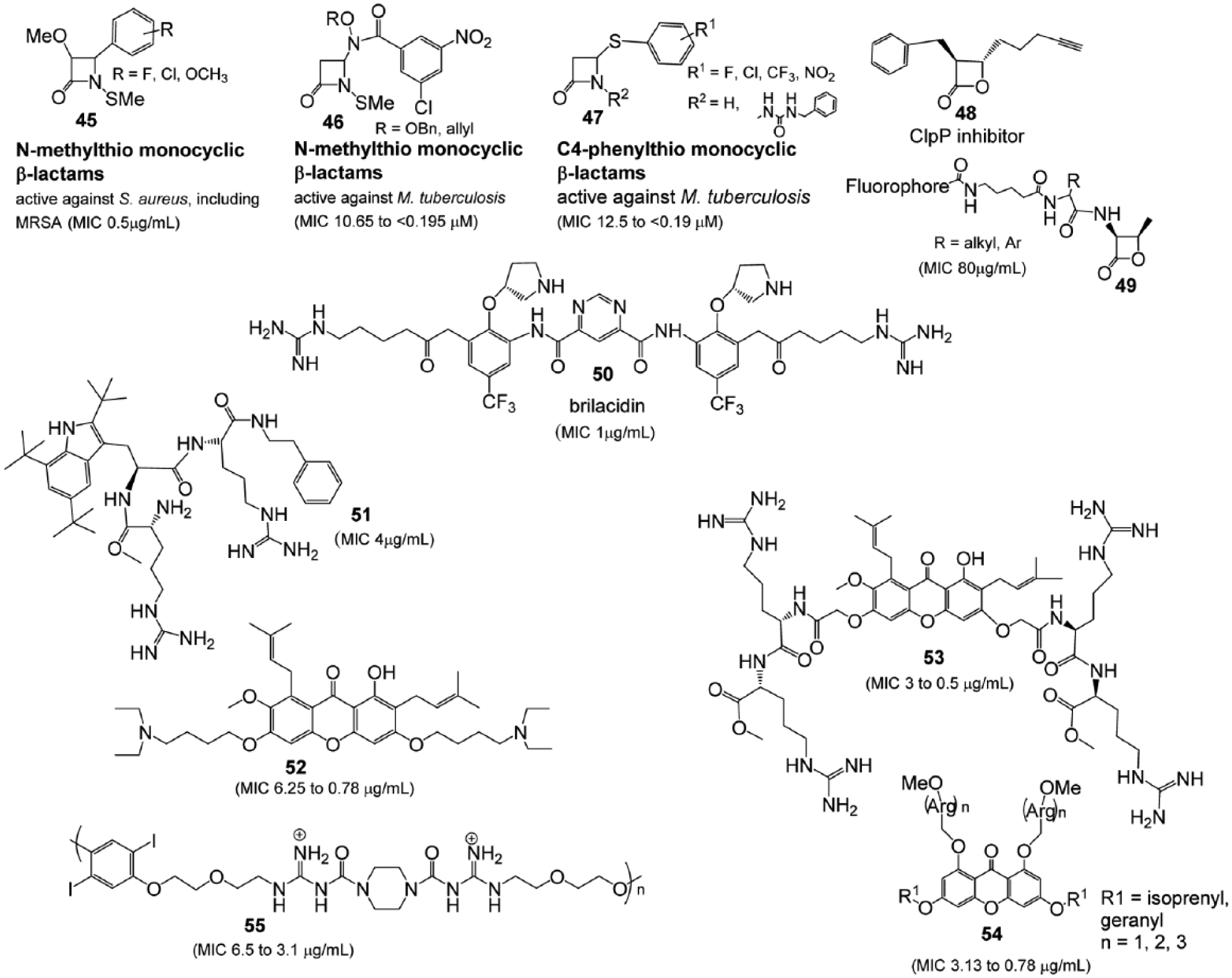
Simplified structures as compared with their naturally occurring counterparts, β-lactam and b-lactones (compounds
Another example that could be considered a design-to-function strategy is the synthetic mimic of first-line host defense mechanisms, such as the antimicrobial peptides (AMPs). These peptides are evolutionary conserved and interact with bacterial membranes, causing cell death. Their main mode of action is to impair the function of the membrane.
172
Most AMPs are amphiphilic, with a positive face interacting with the negatively charged bacterial membrane and a hydrophobic face that allows for insertion into the membrane and interaction with the apolar acyl chains of the bilayer.173–177 Since the membrane is their the primary target, development of resistance is expected to be limited.
178
Several scaffolds, such as D-L peptides, β-amino acid helices, and antimicrobial polymers,179–181 as well as tunable peptide macrocycle assemblies introduced into known AMPs (e.g., buforin II),
182
have been explored to improve bioavailability and proteolytic degradation of the AMPs.
183
Another significant pharmaceutical issue with AMPs is systemic toxicity as well as the difficulty and expense of manufacturing, which have a negative effect on their clinical progress.
