
Abstract
Eleven-nineteen leukemia (ENL) contains an epigenetic reader domain (YEATS domain) that recognizes lysine acylation on histone 3 and facilitates transcription initiation and elongation through its interactions with the super elongation complex (SEC) and the histone methyl transferase DOT1L. Although it has been known for its role as a fusion protein in mixed lineage leukemia (MLL), overexpression of native ENL, and thus dysregulation of downstream genes in acute myeloid leukemia (AML), has recently been implicated as a driver of disease that is reliant on the epigenetic reader activity of the YEATS domain. We developed a peptide displacement assay (histone 3 tail with acylated lysine) and screened a small-molecule library totaling more than 24,000 compounds for their propensity to disrupt the YEATS domain–histone peptide binding. Among these, we identified a first-in-class dual inhibitor of ENL (Kd = 745 ± 45 nM) and its paralog AF9 (Kd = 523 ± 53 nM) and performed “SAR by catalog” with the aim of starting the development of a chemical probe for ENL.
Introduction
The YEATS domain (YAF9, ENL, AF9, TAF41, SAS5) containing paralogs ENL and AF9, also known as MLLT1 and MLLT3, respectively, has long been known for its role in mixed lineage leukemia (MLL) initiation and progression as a fusion partner of MLL. The interaction of the ANC1 homology domain (AHD) with DOT1L drives the overexpression of DNA binding and HOX proteins, and the overexpression of RNA binding and ribosomal synthesis genes via the interaction of ENL/AF9 with p-TEFb.1–4
Native ENL and AF9 are epigenetic reader and scaffold proteins in the super elongation complex (SEC) and partners of the histone methyl transferase DOT1L, and are thus involved in transcription activation and elongation. 5 Even though ENL and AF9 have a high grade of sequence identity (74%), they only share 40% of target genes and show a stark difference in chromatin location. ChIPseq experiments have shown 78.2% of total ENL to be located in promoter regions, compared with only 21.3% of total AF9.5,6
In recent years, several studies have shone a light on the role of functional human YEATS domains in cancer. Native ENL appears to be required for the maintenance of MLL-rearranged acute myeloid leukemia (AML). CRISPR-Cas9-mediated knockout of ENL,6,7 as well as targeted degradation of ENL 7 via the dTAG system, 8 showed a dependence of AML cell lines on the ability of ENL to recognize histone acylation for disease maintenance. It has also been shown that preventing native ENL from recognizing, or lowering its affinity for, acylated histone marks is also responsible for leukogenesis. 9 Impaired binding to histone peptides is also linked to the development of Wilms tumors, a type of infant renal neoplasm. 10
While AF9 has been shown to play a role in embryonal development11,12 and, like ENL, does bind to CBX8 (a component of the Polycomb-group repressive complex 1), 13 no compelling biology for the YEATS domain of AF9 in leukemia has emerged to date.
In order to investigate the druggability of the ENL/AF9 YEATS domain and identify starting points for drug discovery, we developed a peptide displacement assay and screened the Structural Genomics Consortium (SGC) Oxford and Ontario Institute for Cancer Research (OICR) diversity set of compounds. Several genuine inhibitors were identified and followed up with purchased analogs to investigate SAR. Here, we report the identification of a first-in-class inhibitor of ENL/AF9 with submicromolar potency in peptide displacement assays as well as biophysical methods.
Materials and Methods
Protein Expression and Purification
Sequences for the wild-type YEATS domains of ENL, AF9, GAS41, and YEATS2 were cloned into expression vectors (
Cells were harvested by centrifugation and the cell pellet was resuspended in cold binding buffer (50 mM Tris at pH 7.5, 500 mM NaCl, 20 mM imidazole, 2 mM dithiothreitol [DTT]) on a magnetic stirring plate at 4 °C. The suspension was lysed using an EmulsiFlex-C5 (Avestin, Ottawa, Canada) homogenizer (one pass without pressure, four passes with pressure between 1000 and 1500 bar). The lysate was clarified via centrifugation at 16,000 rpm at 4 °C for 1 h (JLA-16 rotor in Avanti J-26S XP centrifuge; Beckmann Coulter, Atlanta, GA). The supernatant was loaded onto a standard NiNTA column (HisTrap FF, 5 mL, GE Healthcare Lifesciences, Buckinghamshire, UK) on an ÄKTAxpress system (GE Healthcare Lifesciences). After washing with binding buffer, the target protein was eluted at 300 mM imidazole. The eluted peak fractions were pooled and concentrated to 5 mL (Amicon concentrators, 10 kDa molecular weight cutoff [MWCO]) and then separated via size exclusion chromatography (buffered with 20 mM Tris at pH 7.5, 500 mM NaCl, 2 mM DTT) using a GE Superdex 75 column on an ÄKTAxpress system. Protein concentration was quantified via extinction at 280 nM (NanoDrop ND-1000 Spectrophotometer; NanoDrop Technologies, Wilmington, DE) and protein identity was verified via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and liquid chromatography–mass spectrometry (LC/MS). Pooled fractions were then again concentrated to at least 2 mg/mL and stored at −80 °C.
