
Abstract
The increasing emergence of multidrug-resistant bacteria is recognized as a major threat to human health worldwide. While the use of small molecule antibiotics has enabled many modern medical advances, it has also facilitated the development of resistant organisms. This minireview provides an overview of current small molecule drugs approved by the US Food and Drug Administration (FDA) for use in humans, the unintended consequences of antibiotic use, and the mechanisms that underlie the development of drug resistance. Promising new approaches and strategies to counter antibiotic-resistant bacteria with small molecules are highlighted. However, continued public investment in this area is critical to maintain an edge in our evolutionary “arms race” against antibiotic-resistant microorganisms.
Impact statement
The alarming increase in antibiotic-resistant microorganisms is a rapidly emerging threat to human health throughout the world. Historically, small molecule drugs have played a major role in controlling bacterial infections and they continue to offer tremendous potential in countering resistant organisms. This minireview provides a broad overview of the relevant issues, including the diversity of FDA-approved small molecule drugs and mechanisms of drug resistance, unintended consequences of antibiotic use, the current state of development for small molecule antibacterials and financial challenges that impact progress towards novel therapies. The content will be informative to diverse stakeholders, including clinicians, basic scientists, translational scientists and policy makers, and may be used as a bridge between these key players to advance the development of much-needed therapeutics.
Introduction
Antibiotic-resistant bacteria are a major public health concern, and the treatment of drug-resistant infections is increasingly problematic. More than 150 small molecule drugs have been approved by the US Food and Drug Administration (FDA) over eight decades for the treatment of bacterial infections. In the context of this article, we will define a “small molecule” based on the FDA’s assessment of a drug substance as a “chemical” according to ISO IDMP 11238. 1 Additionally, we include mixtures, as specified by the FDA, provided that each component of the mixture is classified as a “chemical”. Both the misuse and overuse of antibiotics have been cited as the major driving forces for the development of resistance among human pathogens. Globalization and international travel increases the likelihood that an easily-transmissible antibiotic-resistant infection could result in a pandemic. The lack of effective antibiotic therapies jeopardizes many modern medical practices including transplantation, cancer chemotherapy, surgeries, and care of the critically ill or premature infants. 2 Understanding the mechanisms of antibiotic resistance is critical for developing new and innovative approaches for better and more effective treatments.3–5 This article provides some context and reviews the recent literature describing the development of small molecule therapies to circumvent antibiotic resistance.
US Food and Drug Administration-approved small molecule antibacterial drugs and their targets
