
Abstract
Natural killer (NK) cells are essential players of the innate immune response that secrete cytolytic factors and cytokines such as IFN-γ when contacting virus-infected or tumor cells. They represent prime targets in immunotherapy as defects in NK cell functions are hallmarks of many pathological conditions, such as cancer and chronic infections. The functional screening of chemical libraries or biologics would greatly help identify new modulators of NK cell activity, but commonly used methods such as flow cytometry are not easily scalable to high-throughput settings. Here we describe an efficient assay to measure the natural cytotoxicity of primary NK cells where the bioluminescent enzyme NanoLuc is constitutively expressed in the cytoplasm of target cells and is released in co-culture supernatants when lysis occurs. We fully characterized this assay using either purified NK cells or total peripheral blood mononuclear cells (PBMCs), including some patient samples, as effector cells. A pilot screen was also performed on a library of 782 metabolites, xenobiotics, and common drugs, which identified dextrometorphan and diphenhydramine as novel NK cell inhibitors. Finally, this assay was further improved by developing a dual-reporter cell line to simultaneously measure NK cell cytotoxicity and IFN-γ secretion in a single well, extending the potential of this system.
Introduction
Natural killer (NK) cells are essential players of the immune response against viruses and tumor cells. 1 As group 1 innate cytotoxic lymphoid cells, they are characterized by a potent cytotoxicity and the ability to secrete IFN-γ when activated. In humans, they represent 5%–20% of peripheral blood mononuclear cells (PBMCs). NK cells express a set of membrane receptors allowing them to discriminate normal cells from abnormal, virus-infected, or tumor cells. Tumorigenesis and viral infection are indeed often accompanied by the downregulation of NK cell inhibitory ligands and/or the upregulation of activator ligands at the cell surface, allowing target cell detection by NK cells. Once activated, NK cells release the pore-forming protein perforin 2 and pro-apoptotic factors including granzymes and death ligands (FasL, TRAIL, etc.). Besides, secreted IFN-γ contributes to the activation of other effector cells and the induction of an appropriate immune response. As such, NK cells are prime targets in immunotherapy and defects in their activation are hallmarks of pathological conditions, including cancers, genetic disorders, and viral chronic infections. Functional assays measuring NK cell cytotoxicity in vitro or ex vivo are therefore essential to evaluate the potential impact of small molecules, antibodies, or proteins on this immune cell population.
The reference method to measure the cytotoxic activity of NK cells ex vivo is based on the release of chromium 51 (51Cr) by target cells preloaded with this radioactive isotope. 3 When target cells are co-incubated with NK cells, the permeabilization of their membrane is accompanied by the release of 51Cr in culture supernatant, which reflects the cytolytic activity of NK cells. This method has been widely used for decades, but is progressively abandoned because of the risks and regulatory constraints associated with the manipulation of radioactive elements. A more recent and popular method is based on the use of multicolor flow cytometry to quantify the lysis of target cells in co-cultures. 4 Fluorescent dyes or specific membrane markers are used to discriminate NK cells from target cells at the time of analysis, while the fraction of dead target cells is calculated using viability dies. Alternatively, NK cell cytotoxicity can be indirectly quantified by surface immunostaining of the degranulation marker CD107a (LAMP1), which is correlated with NK cell-mediated lysis. 5 Although widely used, all methods based on flow cytometry require some expensive equipment and trained personal, and are time-consuming at both the acquisition and analysis steps. For these reasons, they are not well adapted to large-scale screening projects, and alternative strategies have been developed to measure NK cell activation in high-throughput settings.
Established strategies include the quantification of secreted IFN-γ by enzyme-linked immunosorbent assay (ELISA) in co-culture supernatants, which is tractable for high-throughput screening (HTS).6,7 However, IFN-γ secretion is not always correlated with the cytotoxic activity, a limitation that adds to the significant cost and multistep protocol inherent to ELISA. Other described tests involve the co-culture of PBMCs or NK cells with adherent target cells. 8 After incubation, effector cells and dead target cells are washed out, and the number of living target cells still attached to culture wells is quantified by different methods. Alternatively, the number of adherent target cells in co-cultures, without the need for washing out NK cells, can be determined by electrical impedance measurements. 9 However, such assays are limited to adherent target cells and rely on the quantification of surviving cells in culture wells rather than a direct quantification of cell lysis. Finally, nonradioactive alternatives to 51Cr have been developed, such as calcein, a fluorescent molecule, which can be loaded as a pro-form (Calcein-AM) in target cells. 10 However, calcein staining relies on endogenous cellular esterase for processing the pro-form of the dye and was criticized for its low signal-to-noise (S/N) ratio. Europium was also used as a nonradioactive substitute to 51Cr, but it never became popular due to some high background observed in many target cell lines. 11 Other cytotoxicity assays are based on the release of endogenous cytosolic enzymes such as lactate dehydrogenase. 12 This enzyme can be detected in culture supernatants with synthetic substrates that are converted into colored or fluorescent products. The sensitivity of these methods has been a matter of debate, in particular in co-cultures, because when dying, not only target cells but also effector cells potentially release such endogenous enzymes in the medium. Therefore, there are still some unmet needs for a sensitive high-throughput assay to measure the cytotoxicity of NK cells.