OICR 24K Diversity Set
The OICR 24K Diversity Set was created from the high-throughput screening (HTS) compound collection of 142K compounds. We have applied three sets of filters that we developed previously in BIOVIA Pipeline Pilot: 14
OICR HTS filters: Developed in-house, these were used to eliminate reactive and undesirable compounds (such as acid halides, aldehydes, catechols, epoxides, haloalkyls, and hydrazines) and also compounds with more than one nitro and more than two nitrile groups.
PAINS filters: Pan-assay interference compounds (PAINS) appear as frequent hitters in biochemical assays. Following Jonathan Baell’s work,15,16 we added 480 PAINS SMARTS to our Pipeline Pilot set of filters.
Physical property filters: We have applied filters based on calculated physical properties to eliminate the undesirable compounds as well as compounds that might have issues with human intestinal absorption. These properties included the molecular weight of the parent structure (300 ≤ MW ≤ 550), polar surface area (6 ≤ PSA ≤ 250), calculated 1-octanol-water partition coefficient 17 (–3 ≤ AlogP ≤ 6.5), number of H-bond donors (0 ≤ HBDon ≤ 10), number of H-bond acceptors (1 ≤ HBAcc ≤ 15), number of freely rotatable bonds (Nrot ≤ 16), number of rings ≥ 1, number of aromatic rings ≤ 7, number of atoms carrying a negative formal charge at pH 7 ≤ 2, number of atoms carrying a positive formal charge at pH 7 ≤ 2, zero metal atoms, F atom count ≤ 7, Cl atom count ≤ 4, Br atom count ≤ 1, I atom count ≤ 1, zero B atom count.
After application of the aforementioned filters, we derived a dissimilarity set of 25,000 compounds using the ECFP_4 substructural fingerprints implemented in BIOVIA Pipeline Pilot. 14 These compounds were visually inspected by experienced medicinal chemists and 411 additional compounds were removed. From the remaining set, we derived a diversity set of 24,320 compounds. The resulting OICR 24K diversity set of high-quality compounds was used in this screening campaign.
Peptide Displacement Assay Development
The assay was set up as a competition assay where the acylated histone tail peptide (purchased from LifeTein, Somerset, NJ; Table 1 ) is displaced from the YEATS domain by an active inhibitor. Detection of YEATS-bound peptide is performed using the AlphaScreen Histidine (Nickel Chelate) Detection Kit (PerkinElmer, Waltham, MA, 6760619M), where hexahistidyl-tagged YEATS domains are bound by the Ni2+-chelating AlphaScreen donor beads and the biotinylated peptide is bound by the streptavidin-coated acceptor beads. Upon excitation with light at a wavelength of 680 nm, the donor beads release a singlet oxygen, which triggers the emission of light between 520 and 620 nm by the acceptor beads. The assay is dependent on the proximity of the beads due to the half-life of the singlet oxygen. Disruption of YEATS-peptide binding causes a loss of signal. Plates were read using a Pherastar FSX plate reader (BMG Labtech, Ortenberg, Germany) with the appropriate AlphaScreen optics module. All steps involving the AlphaScreen beads were performed under low light conditions. As AlphaScreen-based assays have previously generated a very high window between no inhibition and full inhibition of binding with steep dose–response curves in bromodomain inhibitor screening campaigns in our lab (data not shown), we opted for this assay technique over other technologies.
Protein–Peptide Combinations Used in the Peptide Displacement Assays.
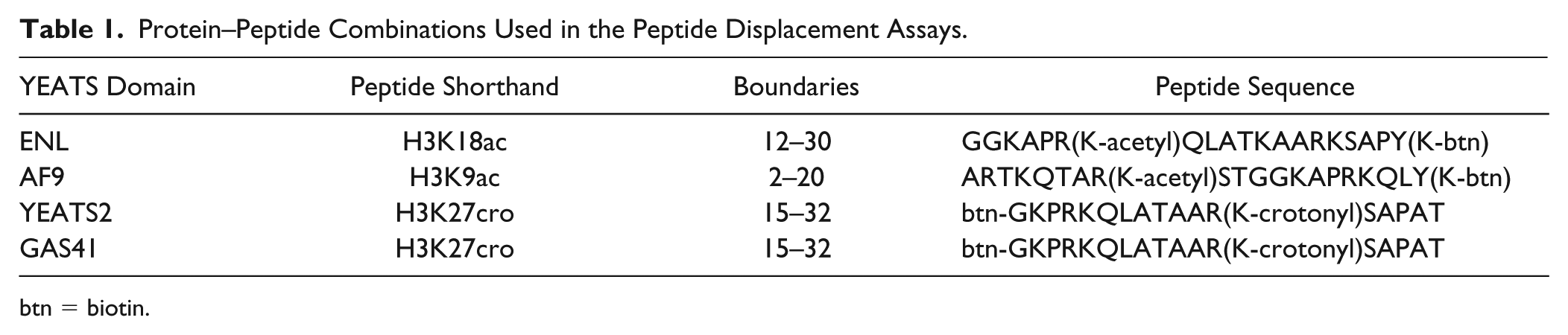
btn = biotin.