183
Because of these limitations, therapeutic development of the peptides has been limited largely to topical or local administrations. One of the clinically advanced (completed phase 1 and two phase 2 clinical trials, with a phase 3 trial currently planned) small mimics of host defense proteins is brilacidin
Another alternative for development of AMPs is to incorporate unnatural functionalities into amino acid side chains to disguise the cationic AMPs.17,25 Amphiphilic AMPs are mimetic with antimicrobial activity against gram-positive bacteria, based on the natural xanthone of α-mangostin and its semisynthesized xanthone-based derivatives, including
Addressing AMR by Other Combinatorial Approaches
Bacteria can adapt to a single antibiotic during treatment, leading to different outcomes: either bacteria become more resistant or more susceptible to other antibiotics. 194 Antibiotic combinations have been used for decades to delay the emergence of resistance. It has been demonstrated that resistance to certain antibiotics in vitro, particularly aminoglycosides, enhances the sensitivity to other antimicrobial agents, as indicated by an increase in the MIC.195,196 However, antibiotic combinations may also promote the evolution of drug resistance by increasing the danger of superinfection due to the coevolution of MDR variants and sensitization to other antibiotics. 197 Therefore, careful design of effective antibiotic combinations is an important approach to delaying the AMR. In addition to the aforementioned antibiotic combinations, which directly inhibit resistance (e.g., β-lactam/β-lactamase combinations), there are other antibiotic combinations that attempt to delay it. Some are directed toward inhibition of the bacterial resistome 198 and others to bypass resistance mechanisms. Several examples of the latter strategy are given below. Loperamide (an antidiarrheal drug, nonantibiotic) enhances the activity of tetracycline by increasing its intracellular concentration. 199 Tetracyclines require a pH gradient to cross the gram-negative outer membrane. Loperamide increases the DpH gradient, which in turn leads to increased uptake of tetracyclines. 199
The flavonoid gallic acid enhances the activity of halogenated quinolones by up to 11,000-fold against S. aureus through an unknown mechanism. 200 Synergistic with kanamycin are synthetic diamines, which, analogously to the aforementioned AMPs, depolarize the cytoplasmic membrane and increase the permeability of the bacterial outer membrane. These diamines are bactericidal against both gram-positive and gram-negative bacteria. 201
Thioridazine in combination with antibiotics to which Mtb isolate was previously resistant for therapy of MDR, extensive drug resistance, and most likely, totally drug resistance, leads to potentiation of the Mtb antibiotic therapy. 202 Thioridazine and methylene blue belong to the phenothiazine class of compounds. Thioridazine is therapeutically safe, and similarly to chlorpromazine, it has been in use for more than six decades to control psychosis. Shortly after the synthesis on methylene blue (1883), Paul Erlich used it for staining live cells and found that it could reduce movement of microorganisms. 203 Thioridazine has an MIC of 8 to 15 μg/mL against Mtb (H37Rv, susceptible strain). Most importantly, it can totally reverse the resistance of Mtb toward isoniazid, 204 mainly but not limited to interference with the overexpressed efflux pump genes of resistant strains. 202
A recent profiling of close to 3000 dose-resolved combinations of antibiotics, human-targeted drugs, and food additives in six strains from gram-negative pathogens (E. coli, Salmonella enterica serovar Typhimurium, and P. aeruginosa) has been accomplished. 205 The goal of this systematic study was to identify general principles for antibacterial drug combinations and understand their potential. More than 70% of detected drug-drug interactions are species specific, and 20% display strain specificity, leading to the conclusion that there is a potential for narrow-spectrum therapies. Most importantly, this study shows that synergies are more preserved and are enhanced in drugs that target the same process. 205 This study has demonstrated that nonantibiotic drugs hold promise as adjuvants, even against MDR clinical isolates. Whether such synergies would have clinical relevance remains to be determined.
Conclusion
The discovery of scaffolds best fitted for antibacterial drug development is challenging. Differences in the requirements for drug development for gram-negative versus gram-positive increases the challenges innate to any drug discovery effort, especially for broad-spectrum antibacterials. The AMR complicates the drug discovery process even further. That alone has reduced the enthusiasm for antibiotic drug development in the past decades. However, there appears to be a resurgence of antibacterial efforts. New antibiotic scaffolds, such as the ones described here, are being synthesized, as well as discovered. More than a century has passed since Paul Ehrlich introduced the discovery of a magic bullet, able to destroy tumor cells and microorganisms, with the help of organic chemistry. Making as many as possible derivatives of a given compound might have a somewhat different interpretation today. There are limited time and resources to synthesize every possible compound. Synthetic chemistry is a prerequisite of antimicrobial research. It has and will always play a key role in this area. The development of new synthetic strategies allows for efficacious preparation of new antibiotics. The existence of an expeditious feedback mechanism between the preparation of a given synthetic organic entity and changes in cell morphology/gene expression that this synthetic product could trigger becomes imperative for future success of the synthetic efforts of antimicrobials. This rapid feedback mechanism between chemistry and biology might allow for delaying of AMR for as long as possible.
Footnotes
Acknowledgements
The author sincerely thanks Drs. Peter Simpson and Robert Campbell for helpful discussion.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by American University infrastructure support from funds provided by the Mellon Fund.