Along with the development of firefly luciferase as a reporter in biological systems, bioluminescent assays have been engineered to measure NK cell cytotoxicity. Decarboxylation of the luciferase substrate luciferin by firefly luciferase leads to light emission that can be quantified. Interestingly, this reaction requires ATP that is essentially present in living but not dead cells. Thus, target cells were engineered to express firefly luciferase, and co-cultured with NK cells. When luciferin is added to culture wells, it penetrates all cells in the co-culture but is only processed by luciferase-expressing target cells that are alive and produce ATP. 13 This method was shown to outperform the standard 51Cr cytotoxicity assay but is dependent on intracellular ATP levels, and the S/N ratio can be low in common target cell lines such as K562. 14 In parallel, other research groups tried to develop cytotoxicity assays based on the release of firefly luciferase in culture supernatants when lysis occurs.15,16 However, this approach was hampered by the short half-life of firefly luciferase in culture medium at 37 °C. Recently, the development of high-performance luciferases derived from marine organisms has provided a new start to this approach.17,18 The best example is NanoLuc, a synthetic luciferase of 19 kDa developed by Promega (Fitchburg, WI) from a luminescent enzyme of the sea shrimp Oplophorus gracilirostris. 19 This enzyme uses furimazine as a substrate, of which decarboxylation is ATP independent. NanoLuc is >150 times brighter than firefly luciferase and, more importantly, is stable for days in culture medium. We took advantage of this new generation of luciferase to develop a high-throughput functional assay to measure the natural cytotoxicity of primary NK cells ( Fig. 1 ). Two clonal cell lines expressing high levels of NanoLuc in their cytoplasm, called K562-NL and twINNE, were engineered, and NanoLuc release in culture supernatant was determined in co-cultures with purified effector cells, including primary NK cells, total PBMCs from healthy donors, treated or not with cytokines or immunomodulatory compounds, and PBMCs from a perforin-deficient patient. We also established that NanoLuc release by target cells is robust enough to screen chemical libraries in high-throughput settings and to identify modulators of NK cell activity. Finally, we further improved this assay by developing the dual-reporter cell line twINNE, where a firefly luciferase reporter gene is used to monitor interferon signaling in target cells while, in parallel, NanoLuc release is reporting their lysis.
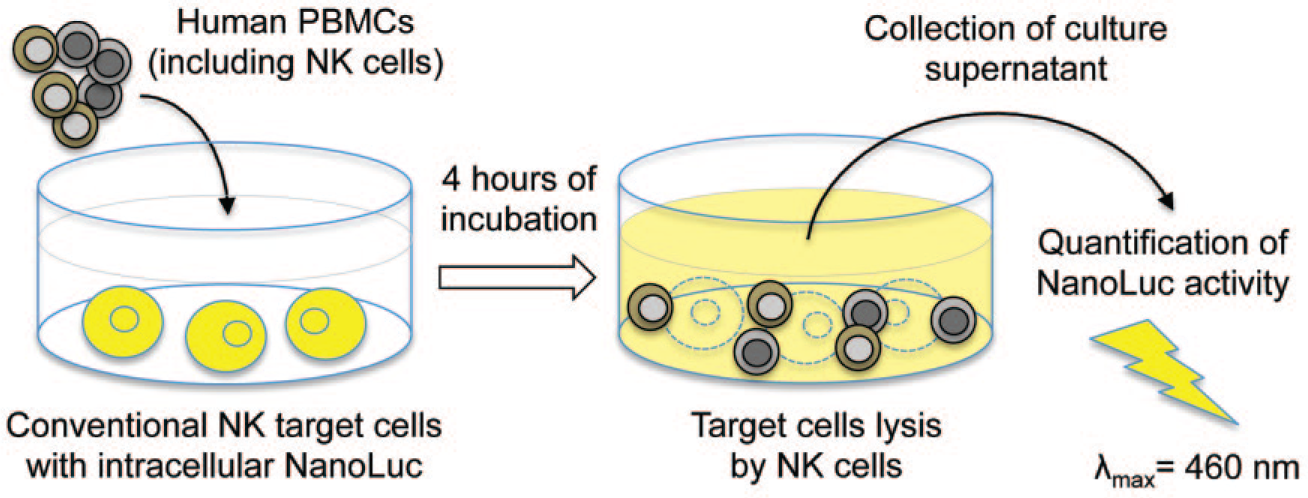
Principle of the luciferase-based cytotoxicity assay. PBMCs are co-cultured with target cells expressing high levels of NanoLuc in the intracellular compartment. After a few hours of incubation, NK cells from PBMCs mediate the lysis of target cells, thus releasing NanoLuc in culture supernatant. NanoLuc activity in culture supernatant reflects target cell lysis.