Based on recent literature,6,18–20 H3K9x, H3K18x, and H3K27x peptides (where x stands for either acetylation or crotonylation) were tested and the strongest binders chosen for the assay. Assay concentrations of protein and peptide were determined via a titration of YEATS domain against peptide in a 16 × 16 well grid on 384-well ProxiPlates (PerkinElmer). Protein and peptide were each titrated from 3.2 µM to 0.2 nM final assay concentration, covering all possible combinations. AlphaScreen donor–acceptor bead mix was added to a final assay concentration of 3.3 µg/mL. Plates were incubated for 1 h at room temperature (RT) before being read on the plate reader. For the final ratio of protein and peptide to use in the assay, the point representing the EC90 in the two-dimensional titration was chosen. Assay buffer throughout was 25 mM HEPES at pH 7.4, 100 mM NaCl, 0.1% bovine serum albumin (BSA), and 0.05% CHAPS.
Single Shot Screen
Compounds were dispensed into 384-well ProxiPlates (PerkinElmer) for a final assay concentration of 50 µM in two replicates. On each plate, two columns were reserved for controls (DMSO addition, no addition). Protein and detection reagents were added in the concentrations determined above and plates were incubated for 30 min with protein–peptide and compounds alone before the addition of detection reagent. Signals were determined for each replicate and expressed as percent change relative to the mean DMSO control signal.
Determination of IC50 Values
Single shot “hits” to be carried forward for IC50 determination were repurchased and solubilized to 50 mM stocks in DMSO. They were dispensed as an 11-point curve in twofold serial dilution steps (top concentration 100 µM) in duplicate, with the difference in dispensing volume between each step backfilled with DMSO to keep the final DMSO concentration consistent at a maximum of 0.5%. DMSO-only (n = 16) and no-addition (n = 16) reference wells were also added on each plate. The assays were performed as described above, and change in signal was also calculated as described above. The inflection point (IC50) of the dose–response curve was calculated in GraphPad Prism 7 (GraphPad, La Jolla, CA) using a sigmoidal dose–response curve with a variable slope:
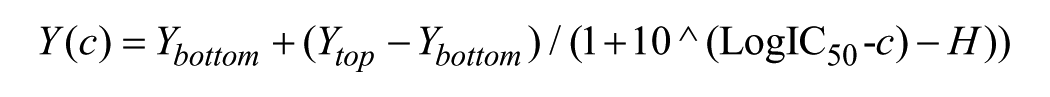
where Y is the percent inhibition; Ytop and Ybottom are the top (total loss of signal) and bottom (no loss of signal) of the curve, respectively; c is the compound concentration; and H is the Hill slope.
At this stage, a counterscreen with a biotinylated hexahistidyl peptide instead of the YEATS domain–histone peptide pair was also performed to eliminate compounds that simply interfere with the assay chemistry.
Thermal Shift Assay
Thermal melting experiments were carried out using a Lightcycler480 (Roche Molecular Systems, Pleasanton, CA), running a temperature gradient from 20 to 95 °C with three acquisitions per cycle (0.19 °C/s). Compounds were dispensed into 384-well PCR plates (20 µL assay volume) at 50 µM in duplicate in three independent experiments. DMSO was backfilled for a final DMSO content of 0.1% (v/v). Measurements from DMSO only and no-addition controls (n = 16 each per assay plate) were also collected. Protein was added at a final concentration of 10 µM buffered with 20 mM HEPES at pH 7.5 and 500 mM NaCl. SYPRO orange (LifeTechnologies, Eugene, OR) was used as fluorescent dye at a 1:1000 dilution from purchased stock solution. The midpoints of the unfolding process were estimated using a sigmoidal Boltzmann equation:
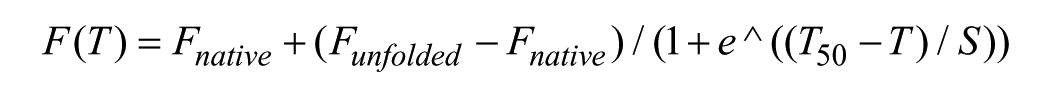
where T50 is the temperature at the midpoint of unfolding, Funfolded and Fnative are the fluorescence intensities of the dye in the presence of native or fully unfolded protein, and S is the slope of the curve.
The shift in unfolding was then calculated as the difference in temperature between the midpoints of unfolding for each compound sample to the mean DMSO reference.