Many antibacterial drugs are natural products that were isolated from microbes in the soil. 6 Therefore, elucidating the antimicrobial mechanisms of action has been a subject of intense interest, and numerous investigations have led to the categorization of these drugs by their effects on the bacterial cell. The vast majority of antibiotics target the disruption of either the bacterial cell wall or membrane, or interfere with essential processes involved in metabolism and replication (Figure 1). While the mechanisms of action for many antibiotics have been described, there are still classes of antimicrobials for which specific binding sites or target(s) remain unknown. These systems continue to be scrutinized for insight into potential areas of exploitation for promising drugs. Studies suggest that the activities of antibacterial drugs are extended beyond the initial drug/target interaction to metabolic and homeostatic networks so that the efficacy of a drug depends on its mode of action in a given cellular context. 7 Table 1 lists the small molecule antibiotics approved by the FDA along with the approval years. Figure 2 shows the number of small molecule antibacterial drugs approved for human use between 1939 and 2017 grouped according to the antibiotic class.
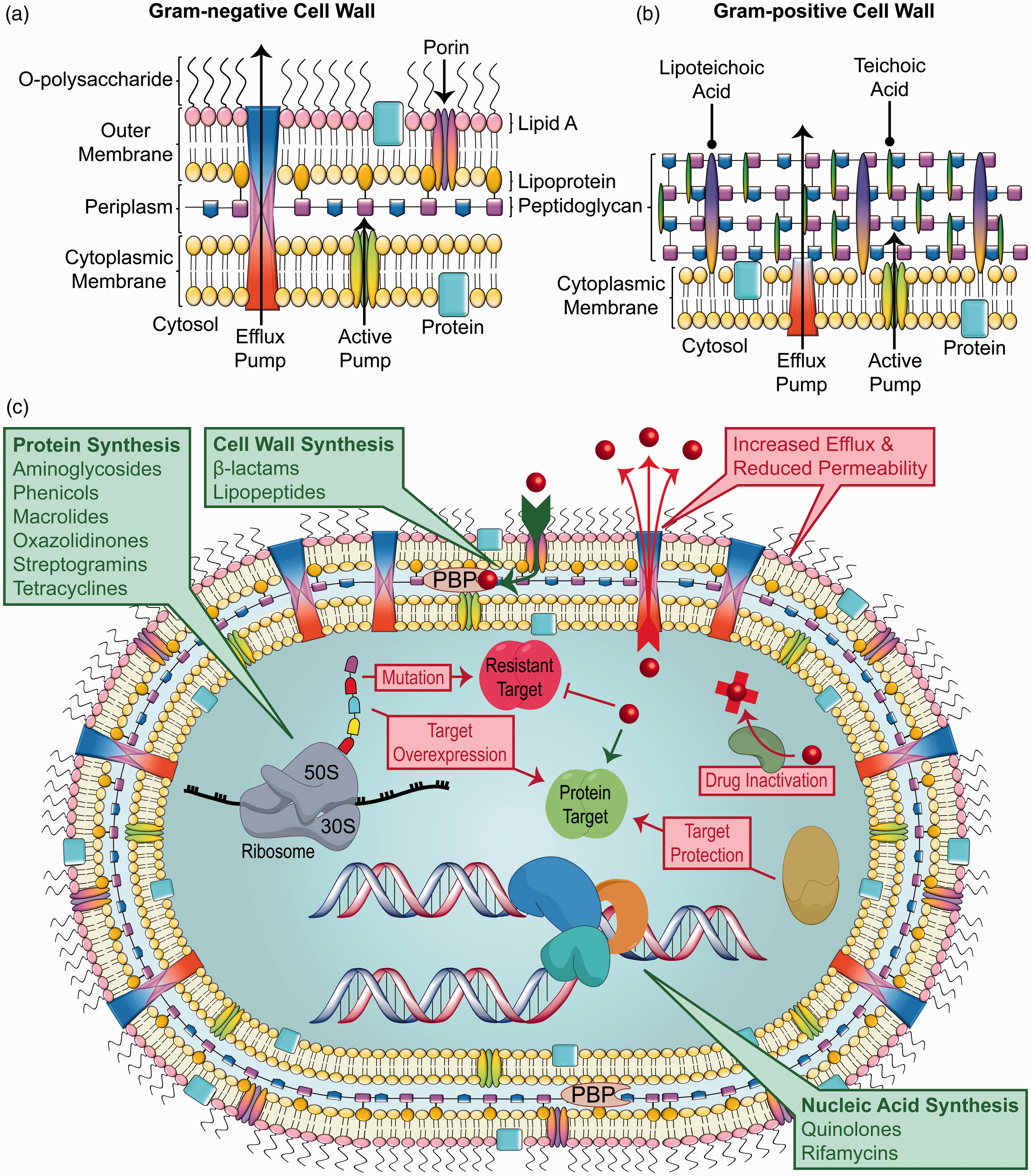
Anatomy of Gram-negative and Gram-positive bacteria and their susceptibility to antibiotics. (a) The cell wall of Gram-negative bacteria includes outer and inner (cytoplasmic) membranes, which form the barriers of the periplasmic space that contains a thin layer of peptidoglycan. The outer leaflet of the outer membrane contains lipopolysaccharide (LPS) consisting of lipid A and O-polysaccharide. The outer membrane contains porins, which provide entrance to the periplasm for some molecules. (b) The outer wall of Gram-positive bacteria contains a thick layer of peptidoglycan that retains the crystal violet stain used in the Gram stain test. Teichoic acids, which are not found in Gram-negative bacteria, are called lipoteichoic acids (LTA) when anchored to the cytoplasmic membrane. Gram-positive bacteria lack an outer membrane and thus are more susceptible to antibiotics. Both Gram-negative and Gram-positive cell walls contain efflux pumps and other active pumps that can export intracellular molecules, including antibiotics. (c) Gram-negative bacteria are intrinsically resistant to many antibiotics due to the architecture of their cell walls. Nevertheless, a variety of antibiotics are effective by targeting the synthesis of protein, the cell wall or nucleic acids. Resistance to antibiotics can be mediated by increased efflux by pumps and reduced permeability through porins in addition to other mechanisms, including target mutation, overexpression or protection and drug inactivation.
Small molecule antibacterials approved by the US Food and Drug Administration (FDA) for use in humans.a
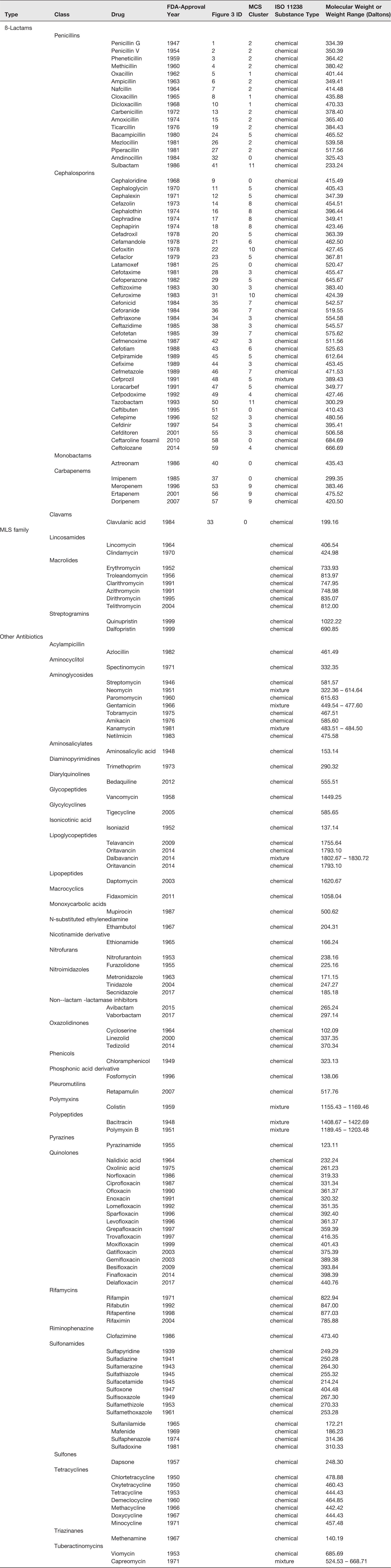
aThe table is organized according to antibiotic type, class, drug name, FDA-approval year, identification number in Figure 3 and MCS cluster designation (β-lactam drugs only), ISO 11238 substance type, and molecular weight or weight range (for mixtures). The maximal common substructure (MSC) clustering analysis was performed with β-lactam drugs as described in the Figure 3 legend and all β-lactams were assigned as either singletons (designated as zero) or one of eleven MCS clusters as indicated in the table. The list of antibacterial drugs was constructed from the NCATS Inxight Drugs database (https://drugs.ncats.io) and eMedExpert (http://www.emedexpert.com/lists/antibiotics.shtml#1) followed by manual curation. Both FDA drug approval databases, Drugs@FDA and the FDA Orange Book, were used to confirm each approval status and date. Effort was made to exclude salts, esters and known pro-drug variants of other approved antibiotic substances. For any active moiety not associated with a definite approval date from FDA's databases, the approval date from the NCATS Inxight Drugs resource was used.