Materials and Methods
Cells and Culture Conditions
Cells were cultured at 37 °C and 5% CO2 in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO; R8758) containing 10% fetal calf serum (FCS). Human K562 cells were kindly provided by Dr. T. Walzer (CIRI, Lyon, France). The reporter cell line STING-37, corresponding to HEK-293 cells stably transfected with the interferon-stimulated response element (ISRE)-luciferase reporter gene, was previously described. 20 Blood from healthy blood bank donors was obtained from Etablissement Français du Sang (Convention 07/CABANEL/106; Paris, France). Human PBMCs were isolated by density centrifugation with Lymphoprep medium (StemCell Technologies, Vancouver, Canada) from total blood or leucocyte concentrates obtained when plateletpheresis is performed. NK cells were purified from PBMCs using the NK cell isolation kit from Miltenyi (Bergisch Gladbach, Germany). Recovered NK cells were pure at >94% as assessed by their CD3–/CD56+ phenotype. A patient with a confirmed homozygous missense mutation in PRF1 was included in this study. A blood sample was obtained upon informed consent.
Cytokines, Small Compounds, and Reagents
Human IFN-γ from Roussel Uclaf (Romainville, France) was a kind gift of Dr. Mounira Chelbi-Alix. IL-2, IL-15, and TGF-β were obtained from Peprotech (Rocky Hill, NJ). R848 was obtained from Sigma-Aldrich, Torin2 was from Tocris (Bristol, UK), and rapamycin was from Calbiochem (San Diego, CA). Luciferase induction in twINNE cells was determined using the Bright-Glo (Promega, Fitchburg, WI) or Britelite plus reagents (PerkinElmer, Waltham, MA), according to the manufacturer’s recommendations. Nanoluc expression was quantified in culture supernatants as previously described. 19 Cellular viability was determined by quantification of ATP in culture wells using the CellTiter-Glo assay (Promega). Bioluminescence was measured for 0.1 s with a luminometer (EnSpire; PerkinElmer). Tergitol NP9 was from Sigma-Aldrich.
Establishing K562-NanoLuc and twINNE Cell Lines
The NanoLuc gene was introduced in target cells by transduction with the pLVX-Puro lentiviral vector (Clontech, Mountain View, CA). First, the NanoLuc sequence was cloned in the pLVX-Puro vector using the Gateway cloning system (Thermo Fisher Scientific, Waltham, MA). Briefly, the pLVX-Puro plasmid was made compatible with the Gateway system by inserting the Gateway cassette C1 at the SmaI site to generate the new destination vector called pLVX-Puro-GW. A pDONR207 plasmid containing the coding sequence for the NanoLuc enzyme (Promega) was kindly provided by Dr. Y. Jacob (Institut Pasteur, Paris, France). The NanoLuc sequence was transferred by in vitro LR recombination from pDONR207 to pLVX-Puro-GW vector following the manufacturer’s recommendations to obtain pLVX-Puro-NanoLuc. This vector was used to produce lentiviral particles by co-transfection with packaging plasmids pVSV-G and pGag-Pol (kindly provided by Dr. S. Nisole, IRIM, Montpellier, France) into HEK-293T cells. Culture supernatants containing lentiviral particles were collected 2 days later and stored at –80 °C.
K562 and STING-37 cells were transduced with the lentiviral vector expressing NanoLuc, and puromycin at 1 µg/mL was applied for 4 weeks to enrich the cellular pool for transduced cells. Using a cell sorter, 576 and 63 cells were cloned from the transduced populations of K562 and STING-37 cells, respectively. After 4 weeks, culture wells were tested for the expression of NanoLuc to identify best-growing clones expressing high levels of the enzyme. Top candidates were amplified for establishing the K562-NL and twINNE cell lines, which respectively correspond to K562 and STING-37 clones expressing high levels of NanoLuc.