Peptide Affinity
The affinities of peptides for the individual YEATS domains were determined using an Octet RED384 system (FortéBio). Peptides ( Table 1 ) were immobilized onto eight streptavidin-coated sensors each to saturation of the sensor. An additional set of eight “free” reference sensors was used. Proteins were titrated in 1:3 dilutions from 100 µM down to 137 nM (7 data points plus one “blank”) in 25 mM HEPES at pH 7.5, 150 mM NaCl, 0.05% (v/v) Tween-20. Each peptide was dipped into each protein (baseline in buffer 60 s, association in protein 120 s, dissociation in buffer 120 s). Data were analyzed using the FortéBio Data Analysis software (version 9.0) supplied with the instrument. The data from the reference sensograms were subtracted from the peptide-bound sensograms to correct for unspecific binding. Report points for each sensor were exported to GraphPad Prism and fitted to a one-site binding model:
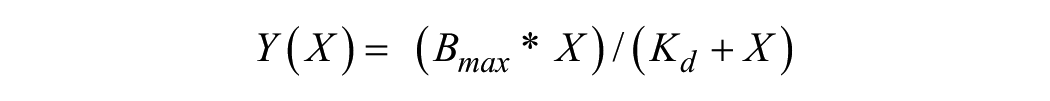
where Y is the response, Bmax is the maximum response, and X is the concentration of protein.
Isothermal Titration Calorimetry
Protein was prepared for isothermal titration calorimetry (ITC) by dialysis against a ~2000 times excess of ITC buffer (20 mM Tris at pH 7.5, 500 mM NaCl, 5% [v/v] glycerol, 2 mM DTT) using SnakeSkin Dialysis Tubing (Thermo Scientific, Waltham, MA) with a 7 kDa MWCO and then concentrated to 300 µM with DMSO added to 0.1% (v/v) to minimize buffer mismatch between cell and syringe. Compounds were diluted to 50 µM in dialysis buffer. ITC was performed on a NanoITC Standard Volume device (TA Instruments, Newcastle, DE) using reverse titration (compound in cell, protein in titration syringe) at 20 °C with an initial injection of 3.7 µL and 30 injections of 7.96 µL at a stir rate of 350 rpm. Data were analyzed with the NanoAnalyze software (TA Instruments) using an independent fit model.
NanoLuciferase Bioluminescent Resonance Energy Transfer Assay
Cellular activity against AF9 was assessed using a NanoLuciferase Bioluminescent Resonance Energy Transfer (NanoBRET) assay. 21 HEK293 cells (8 × 105) were plated in each well of a six-well plate; after 6 h cells were co-transfected with C-terminal HaloTag-histone 3.3 (NM_002107) and an N-terminal NanoLuciferase fusion of AF9 (original AF9 WT sequences from Promega HaloTag human ORF in pFN21A and AF9 MUT [Madison, WI]; Y78A tyrosine is changed to an alanine) at a 1:10 (NanoLuc to HaloTag) ratio, respectively, with FuGENE HD transfection reagent.
Sixteen hours posttransfection, cells were collected, washed with PBS, and exchanged into media containing phenol red-free DMEM and 4% FBS in the absence (control sample) or presence (experimental sample) of 100 nM NanoBRET 618 fluorescent ligand (Promega). Cells were then replated in a 96-well assay white plate (Corning Costar 3917, Corning, NY) at 2 × 104 cells/well. Compounds were then added directly to media (in the presence of SAHA 2.5 µM) at final concentrations of 0-–0 μM or an equivalent amount of DMSO as a vehicle control, and the plates were incubated for 24 h at 37 °C in the presence of 5% CO2.
NanoBRET Nano-Glo substrate (Promega) was added to both control and experimental samples at a final concentration of 10 µM. Readings were performed within 10 min using a ClarioSTAR (BMG Labtech) equipped with 460 and 610 nm filters. A corrected BRET ratio was calculated and is defined as the ratio of the emission at 610 nm/460 nm for experimental samples minus the emission at 610 nm/460 nm for control samples (without NanoBRET fluorescent ligand). BRET ratios are expressed as milliBRET units (mBU), where 1 mBU corresponds to the corrected BRET ratio multiplied by 1000.
Results and Discussion
Protein Production
After initially expressing ENL/AF9 and YEATS2 in TEv-cleavable N-terminally hexahistidyl-tagged form, switching to noncleavable C-terminally tagged protein greatly increased the yield for ENL/F9 (~4-fold from ~10 mg/12 L culture), allowing for easier conduction of high-protein-consumption assays such as ITC. No notable change was observed for YEATS2 (yielding ~10 mg/12 L culture), whereas GAS41 could only be expressed in sufficient quantities for any assay in C-terminally tagged form (~50 mg/12 L culture). Differences in behavior in the assays between the N- and C-terminally tagged proteins could not be observed. The affinities of the produced proteins to histone peptides as determined by biolayer interferometry were in good agreement with the literature data obtained by ITC (
Initial Screening Campaign and Biophysical Characterization of Hits
We established assay conditions for all four human YEATS domains, resulting in a robust assay with a substantial assay window and Z′ = 0.94 (
Final Assay Concentrations (FACs) of YEATS Domains and Histone Peptides.