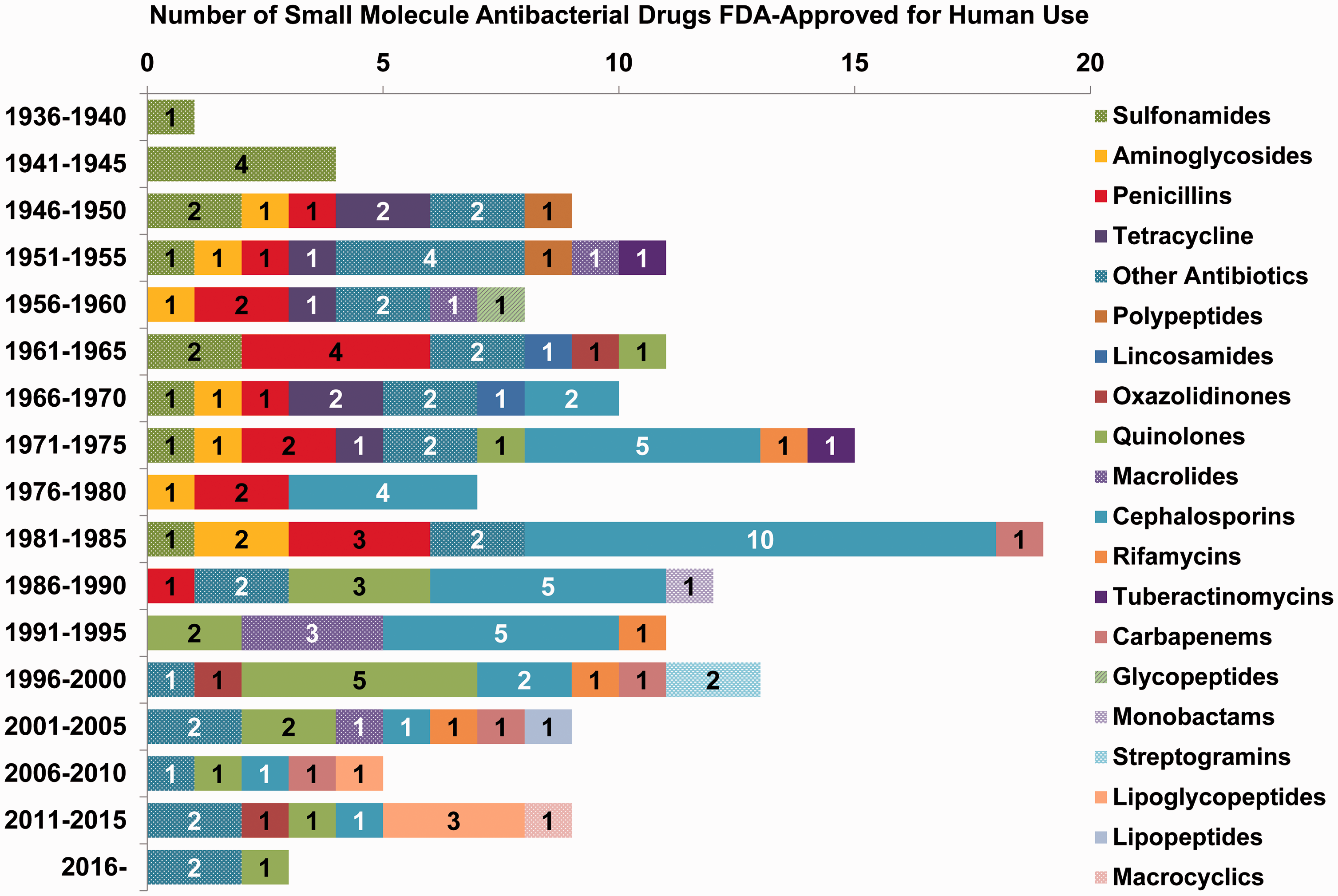
Small molecule antibacterial drugs approved by the US Food and Drug Administration (FDA) for use in humans. This plot shows the number of drugs approved within five-year periods from 1939 until 2017 (all drugs are listed in Table 1). The 157 drugs are grouped according to 20 classifications as shown by the legend. The greatest number of drugs were approved between 1981 and 1985, consisting of 19 antibacterials that span more than six classifications. During the past three decades, the number of approved small molecule antibacterial drugs has steadily decreased.
Cell wall synthesis
Peptidoglycan provides the bacterial cell with its fundamental structural integrity and essential reactions can be inhibited when antibiotics bind to penicillin binding proteins (PBPs) that are crucial for forming peptidoglycan. Upon the arrest of peptidoglycan production, the cell wall loses its integrity, causing lysis of the bacterium. 8 Antibiotics that include the β-lactams and glycopeptides are effective against bacteria due to their ability to interfere with bacterial peptidoglycan synthesis. 9 Subclasses of the β-lactams include penicillins, cephalosporins, monobactams, carbapenems, and clavams. Vancomycin is a glycopeptide class antibiotic that has been FDA-approved. Peptidoglycan synthesis is also inhibited by the oxazolidinone class antibiotic cycloserine and the polypeptide class antibiotic bacitracin.
Cell membrane function
The lipopeptide class antibioticdaptomycin targets the cell wall in Gram-positive microorganisms. While the exact mechanism responsible for the membrane disruption remains unknown, it has been shown that the presence of both phosphatidylglycerol (PG) and calcium are necessary for daptomycin to be effective. Comparisons among membranes that contain PG and those made entirely of phosphatidylcholine have shown that daptomycin is less effective on membranes without PG. Calcium levels are also important, as the concentration of daptomycin needed to penetrate the cell membrane decreases by as much as 50-fold in the presence of calcium ions. 10 The polypeptide class antibiotic polymyxin B is active against Gram-negative organisms, destabilizing both the outer and inner membranes. 11
Protein synthesis
Several classes of antibiotics target ribosomes in order to disrupt protein synthesis in bacteria. These classes of antibiotics include the phenicols, macrolides, lincosamides, and streptogramins which bind to the 50S ribosomal subunit and hinder translation. It is thought that the structures of lincosamides might resemble the 3ʹ-ends of L-Pro-Met-tRNA and deacylated-tRNA, allowing them to bind to and subsequently inhibit the ribosome. 12 The oxazolidinone class antibiotics linezolid and tedizolid also inhibit protein synthesis by binding the 50S ribosomal subunit.
In the bacterial ribosome, the 30S subunit is a target for other classes of antibiotics such as the tetracyclines and aminoglycosides. Examples of aminoglycosides include kanamycin, gentamicin, neomycin, and streptomycin. These drugs bind to the 16S ribosomal RNA within the 30S subunit and do not allow the binding of charged aminoacyl-tRNA to the ribosome, thereby arresting protein synthesis. While the exact site of tetracycline binding has yet to be identified, a recent study suggested that the ribosomal RNA structure is more important than the actual ribonucleotide sequence for binding, as tetracyclines can bind to random sequences of double-stranded RNA. 13
Nucleic acid synthesis
Bacterial enzymes are targets of antibiotics, including those that play roles in nucleic acid synthesis, DNA replication and regulation. Sulfonamide drugs were the first class to be FDA-approved beginning in 1939 and act as competitive inhibitors of the enzyme dihydropteroate synthase, which is required for the synthesis of tetrahydrofolic acid and nucleotides. The diaminopyrimidine trimethoprim targets the same pathway by inhibiting dihydrofolate reductase. Enzymes like DNA gyrase, type IIA topoisomerases, and topoisomerase IV serve as targets of the quinolone class of antibiotics. Topoisomerases form covalent bonds with DNA and catalyze DNA breakage and re-ligation. The quinolones inhibit these topoisomerases, resulting in the formation of ternary complexes that cause strand breakage and block DNA re-ligation. Quinolones were also shown to lower the threshold of magnesium ions required for topoisomerases to initiate strand breakage, allowing for increased rates of cleavage and the formation of ternary complexes. 14 The rifamycin class of antibiotics inhibit RNA polymerase and prevent RNA synthesis. 15
Recent FDA approvals
There were no new approvals of small molecule antibacterial drugs by the FDA in 2016; however, three were approved in 2017 as of 10 October 2017. Secnidazole (Symbiomix Therapeutics, approved 15 September 2017) is a nitroimidazole antibiotic reported as early as 1976, but only now approved for bacterial vaginosis in adult women. 16 Also, a combination of the broad-spectrum β-lactam antibiotic meropenem with the new β-lactamase inhibitor vaborbactam (trade name Vabomere, Rempex Pharmaceuticals, approved 29 August 2017) was approved as an injection formulation for treating complicated urinary tract infections. Vaborbactam is a new chemotype containing a boronic acid that covalently inhibits serine-containing carbapenemases at the active site. 17 While vaborbactam inhibits by a new mechanism of action, it does not act against a new target class and it is an adjuvant to the antibiotic. Finally, delafloxacin is a fluoroquinoline (Melinta Therapeutics, approved 19 June 2017) used to treat patients with acute bacterial skin and skin structure infections (ABSSSI). 18
Unintended consequences of antibiotic use
Alexander Fleming’s discovery of penicillin in 1928 and its release for use in 1942 marked the era of antibiotics and the foundation for the treatment of diseases that had previously been lethal, such as staphylococcal and streptococcal infections. 19 However, over the last several decades we have seen an overwhelming increase in resistance to various antimicrobials. Further, there are a number of undesirable outcomes that occur during and after antibiotic treatment that can exacerbate the duration and severity of infectious diseases.