Cell Cytotoxicity Assay
PBMCs from healthy donors were incubated overnight in the absence or presence of compounds to be tested. Unless specified otherwise, PBMCs were plated in 96-well, round-bottom plates at a concentration of 400,000 cells/well in a final volume of 100 µL. After 16 h, 16,000 K562-NanoLuc or 100,000 twINNE cells were added to culture wells (100 µL of a cell suspension at 160,000 cells/mL or 1 × 106 cells/mL, respectively). The total volume in culture wells was 200 µL, and plates were centrifuged briefly for 2 min at 1200 rpm. Unless specified otherwise, 20 µL of culture supernatants was collected after 4 or 7 h of co-culture for K562-NL or twINNE, respectively, and NanoLuc activity was determined by adding 40 µL of RPMI-1640 and 30 µL of NanoLuc reagent (as described in Hall et al. 19 Promega) in black, flat-bottom, 96½-well plates. Bioluminescence was measured for 0.1 s with a luminometer (Enspire; PerkinElmer).
To quantify the death of target cells by flow cytometry, K562-NL cells were labeled with the VDP450 dye at 1 µM in PBS (BD Biosciences, Franklin Lakes, NJ). After 10 min of incubation at 37 °C, cells were washed thoroughly in culture medium and co-cultured with PBMCs as described above. After 4 h, cells were collected and dead cells were labeled using the LIVE/DEAD fixable green stain reagent from Invitrogen (Waltham, MA) following the manufacturer’s recommendations. Cells were analyzed by flow cytometry, and the percentage of dead K562-NL cells was determined by gating on the VDP450+ cells.
Screening of the TMIC Human Metabolome Library
The TMIC Human Metabolome Library was obtained from The Metabolomics Innovation Center in Canada (TMIC; www.metabolomicscentre.ca). It is a comprehensive library of compounds representing a broad range of metabolites, xenobiotics, and drugs commonly found in body tissues. Briefly, 5 mg of each compound was dissolved in 2 mL of either pure water or a 50% mix of water and DMSO to achieve a final stock concentration of 2.5 mg/mL. Compounds were rearrayed in deep-well plates, and daughter plates were prepared by spotting 2 µL in round-bottom, 96-well plates for screening. Culture wells were filled with PBMCs at 400,000 cells/well in a final volume of 100 µL as described above, so that test compounds were at 50 µg/mL. For each screening plate, negative control wells were filled with culture medium alone, whereas positive control wells were filled with PBMCs plus 2 µL of a 50% water–DMSO mixture. After 16 h, 16,000 K562-NanoLuc cells were added to all culture wells (100 µL of a cell suspension at 160,000 cells/mL). NanoLuc activity in culture supernatants was determined after 4 h of co-culture as described above, and the results were expressed as a percentage relative to positive control wells. The Z′ factor that reflects the robustness and quality of the assay was calculated from means (μ+ and μ–) and standard deviations (σ+ and σ–) of negative and positive controls such as Z′ = 1 – 3*(σ+ + σ–)/(μ+ – μ–), as described by Zhang et al. 21 Very good assays have Z′ factors that are close or superior to 0.5. The signal-to-background (S/B) ratio corresponds to μ+/μ–, and the S/N ratio to (μ+ – μ–)/σ–, as previously defined. 21
Results
Development and Assessment of the K562-NL Reporter Cell Line
Myeloid leukemia cells K562 were transduced with a lentiviral vector to express NanoLuc, and cell clones were carefully screened to select those expressing this bioluminescent enzyme at high levels. The best cellular clone was amplified to establish the K562-NL reporter cell line. First, we tested if NanoLuc is released in culture supernatants when membrane permeabilization occurs. K562-NL cells were treated with 1% Tergitol as a detergent to lyse cells, and culture supernatants were harvested at different time points. Cell lysis was confirmed by standard light microscopy (data not shown). As presented in Figure 2A , high levels of NanoLuc activity were detected in culture supernatants and were maximal after only 3 min. As expected, ATP in culture wells, which is a common biomarker of cellular metabolic activity and used to determine viability, was gradually degraded in the 0–30 min time window, showing that NanoLuc release is concomitant with cell death.