In the first instance, we screened two libraries from the OICR as well as internal libraries of acetyl lysine mimetic compounds against the YEATS domain of ENL at a single concentration (50 µM) and obtained a large number of hits that included a sizable portion of known PAINS. After eliminating compounds that interfere with the assay chemistry, such as obvious metal chelators, we followed up 16 compounds with full dose–response curves for all four YEATS domains to experimentally determine potency and selectivity and obtained IC50 values for 7 compounds (
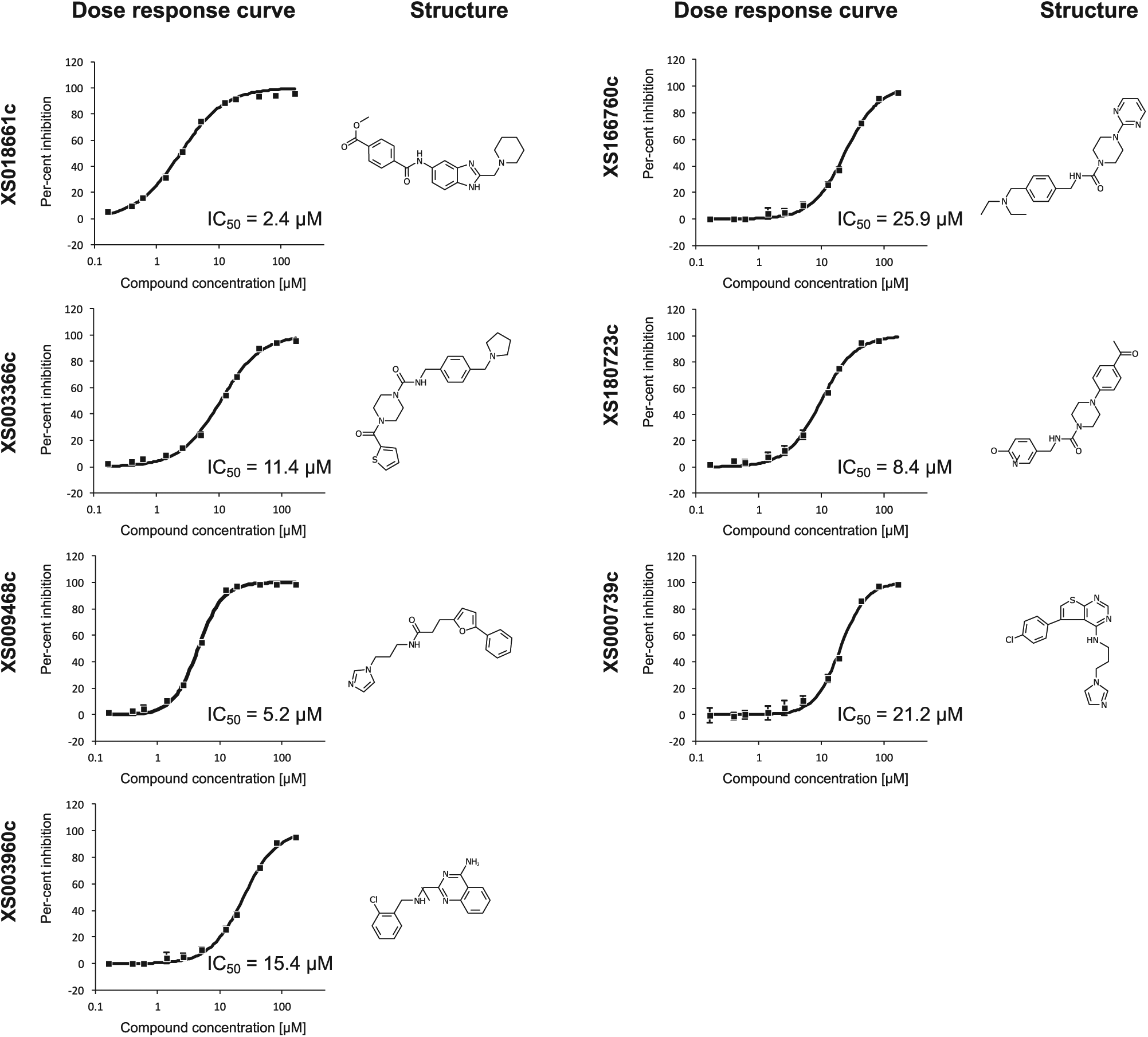
Structures and representative dose–response curves of the initial hits of the single shot campaign against the YEATS domain of ENL.
IC50 (µM) and ΔTm (°C) Values of Original Library Hits Tested.