Alteration of the microbiome
The human microbiome plays a vital role in host health. It is comprised of trillions of microbial cells encompassing as many as 500 species,20,21 maintains a crucial symbiotic relationship with the host and plays a fundamental role in educating the immune system. 22 The components of one’s own microbiota are unique enough to be used for self-identification, and success in using the community structure of the human gut to distinguish individuals in a group has been demonstrated. 23 The commensal bacteria of the human microbiota maintain over two orders of magnitude more genes than their host, 24 including those that encode enzymes that facilitate host digestion, lipid absorption via bile acid conversion, vitamin production as well as the protection and maintenance of the intestinal mucosa.25–28 Along with providing essential functions to the host, commensals also compete with pathogenic microorganisms for nutrition and attachment sites and therefore help prevent infection. Bohnhoff and Miller first reported in 1962 what is now known as “colonization resistance” by determining that mice were significantly more susceptible to Salmonella infection after treatment with streptomycin, which decreased the normal gut microbiota. 29 Antibiotic treatment eliminates many commensal organisms in the gut, enabling enteric pathogens such as Clostridium difficile, vancomycin-resistant Enterococcus faecalis, and Salmonella enterica serovar Typhimurium to colonize and infect the host.30–32 Antibiotic-mediated disruption of the normal microbiota has also been implicated in liver disease and colorectal cancers.33–35 Further, these changes in the gut flora can lead to significantly elevated levels of resistance genes in the microbial population, the increased carriage of which might last for many years following a single course of antibiotic treatment. 36 Changes in the microbiota of the upper respiratory tract, skin and genitourinary tract can likewise affect susceptibility to infections. A recent systematic review of the potential for exposure to antibiotics to increase the risk of community-acquired infections found that the collateral damage to the patient’s microbiome could be associated with upper respiratory, urinary tract and soft tissue infections as well as infectious diarrhea, and that this association decreased with increasing time since antibiotic exposure. 37
Activation of toxin–antitoxin systems
Another consequence of antibiotic use is the induction of growth arrest in bacteria subsequent to the activation of toxin–antitoxin (TA) modules. Five unique TA families have been identified (I-V); the toxin is always a protein but the antitoxin can be a protein or nucleic acid, and this characteristic determines to which family a module belongs. These gene pairs are found in hundreds of bacteria as well as in the archaea. 38 The type II TA systems have protein antitoxins and are the best characterized. Upon translation, the toxin and antitoxin interact to form a non-toxic complex. When bacteria experience stress in their microenvironment, such as exposure to reactive oxidative species, nutrient limitation, temperature, pH or pressure changes and antibiotic therapy, induced proteases such as Lon and ClpXP degrade the labile antitoxin, freeing the more stable toxin to carry out its function.39,40 In the type II TA modules, the toxin is often a ribonuclease enzyme which when released from its cognate antitoxin cleaves bacterial mRNA, eliciting a state of reversible growth arrest. It is thought that many bacterial species in the environment have maintained these systems to enhance their ability to survive nutrient-poor and harsh conditions over hundreds (and, in some cases, thousands) of years. 41 For bacterial pathogens, this state of stasis results in nonspecific tolerance to antimicrobials that target enzymes and pathways essential to growth and replication, allowing a subpopulation to survive antibiotic therapy and resume growth later when conditions improve.
Livestock use generating resistance
Antimicrobial resistance is a problem in veterinary medicine and the inappropriate use of these agents in agriculture continues to have a significant impact on human health. Antibiotics are routinely used in animal production to promote growth by preventing and treating infections. 42 The preventative treatment of entire flocks or herds over time has facilitated the evolution of increased numbers of resistant pathogens. For example, there has been a marked rise in multidrug-resistant (MDR) Escherichia coli, Salmonella, Campylobacter, and methicillin-resistant Staphylococcus aureus (MRSA) emerging from commercial animal production across the globe.43–45 In 2014, approximately 9.7 million kilograms of medically-important antibiotics were sold for use in food-producing animals in the United States (including almost 7 million kilograms of the tetracycline drug class alone), compared with approximately 3.5 million kilograms of antibiotics sold for human use. 46
On large farms with poor living conditions, livestock are treated with high doses of various antibiotics to decrease disease until slaughter. This practice selects for resistant bacteria which can be spread to packaging plants and end up in the kitchens of consumers. 47 Improper cooking and poor hand hygiene can facilitate dissemination, resulting in difficult-to-treat infections. Farm wastewater contaminated with fecal matter harboring resistant pathogens can accumulate as runoff in nearby rivers, streams, and ground water. 48 These resistant pathogens can exchange DNA with the existing organisms of that environment and cause life-threatening bacterial infections in human hosts downstream. 49 Manure from treated animals used for fertilization results in the selection for increased antibiotic resistance in soil microorganisms, which are ultimately transferred to the crop at harvest and then on to consumers.48,50,51
Establishment of environmental reservoirs
In many parts of the world, over-the-counter sales resulting in inappropriate and/or excessive use, payback mechanisms, and lack of oversight plague antibiotic production. 52 A related concern is the potential for the release of drug-containing effluent from pharmaceutical manufacturing centers into the environment, resulting in selection pressure that facilitates the evolution of antibiotic resistance. For example, an LC-MS analysis of samples from 28 different sites around a bulk drug producing area in Hyderabad, South India, resulted in the identification of high concentrations of three antimicrobials (and increased concentrations of eight others) from each site. Further, tandem microbiological studies found the presence of extended-spectrum β-lactamase (ESBL) and carbapenemase-producing Enterobacteriaceae and non-fermenters in 95% of the samples. 53 The release of drugs into the environment has global consequences in terms of increasing the potential for infections by pathogens that are resistant to the very antimicrobials that we are producing on an industrial scale.