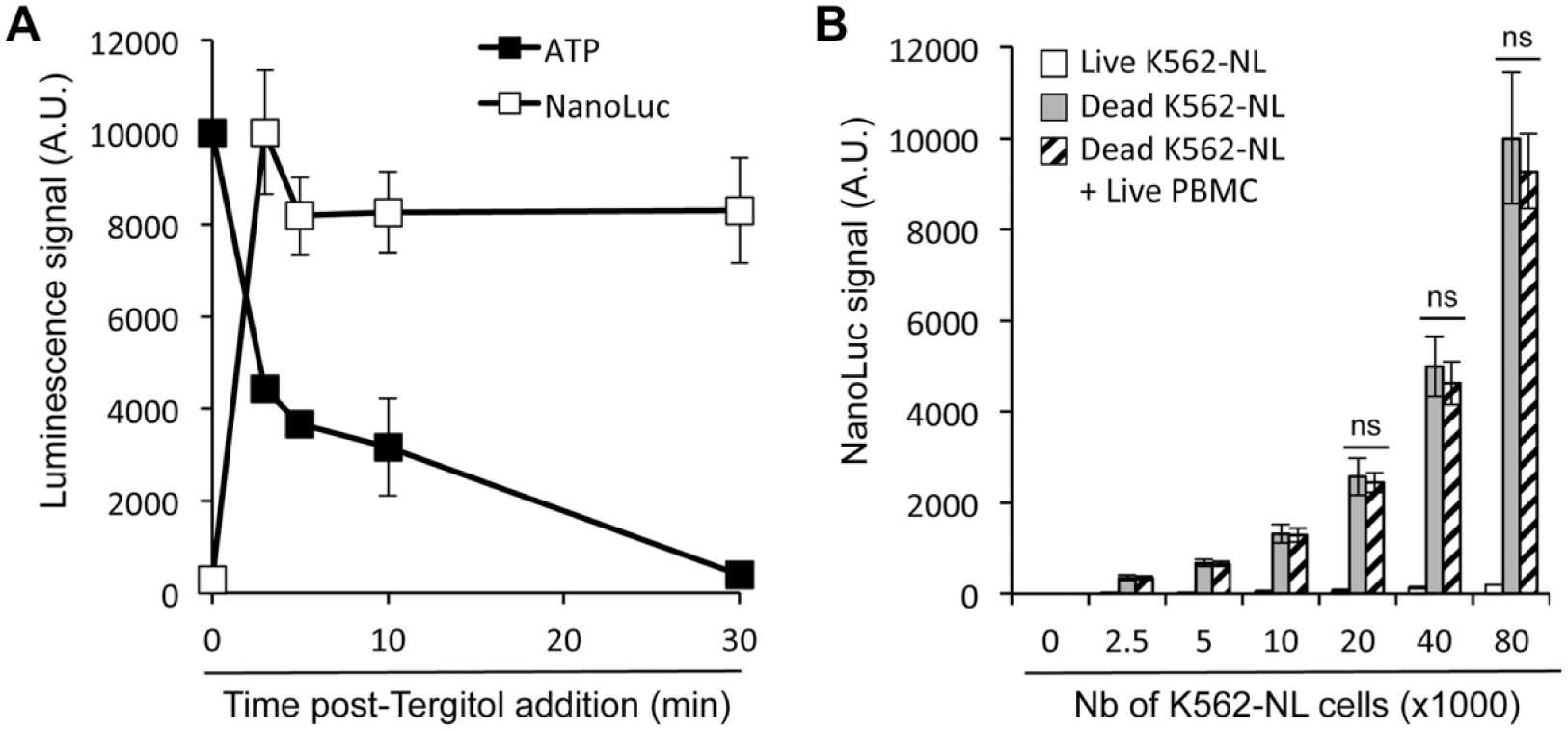
Nanoluc accumulates in culture supernatants of K562-NL cells when killed by chemical or physical treatments. (
Since our goal was to determine whether K562-NL cells can be used as target cells to measure the cytotoxic activity of NK cells in PBMCs, we verified that NanoLuc activity in culture supernatants is not impaired by the addition of a large number of PBMCs in the culture wells. Increasing numbers of K562-NL cells—ranging from 2500 to 80,000 cells—were dispensed in culture wells of a 96-well plate and killed by freezing and thawing. Then, 400,000 PBMCs were added or not to culture wells, and after centrifugation, culture supernatants were immediately collected to quantify NanoLuc activity. As expected, the NanoLuc activity dramatically increased in culture supernatants when K562-NL cells were killed, and more importantly, the signal was not affected by the addition of PBMCs to culture wells ( Fig. 2B ). We also verified that the NanoLuc signal in culture supernatants of killed K562-NL cells mixed with PBMCs was stable in time, without any significant loss over 24 h (data not shown). Altogether, this demonstrated that NanoLuc release in culture supernatants of K562-NL cells is suitable to monitor their lysis by NK cells.