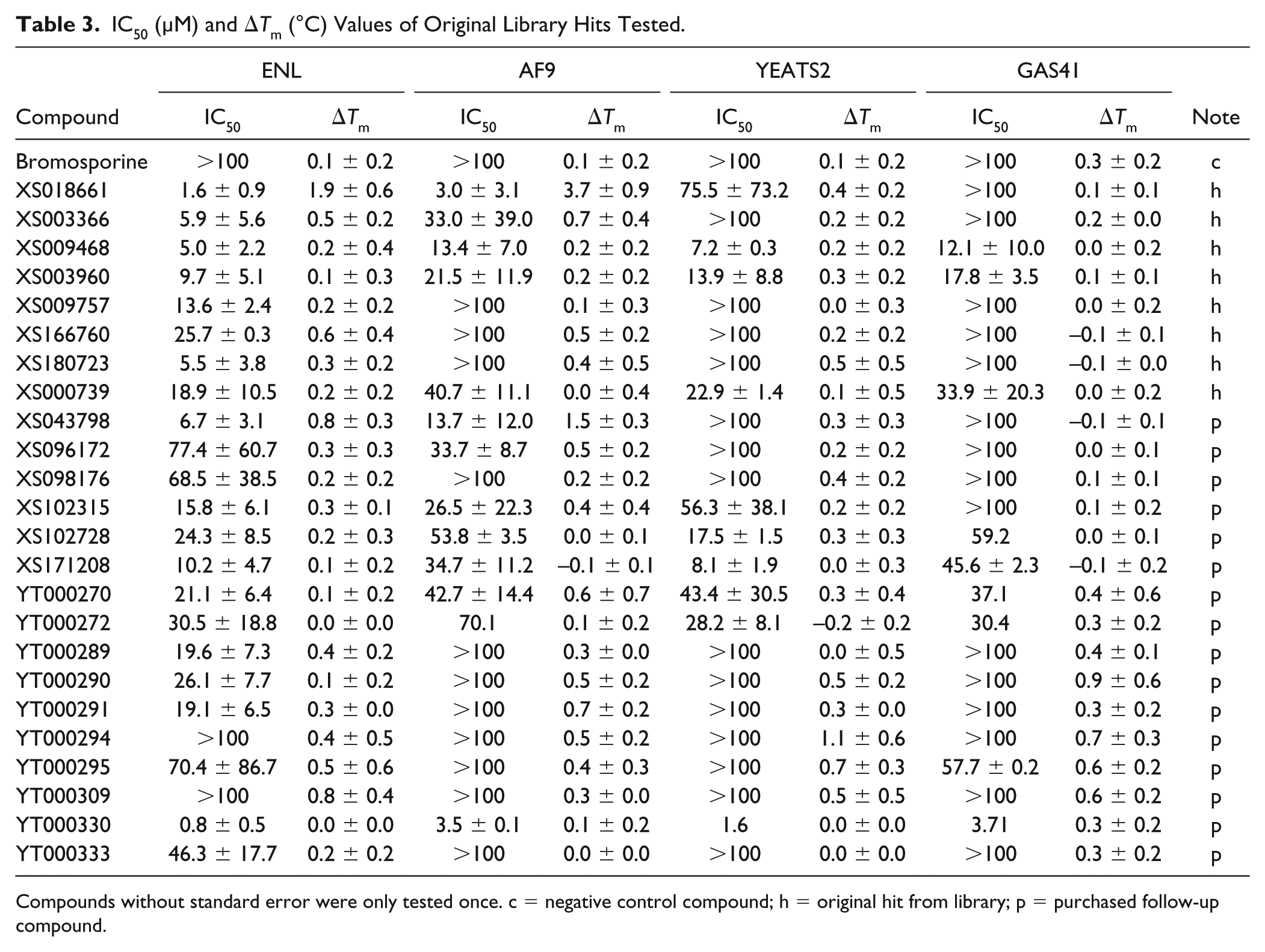
Compounds without standard error were only tested once. c = negative control compound; h = original hit from library; p = purchased follow-up compound.