Mechanisms of antibiotic resistance
Antibiotic resistance is a natural phenomenon that predates the discovery of antibiotics and the selective pressures of antibiotic use, as genes providing resistance to the β-lactam, tetracycline, and glycopeptide antibiotics have been identified from 30,000-year-old Beringian permafrost sediments. 54 This natural resistance can be a direct result of competition between microorganisms in the environment but can also stem from spontaneous mutations that confer an adaptive benefit. 55 The dissemination of resistance can occur horizontally, carried between species via mobile genetic elements such as prophages, conjugative plasmids, or insertion sequences. 56 It can also be inherited vertically by genetic mutations that are subsequently propagated in each daughter cell. 57 Bacteria can be resistant to certain antibiotics by virtue of their physiology as well. For instance, the low permeability barrier of the Gram-negative outer membrane (Figure 1(a)) serves as the first line of defense and provides intrinsic resistance to many antibiotics. 58 As an example, Gram-negative bacteria are not susceptible to daptomycin, likely due to a lower abundance of anionic phospholipids that are required for the Ca2+-mediated insertion of the lipopeptide antibiotic. 59 Thus, daptomycin is limited to the treatment of Gram-positive organisms (Figure 1(b)). The widespread use of antibiotics is thought to have provided the selective pressure to accelerate development of antibiotic resistance and MDR in previously susceptible clinically-relevant microorganisms. 60 Acquired resistance to essentially all antibiotics can be achieved by genetic mutations, the acquisition of genes or modulation of gene expression, and new resistance mechanisms continue to be characterized.61,62 Generally, these mechanisms include lowering the antibiotic concentration in the cell (either by reducing entry or increasing efflux 63 ), protecting the target or altering the target structure (through mutations, enzymatic modifications, or protein binding 64 ), over-expression of the target 65 or modifications to inactivate the antibiotic 66 (Figure 1(c)). Resistance can also be due to multiple mechanisms that are not mutually exclusive. In addition to target mutations, other mutations that result in reduced drug accumulation by either increased efflux or decreased uptake, as well as horizontal transmission by plasmids carrying genes for target protection proteins or drug modifying enzymes can give rise to resistance. 67
Porins
Hydrophilic antibiotics (such as the β-lactams) can cross Gram-negative and mycobacterial membranes through aqueous channels called porins that normally provide the organism with nutrients. 63 Porins are classified according to their structure, selectivity and expression regulation. Antibiotic uptake can be decreased by altering the type or level of porin expression or altering the selectivity/function of these channels.61,62 As an example, studies of Klebsiella pneumoniae clinical isolates revealed that porin integrity was more of a determinant for carbapenem resistance than the presence of ESBL. 68 The study did not detect carbapenemases among 61 Chilean clinical isolates of carbapenem-resistant Enterobacteriaceae, so resistance was attributed to a combination of porin loss or alteration and β-lactamase activity. Another study characterized clinical isolates of Enterobacter aerogenes from patients treated with the carbapenem drug imipenem and linked altered porin expression with the adaptive response of E. aerogenes to the antibiotic. 69
Efflux pumps
While alterations to porins can reduce the penetration of antibiotics, efflux pumps actively transport many antibiotics out of Gram-negative and Gram-positive organisms. 70 Among the first efflux pumps to be described was the system that pumps tetracyclines out of E. coli. 71 The efflux pumps can have a narrow or broad range of specificity for antibiotic substrates and increased expression can enable higher levels of resistance to an antibiotic. Pumps able to transport different classes of antibiotics are associated with MDR. The five major families of efflux pumps include the ATP-binding cassette (ABC) superfamily, the major facilitator superfamily (MFS), the multidrug and toxic-compound extrusion (MATE) family, the small multidrug-resistance (SMR) family, and the resistance nodulation division (RND) family. 70 Classification into a family is based on the number of components comprising the pump, the number of transmembrane-spanning regions, the energy source for active transport and the substrate type. The efflux transporters with the highest clinical relevance from Gram-positive bacteria are members of the MFS, whereas for Gram-negative organisms the transporters are from the RND family, which are able to extrude a wide range of antibiotics. 70 Recently, genomic analysis of pre- and post-therapy MDR clinical isolates of Salmonella typhimurium were compared from a patient that failed antibacterial therapy and a G288D mutation was identified in the Na+ regulatory domain (NRD) transporter acriflavine resistance protein (AcrB) of the AcrAB-TolC tripartite MDR efflux pump system. 72 Characterization studies indicated that the mutation increased the efflux of the fluoroquinolone antibiotic ciprofloxacin but decreased the efflux of other drugs. While this study demonstrates how mutations to a transporter can alter bacterial susceptibility to antibiotic therapy, mutations can also alter the regulatory network controlling the expression of efflux pumps to result in increased antibiotic resistance. 62 Resistance to antibiotics has also been linked to a combination of efflux pump overexpression and porin downregulation in clinical isolates from Pseudomonas aeruginosa, K. pneumoniae, and E. coli.73,74
Protecting the target
A general strategy to achieve antibiotic resistance is to prevent an antibiotic from binding its target, which can be accomplished by different means. One paradigm of this is a target-protection mechanism, whereby a protein can associate with the drug target to prevent association with the antibiotic. An example is given by the FusB fusidic acid resistance protein, which protects elongation factor G (EF-G) from the antibiotic fusidic acid (FA). FA is a steroid antibiotic that is effective against Gram-positive bacteria, including MRSA, and acts by inhibiting the release of EF-G from the ribosome, thereby halting protein synthesis and inducing growth arrest. 75 Recent structural studies of the FusB-EF-G complex by solution NMR revealed dynamic changes in EF-G following FusB binding that facilitates disassembly of the stalled post-translational complex to rescue protein synthesis. 76 Another mechanism to prevent target engagement by an antibiotic is genetic mutations to the target gene. An example of this is fluoroquinolone resistance, which can result from mutations to DNA gyrase and topoisomerase IV that are targets of the antibiotic. 77 Mutations to the topoisomerases alter their structures to reduce the binding efficiency of quinolones.