Measuring NK Cell Cytotoxicity with K562-NL Target Cells
A fixed number of K562-NL cells—herein referred as the target cells—were co-cultured with PBMCs, the effector cells, at different effector-to-target (E/T) ratios ranging from 0.8 to 50. After 4 h of incubation, culture supernatants were harvested and tested for NanoLuc activity. Results were expressed as the percentage of NanoLuc released by K652-NL cells permeabilized with 1% Tergitol. As shown in Figure 3A , NanoLuc activity increased proportionally with E/T ratios. We also performed the same experiment quantifying the number of dead K562-NL cells in co-cultures by chemical staining (LIVE/DEAD reagent) and conventional flow cytometry analysis. NanoLuc accumulation in culture supernatants appeared to be more sensitive than flow cytometry, as the killing of K652-NL cells was detectable at lower E/T ratios ( Fig. 3A ). We also performed the same co-culture experiment at a 25/1 ratio and quantified NanoLuc in supernatants at different time points ( Fig. 3B ). As expected, the signal continuously increased over 8 h of co-culture. After 24 h of co-culture, 100% lysis was reached in these co-culture conditions (data not shown). However, 4 h was sufficient to obtain a strong signal that is statistically distinct from K562-NL cells alone. Finally, donor-to-donor variation was assessed by testing PBMCs from 13 different donors at an E/T ratio of 25/1 and measuring lysis of target cells after 4 h of co-culture ( Fig. 3C ). Altogether, this demonstrated the efficiency and robustness of this assay to detect the cytotoxic activity of PBMCs.
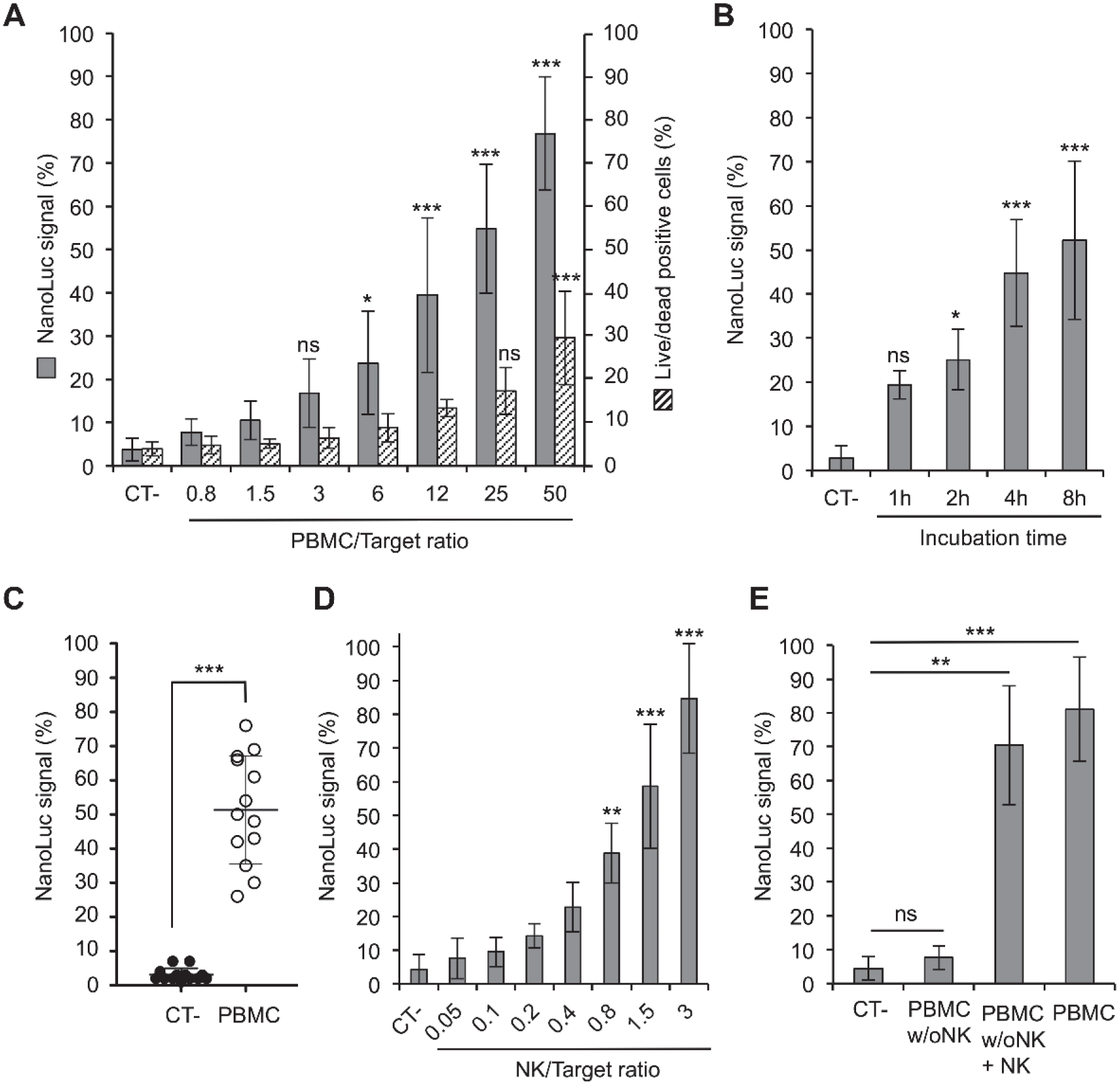
Measuring NK cell cytotoxicity with K562-NL target cells. (
The percentage of NK cells in PBMCs varies from 5% to 20%. We thus performed the same assay with NK cells purified from PBMCs. As expected, NanoLuc activity increased in culture supernatants of K562-NL cells when co-cultured with purified NK cells ( Fig. 3D ) and reached high levels at lower E/T ratios than previously reported with total PBMCs. Furthermore, we showed that when PBMCs were depleted of NK cells, NanoLuc did not accumulate anymore in co-culture supernatants ( Fig. 3E ). Reconstitution with purified NK cells restored the cytotoxic activity of PBMCs and the accumulation of NanoLuc in culture supernatants. Altogether, these data demonstrated the key role of NK cells in the cytotoxicity of PBMCs toward K562-NL cells and the accumulation of NanoLuc in culture supernatants.