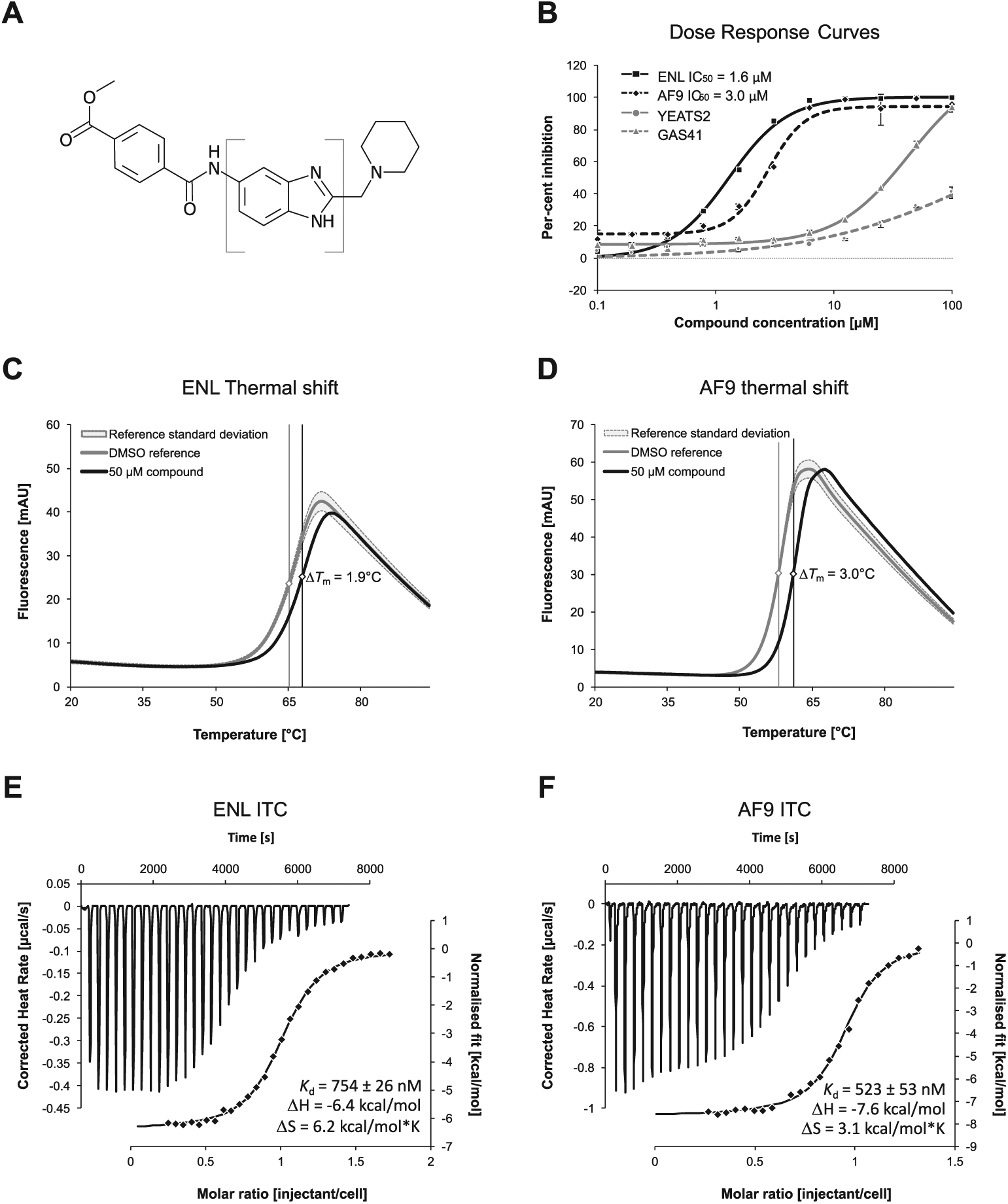
Biophysical characterization of most potent compound. (
For this compound, we then determined a Kd value of 754 ± 26 nM for ENL and Kd value of 523 ± 53 nM for AF9 via ITC in three independent experiments (
SAR by Catalog
We purchased a number of additional compounds to investigate the SAR of the scaffold of XS018661. However, none of the purchased compounds showed inhibition of protein–peptide binding or a shift in thermal unfolding greater than that of the originator (denoted with “p” in
Intracellular Activity
We were able to demonstrate moderate cellular target engagement of XS018661 against AF9 in the micromolar range (Fig. 3). A mutant of AF9 with a deficiency in the binding pocket (Y78A) was not significantly affected by the inhibitor. The effect of SAHA on the binding of histone peptides to YEATS domains was tested in a dose–response curve in the AlphaScreen assay (top concentration 50 µM) and no inhibition was observed.