Altering the target structure
Alternatively, modifications to antibiotic target structures can be achieved enzymatically. For example, the chloramphenicol-florfenicol resistance (Cfr) ribosomal RNA (rRNA) methyltransferase of Gram-positive and Gram-negative bacteria modifies the 23S rRNA at position A2503, providing MDR to a range of antibiotics including: phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A. 78
Altered gene expression
Resistance to antibiotics can be due to the modulation of gene expression. For example, resistance to methicillin in S. aureus results from the mecA gene that encodes PBP2a, a PBP that has low affinity for many β-lactams including penicillins, cephalosporins, and carbapenems. 79 While this is an example of replacing a target with another protein of similar biochemical function that is not susceptible to antibiotics, resistance has also been achieved by overexpression of a target to a concentration beyond what the antibiotic can inhibit. For instance, a clinically isolated strain of E. coli was shown to have mutations that resulted in overexpression of dihydrofolate reductase by several hundredfold, which resulted in a three-fold increase in the Ki for the antifolate drug trimethoprim. 80
Modifications to the antibiotic
Antibiotic resistance can also be achieved through biochemical modifications to the antibiotic, including hydrolysis, group transfer, and redox mechanisms. 66 The β-lactamase enzymes provide a classic example of a hydrolysis mechanism, as the first β-lactamase termed “penicillinase” was identified in, 1940 from E. coli prior to the clinical use of penicillin.81,82 Since that time, more than 850 β-lactamases have been identified, which are able to hydrolyze β-lactam antibiotics (including penicillins, cephalosporins, monobactams, and carbapenems). 83 Alternatively, antibiotics can be inactivated due to various chemical modifications, including acylation, phosphorylation, glycosylation, nucleotidylation, ribosylation, and thiol transfer. 66 For example, the aminoglycoside antibiotic kanamycin B can be enzymatically modified by acylation, phosphorylation, and nucleotidylation. 84 A well-studied example of the redox mechanism of antibiotic inactivation is the enzyme TetX, which catalyzes the monohydroxylation of tetracyclines at position 11a, disrupting the antibiotic Mg2+-binding site which is required for antibacterial activity. 66
From a perspective of evolving drug resistance, it is interesting to consider how the structures of approved antibiotics have changed over time. Fifty-nine β-lactam drugs were approved by the FDA between 1947 and 2014 (Table 1). To capture the evolution of these molecules since penicillin G was approved, structural similarities were evaluated among all 59 β-lactam drugs. The method of maximal common substructure (MCS) clustering was applied to detect various chemotypes through the identification of common structural elements among similar molecules.85,86 The expected outcome from such an analysis is that molecules in a given cluster are more similar to each other than to molecules of another cluster. The resulting chemical space–time analysis is shown in Figure 3. The 2-dimensional transformation reveals closely related molecules and the overall structural diversity among the β-lactam antibacterial drugs. To highlight the evolution of β-lactam chemotypes, selected structures are shown. The plot clearly shows a trend towards new scaffolds over the course of time.
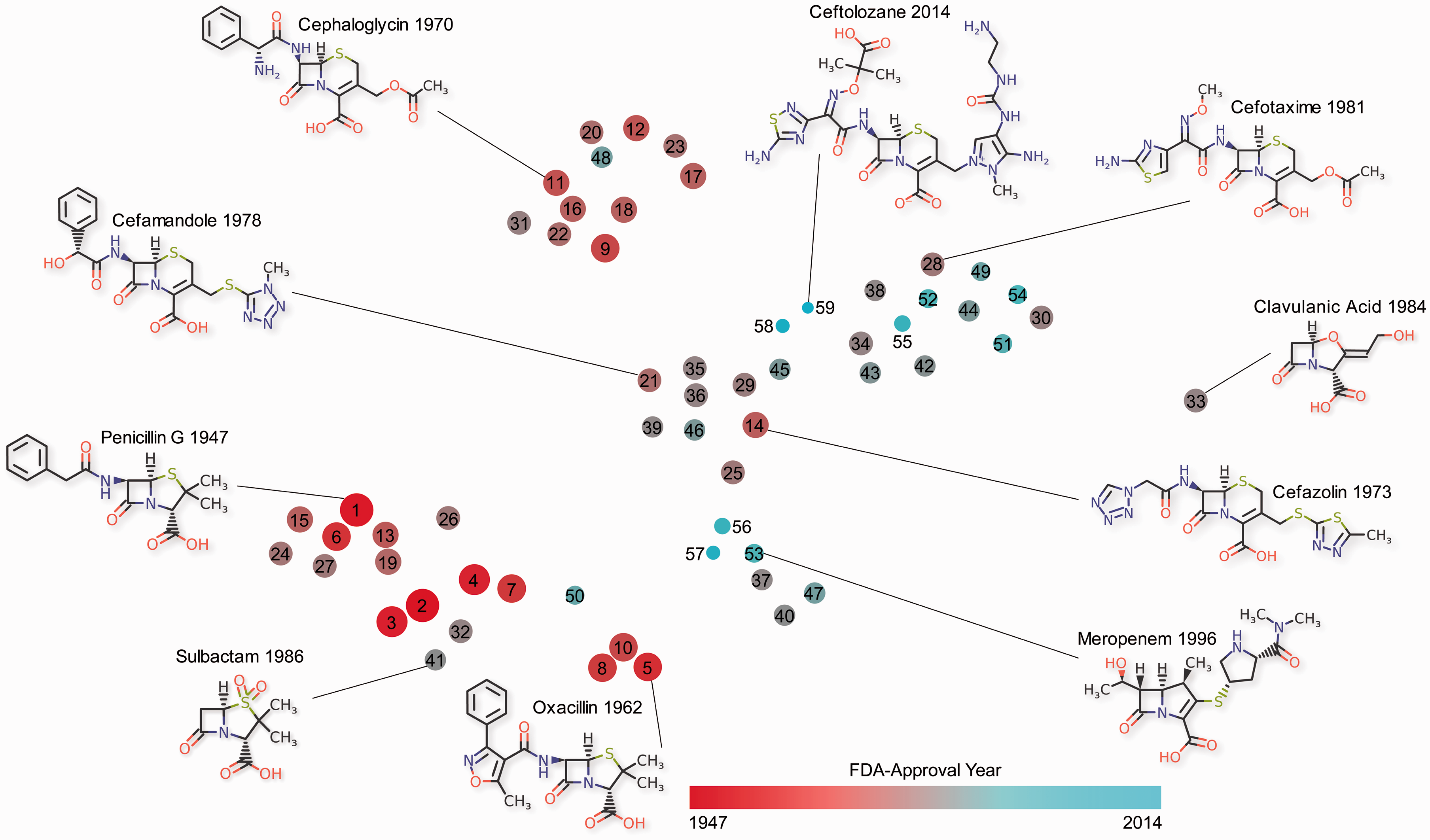
Chemical space-time of β-lactam antibacterial drugs. The dots indicate structures of β-lactam antibacterial drugs in a 2-dimensional chemical space. The distance between dots reflects the extent of structural similarity, with similar structures in closer proximity. The color and size of the dots corresponds to the year of FDA-approval, with more recent approvals indicated by a color closer to the blue end of the spectrum and a smaller size. The maximal common substructure (MSC) clustering was performed with a similarity threshold of 0.60 using the StarDrop scientific software suite (version: 6.2.0, Optibrum Ltd, Cambridge, UK). The clustering resulted in 11 MCS clusters and eight singletons (Table 1). In oder to characterize the diversity among β-lactam structures, they were embedded in a 2-dimensional space using the t-distributed stochastic neighbor embedding algorithm of StarDrop.,87
New approaches for small molecule antimicrobials
Due to the crisis of antibiotic resistance, it is necessary to explore alternate or adjunctive therapies to enhance our current defenses against infectious diseases. Analyzing the role of small molecules to advance our strategies against antibiotic-tolerant and -resistant bacterial infections is of great interest and has afforded a number of successful new approaches.