Testing the Effects of Known Modulators of NK Cell Cytotoxicity
We then tested in this assay the effect of small compounds or cytokines that are known to modulate NK cell cytotoxicity, either directly or indirectly. First, PBMCs were stimulated with R848 (or resiquimod), a small agonist of TLR7 and TLR8. R848 mimics the immunostimulatory effects of single-stranded viral RNA molecules and stimulates NK cell cytotoxicity through the activation of antigen-presenting cells. PBMCs were pretreated overnight with R848, and K562-NL target cells were added. After 4 h of co-culture, NanoLuc activity in culture supernatants was measured. As shown in Figure 4A , R848 increased NanoLuc levels in co-culture supernatants as a consequence of NK cell stimulation. Similar results were obtained when pretreating PBMCs with IL-15, a cytokine known to increase the cytotoxicity of NK cells ( Fig. 4B ). 22 Alternatively, PBMCs were pretreated overnight with TGF-β, a cytokine known to inhibit NK cell functions. 23 As expected, NanoLuc accumulation in co-culture supernatants was reduced ( Fig. 4B ). Similar results were obtained with Torin2, a potent inhibitor of the mTOR pathway that is essential to NK cell cytotoxicity ( Fig. 4B ). 22 Altogether, these results demonstrated that our assay is suitable to screen small compounds or cytokines to identify new inhibitors or activators of NK cells.
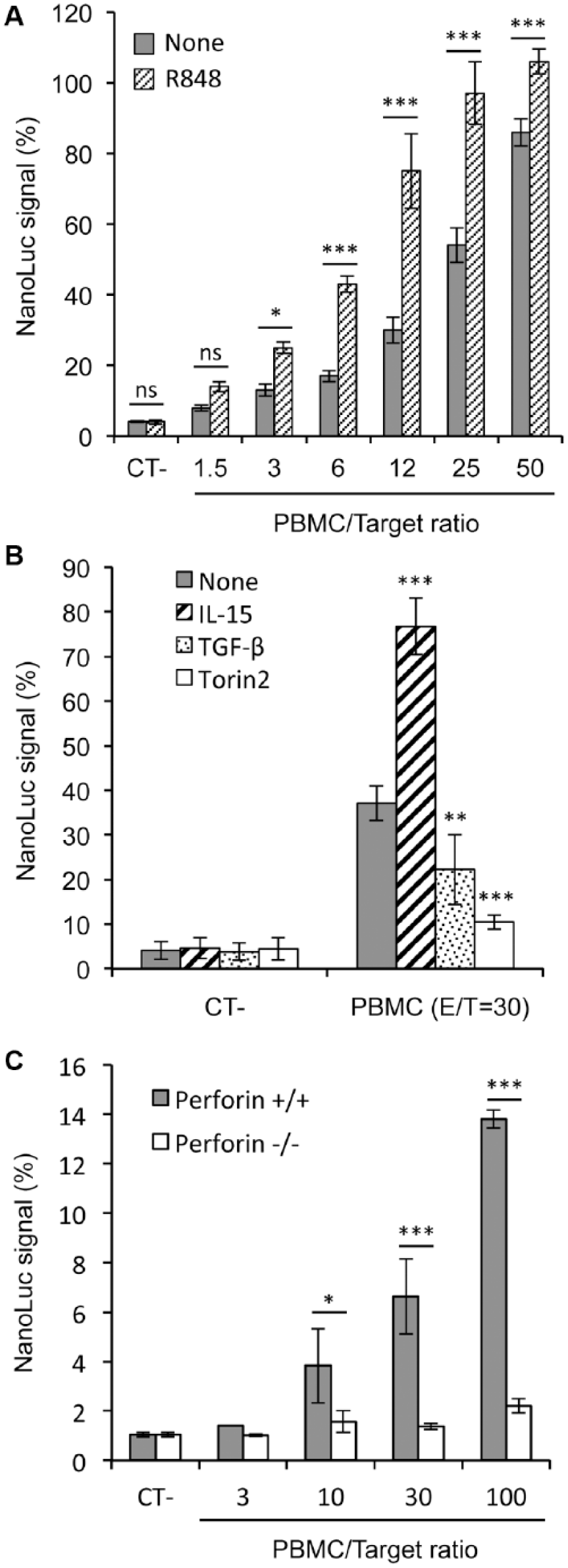
Testing the effects of known modulators of NK cell cytotoxicity. (
Familial histiolymphocytosis (FHL) is a severe inflammatory condition characterized by abnormally activated macrophages infiltrating multiple tissues, hepatosplenomegaly, nonremitting high fever, cytopenia, hypertriglyceridemia, and hyperferritinemia. It is caused by gene mutations that impair NK and CD8 T-cell cytotoxic function. In this group of diseases, perforin (PRF1) deficiency is responsible for FHL type 2 (FHL2). Quick diagnosis of FHL and potent treatment are essential to prevent fatal evolution. To determine if our assay could help in the diagnosis of such a pathological condition, PBMCs from a perforin-deficient patient were tested. Because of technical constraints, PBMCs from this patient and the matching healthy control were conserved overnight at 4 °C before the assay could be performed. This probably accounts for the low cytotoxic activity detected with control PBMCs even at a high E/T ratio. Nevertheless, the results obtained clearly showed that PBMCs from the perforin-deficient patient completely lost their cytotoxic activity as assessed by the absence of NanoLuc accumulation in culture supernatants ( Fig. 4C ). Therefore, this assay can be used to monitor ex vivo the cytotoxic activity of NK cells from patients while starting from a blood sample of only a few milliliters and could be used as a screening test in the diagnosis of diseases caused by defects in lymphocyte cytotoxicity.