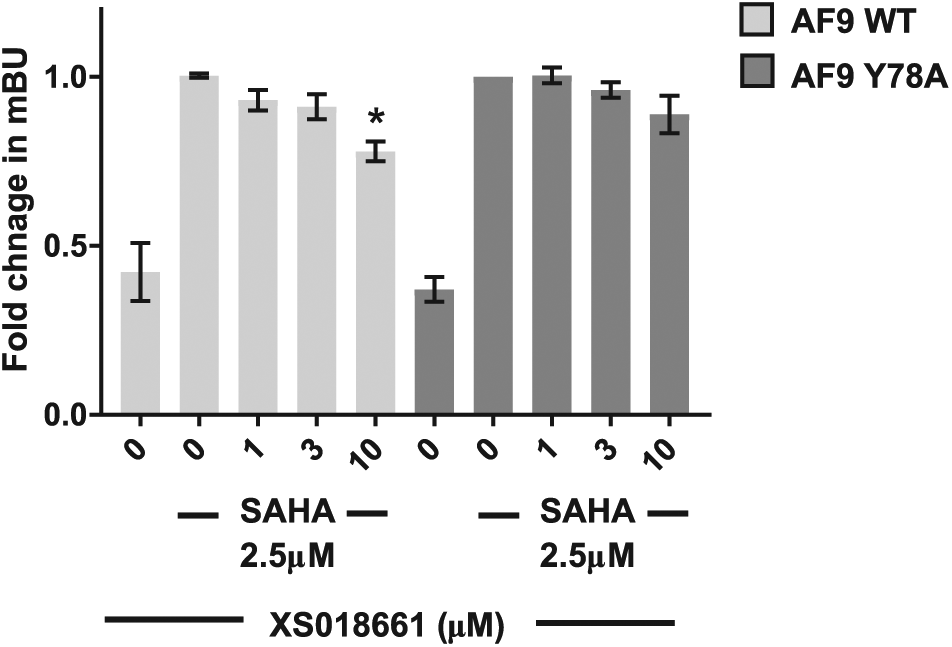
NanoBRET assay with wild-type and mutant AF9. Bar chart represents fold change in mBU NanoBRET showing the displacement of N-terminal nanoLuc-WT AF9 (AF9 WT) or N-terminal nanoLuc-MUT AF9 (AF9 Y78A) from C-terminal HaloTag-histone 3.3 after 24 h treatment with XS018661 (1–10 µM) in the presence of 2.5 µM SAHA. Graph represents n = 3 independent biological replicates, with n = 4 technical replicates. Mean ± SD, mBU – BRET units. One-way ANOVA, AF9 WT, p ≤ 0.003; post hoc Dunnett’s multiple comparisons test (using SAHA 2.5 µM control from AF9 WT or Y78A, respectively), *p = 0.0073; AF9 Y78A, not significantly different.
Summary and Conclusions
Here we report the discovery of a potent first-in-class inhibitor, XS018661, of the YEATS domains of ENL (IC50 = 1.6 ± 0.9 µM, Kd = 754 ± 26 nM) and AF9 (IC50 = 3.0 ± 3.1 µM, Kd = 523 ± 53 nM) through a library screen that already displays moderate cellular activity. The identified scaffold will serve as the template for the development of a chemical probe. Knockout of AF9 has shown no major impact on cell viability and invasiveness in any AML cell line to date.6,7 This, taken together with the lack of biology of AF9 in cancer and the absence of AF9 on promoter regions relative to ENL, should make even a dual inhibitor of ENL and AF9 a useful tool compound. Indeed, CRISPR-Cas9-mediated knockout of AF9, used as a control experiment by Wan et al., 6 showed no adverse effect on cell viability in a number of MLL-rearranged leukemia cell lines as well as HeLa and U2O cell lines.
While the biology of ENL/AF9 as a fusion partner of MLL has been intensively investigated in MLL/AML, there is no consensus about the role of the native protein in leukemia, yet. It has recently been shown that binding of ENL to PAF1 might be responsible for leukemogenesis in non-MLL-ENL-rearranged leukemia, 9 slightly contradicting the evidence for the role of its canonical activity of ENL in the SEC and with DOT1L in AML.6,7 There is also evidence that ENL/AF9 is involved in transcriptional repression in double-strand repair processes. ENL/AF9 is phosphorylated by ATM and can then interact with the polycomb group complex PRC1, whose E3–ubiquitin ligase RING1B ubiquitinates H2A, thereby stalling transcription until the double-strand break has been repaired. 24 Based on this information, one could conceivably envision that inhibition of the ENL/AF9 function might sensitize cancer cells with impaired double-strand break repair machinery to the conventional chemotherapy. Furthermore, all studies investigating the role of ENL in AML so far have relied on techniques involving the complete abolishment of ENL from the cellular context and/or ectopic expression of native or mutant protein.
In light of this seemingly contradictory background, a potent small-molecule inhibitor will enable probing the biology of ENL and AF9 without perturbing the interaction of the protein with its partners in vivo, other than the recognition of histone marks. The compound we present here will make an excellent starting point for the further development of highly active and selective inhibitors for the YEATS domains of ENL and AF9.
Supplemental Material
Christott_et_al,_Supplementary_Data – Supplemental material for Discovery of a Selective Inhibitor for the YEATS Domains of ENL/AF9
Supplemental material, Christott_et_al,_Supplementary_Data for Discovery of a Selective Inhibitor for the YEATS Domains of ENL/AF9 by Thomas Christott, James Bennett, Carmen Coxon, Octovia Monteiro, Charline Giroud, Viktor Beke, Suet Ling Felce, Vicki Gamble, Carina Gileadi, Gennady Poda, Rima Al-awar, Gillian Farnie and Oleg Fedorov in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The SGC is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, the Canada Foundation for Innovation, the Eshelman Institute for Innovation, Genome Canada, the Innovative Medicines Initiative (EU/EFPIA; ULTRA-DD grant no. 115766), Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, the Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome (106169/ZZ14/Z).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.