New chemotypes
As indicated in the previous section, the development of resistance against new antibiotics by pathogenic organisms is essentially inevitable due to a range of mechanisms. However, resistance is not anticipated to be easily developed against the recently discovered antibacterial molecule pseudouridimycin (PUM), which is a nucleoside analog. 88 Because PUM binds directly and specifically to the nucleotriphosphate-binding active site of bacterial RNA polymerase (RNAP), mutations that reduce PUM binding would likely also reduce or abolish enzymatic activity. The compound was identified from extracts of soil microbes in a screen for inhibitors of bacterial RNAP. PUM inhibited Gram-negative and Gram-positive bacterial growth, including drug resistant Streptococcus strains, at low micromolar concentrations and cleared infection in a mouse model of Streptococcus pyogenes peritonitis. The antibiotic rifampin binds RNAP at a distinct site and additive antibacterial activity was observed when PUM and rifampin were co-administered. 88
New chemotypes are required to stay ahead of the rapid evolution of antibiotic resistance. The fatty acid biosynthesis type 1 enzyme (FabI) has been known as an antibacterial target for many years, but several new inhibitors are currently in clinical development that appear to offer genuinely new chemotypes, including afabicin (Deniopharm International), and CG400549 (CrystalGenomics), both of which are in phase II clinical trials. 89 As well, the mycolic acid transporter MmpL3 in Mycobacterium tuberculosis has been targeted by novel diamine- and indolamide-based compounds, and these have been determined to be synergistic or additive with the activity of other antimicrobials, increasing their utility and effectiveness. 90
Chemotypes for broad-spectrum activity
A recent advancement in the understanding of ideal physicochemical properties for small molecule antibacterials was made by Richter and colleagues, who studied a diverse set of compounds and used structure–activity relationship studies with computational analyses to develop predictive guidelines for small molecule accumulation in Gram-negative bacteria. 91 Notably, the results indicated that small molecules most likely to accumulate contain an amine, are both rigid and amphiphilic and have low globularity, which differed substantially from the properties thought to be important based on a retrospective analysis of known antibiotics, particularly polarity and molecular weight. The authors note that compounds with primary amines are rare in standard compound libraries used for high-throughput screening and the assembly of compound collections that meet the guidelines for accumulation might enable the discovery of novel chemical leads for the development of broad-spectrum antibiotics.
New targets
A potential target for new antibiotics is the membrane enzyme phospho-MurNAc-pentapeptide translocase (MraY), which is essential for the synthesis of the bacterial envelope. The role of MraY is to catalyze the initial processes in peptidoglycan synthesis by generating uridine-monophosphate (UMP) and lipid 1. There are MraY inhibitors currently in development, but some issues remain, not the least of which is the challenge of bacterial cell entry and the problem of the inhibitor reaching effective levels within the cell. 9
Riboswitches, which are segments of mRNA that can bind to small molecules and metabolites, are also potential targets for antimicrobial molecules. Engineered small molecules can inhibit the function of riboswitches and some were shown to be effective in bacteria. Ribocil is a selective chemical modulator that binds to riboflavin riboswitches and prevents riboflavin production in bacteria. The use of ribocil was shown to lower the E. coli bacterial burden in a mouse model of septicemia by two to three orders of magnitude. 92 While further research on riboswitches as drug targets is warranted, caveats include the potential for gain of mutations as well as the observed difficulty for some modulators to cross the bacterial cell wall. Nevertheless, these molecules represent unique targets and an opportunity for novel antimicrobials. 93
A promising approach for new antibacterials is antivirulence, which focuses on targeting pathogenic bacteria by binding to and inhibiting virulence factors such as toxins, particularly those with enzymatic activity.94,95 For example, a small molecule was identified in silico and confirmed in vitro and in vivo to target the enzymatic site of Shiga toxin from Shigella dysenteriae. 96 In theory, because these virulence factors are not necessary for bacterial survival outside the context of a host infection, this approach will not apply direct selective pressure and therefore resistance should not rapidly emerge. Antivirulence treatments are anticipated to supplement small molecule therapeutics and improve their efficacies, resulting in optimal clinical treatment regimens.
Drug repurposing
The approval of new antibiotics has declined during the past three decades (Figure 2) while resistance to the approved antibiotics continues to increase. Despite an urgent need for new antibiotics, most major pharmaceutical companies have moved away from the antibacterial market thereby drying up the pipeline for new treatments. 2 Therefore, repurposing of approved drugs to treat infectious diseases might provide an alternative approach for effective new therapies. 98 Approved drugs repurposed for a new indication can be rapidly advanced to phase II clinical trials without a phase I clinical trial due to the existing preclinical, human pharmacokinetics, and drug safety data. An example of repurposing a drug to treat bacterial infections is provided by the gold-containing compound auranofin. This drug is FDA-approved for the treatment of rheumatoid arthritis, but has also demonstrated antimicrobial activity against a range of Gram-positive and Gram-negative pathogens.98–105 The thiol ligand of auranofin can form irreversible adducts with selenol or thiol groups, and auranofin is a well-known inhibitor of reduction/oxidation enzymes including thioredoxin reductase (TrxR). 98 The antibacterial activity of auranofin has been linked to the disruption of selenium metabolism and interference with a number of selenoproteins including glycine reductase, proline reductase, and xanthine dehydrogenase.99–101 Biochemical studies have demonstrated that auranofin is a potent inhibitor of bacterial TrxR, which is not targeted by other antibiotics and is involved in a variety of processes required for survival and proliferation, such as DNA synthesis and protein repair. 104 The ability of auranofin to interfere with multiple bacterial enzymes might reduce the development of resistance.
Synergistic combinations
Drug combinations have been approved for the treatment of bacterial infections, such as the combination of amoxicillin (a β-lactam) with clavulanic acid (a β-lactamase inhibitor). 106 Advantages of drug combinations include extending the spectrum of susceptible organisms, overcoming drug resistance, reducing the development of antibiotic resistance, and the potential for drug synergies. 97 New combinations of antibiotics have been successfully applied to treat drug-resistant pathogens. For example, a combination of three β-lactams (meropenem, piperacillin, tazobactam) was reported to act synergistically against MRSA and showed similar in vivo activity to the oxazolidinone antibiotic linezolid. 107 While Gram-negative bacteria are intrinsically resistant to the antibiotic vancomycin, a study showed strong synergies against E. coli when combining vancomycin with antibiotics such as trimethoprim or nitrofurantoin. 108 The drug fosfomycin, which has been registered in the United States for oral treatment of uncomplicated UTIs since 1995, has been tested as a participant in a cocktail with polymyxin B, tobramycin or ciprofloxacin to target P. aeruginosa, although issues with resistance remain.109,110 Other novel combinations of antibiotics have shown synergy against Gram-negative pathogens, including Acinetobacter baumannii. 111 Progress has also been made in identifying synergistic drug combinations that combine antibiotics with approved drugs not previously used to treat infections. As an example, the antibiotic penicillin G is primarily only active against Gram-positive organisms, but showed synergistic activity with the non-antibiotic drug promethazine against E. coli. 112 The opioid receptor agonist loperamide (Imodium) is an approved anti-diarrheal that was shown to synergize with tetracyclines due to its ability to dissipate bacterial membrane potential and enhance antibiotic uptake. 113 Recently, a drug combination screen identified 17 synergistic three-drug combinations that were effective against K. pneumoniae at concentrations which are clinically achievable. Additionally, three sets of three-drug combinations, two of which contained auranofin, were active against a panel of 10 MDR clinical isolates, including K. pneumoniae, A. baumannii, P. aeruginosa, Citrobacter freundii, Enterobacter cloacae, and E. coli. 105 While the previous examples of combinations utilized antibiotics, an interesting strategy was recently applied to completely replace an antibiotic pair with non-antibiotic drugs. The aim was to identify synergistic pairs based on the antibiotics trimethoprim and sulfamethoxazole that have been used in combination for decades to treat bacterial infections. First, the antiviral drug azidothymidine (AZT) was found to synergize with trimethoprim or sulfamethoxazole against MDR E. coli and K. pneumoniae. The development of a mechanistic understanding of the underlying interaction enabled the discovery of synergy between AZT and the cancer drug floxuridine, which reduced the median bacterial burden by 10,000-fold compared with the traditional pair of trimethoprim/sulfamethoxazole in a zebrafish model. 114
The potential opportunities to repurpose drugs, although exciting, are not without challenges.115–117 There are safety concerns associated with utilizing drugs known to target host tissues, such as oncology medications. 116 Additionally there are currently major economic barriers to repurposing because there are insufficient market incentives to advance drugs that are off-patent. It has been estimated that repurposing a drug costs around $300 million and takes about 6.5 years. 117 Other current challenges include intellectual property rights and other legal concerns, regulatory issues and timelines, return on investment, as well as the design and execution of proof-of-concept clinical trials.
Financial concerns
Another unsolved but important issue in the search for effective small molecule antibiotics is that of return on investment. Most pharmaceutical companies are in business to make a profit for their shareholders, and antimicrobial drug discovery is currently not profitable. It was recently estimated that the cost of a new drug with market approval is $2.6 billion and takes more than a decade to develop. 118 When a new antibiotic is finally available to the public, it is prescribed only transiently for infections and not needed by otherwise healthy people on a regular basis, unlike medications that lower cholesterol or treat high blood pressure. To address these concerns and incentivize antibacterial drug development, in 2012 the 112th United States Congress passed the Food and Drug Administration Safety and Innovation Act, 119 which included as Title VIII the “Generating Antibiotic Incentives Now” (GAIN) Act. As a commercial inducement, the GAIN Act extended the market exclusivity period, which is the length of time that a pharmaceutical company is allowed to be the sole provider of an antimicrobial by law. At the same time, an FDA Task Force was launched to identify challenges and provide guidance for antibiotic drug resistance.
A novel solution to this problem has been put forward which consists of incentivizing antibacterial research and development by drug companies using a model that is either fully or partially delinked from drug sales through milestone payments and other financial encouragements. 120 The successful implementation of this model would require both multinational coordination and cooperation as well as regional and global banking institutional support. However, because the health threat posed by antibiotic-resistant organisms affects the population worldwide, this approach might have merit and should be subject to serious consideration.
Conclusions
It is not an exaggeration to describe our current situation as an evolutionary “arms race” against antibiotic-resistant microorganisms. There is not likely to be a single compound or class of drugs that will continue to be effective for the foreseeable future against bacterial infections. Rather, the prediction is that we will discover novel treatments (natural, man-made or new combinations) that will initially be valuable additions to our arsenal of antibiotics, but which will eventually lose efficacy due to bacterial resistance mechanisms. This is a clear indication that academia, industry and government must be called upon to invest in a concerted and unified effort to fill the drug development pipeline with new antimicrobials. The first antibiotic was released to the public more than 80 years ago, which means that there are people alive today that were born in the pre-antibiotic era. For those of us that were raised in a world with available and effective treatments, imagining a future without antibiotics is daunting indeed.
Footnotes
Authors’ Contributions
All authors participated in the writing and review of the manuscript.
Acknowledgments
Minireview space limitations prevented citation of all relevant literature. The authors wish to thank Noel Southall for helpful discussions.
Declaration of Conflict of Interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: in part by U01 DC014756 (DAD) and the Intramural Research Program of the National Institutes of Health, National Center for Advancing Translational Sciences (NPC, TP, GZ-K and MDH).