Screening a Chemical Library of Metabolites, Xenobiotics, and Common Drugs
Our next objective was to test if our NanoLuc-based functional assay was suitable to rapidly screen hundreds of molecules using fresh PBMCs. To establish a proof of concept, we selected a chemical library from TMIC that aggregates 782 metabolites, xenobiotics, and frequently used over-the-counter medications: the TMIC Human Metabolome Library. Our objective was to determine if some of these molecules might impact the function of NK cells, with potential consequences on the organism’s ability to fight against virus infections or tumors.
PBMCs from two healthy donors were incubated overnight with compounds at 50 µg/mL, and K562-NL target cells were added to evaluate the cytotoxic activity of NK cells. Each screening plate included either four (donor 1) or seven (donor 2) positive and negative controls that corresponded to PBMCs + K562-NL cells or K562-NL cells alone, respectively. After 4 h of co-culture, NanoLuc activity was measured in culture supernatants. On average, the S/B ratio, which corresponds to the mean value of positive controls (μ+) divided by the mean value of negative controls (μ–), was 8.6 for donor 1 and 19.2 for donor 2. The S/N ratio, which corresponds to the difference of μ+ minus μ– relative to the standard deviation of negative controls (σ–), was 22.2 for donor 1 and 66.5 for donor 2. The Z′ factor, which reflects the overall quality of the assay and is calculated with the formula 1 – 3*(σ+ + σ–)/(μ+ – μ–), was 0.49 for donor 1 and 0.61 for donor 2. Altogether, these statistical parameters qualify this assay for HTS. 21 In parallel, PBMCs were incubated in the same conditions with test compounds, and ATP levels in culture wells were determined to identify toxic molecules ( Fig. 5A ). Eighty compounds reduced ATP levels by more than 25%, thus supporting some significant impact on cellular metabolic activity and viability. Of the remaining 702 compounds, 12 compounds were found to inhibit the lysis of target cells by >75% in the two screens, supporting the inhibition of NK cell cytotoxicity toward target cells ( Fig. 5A ).
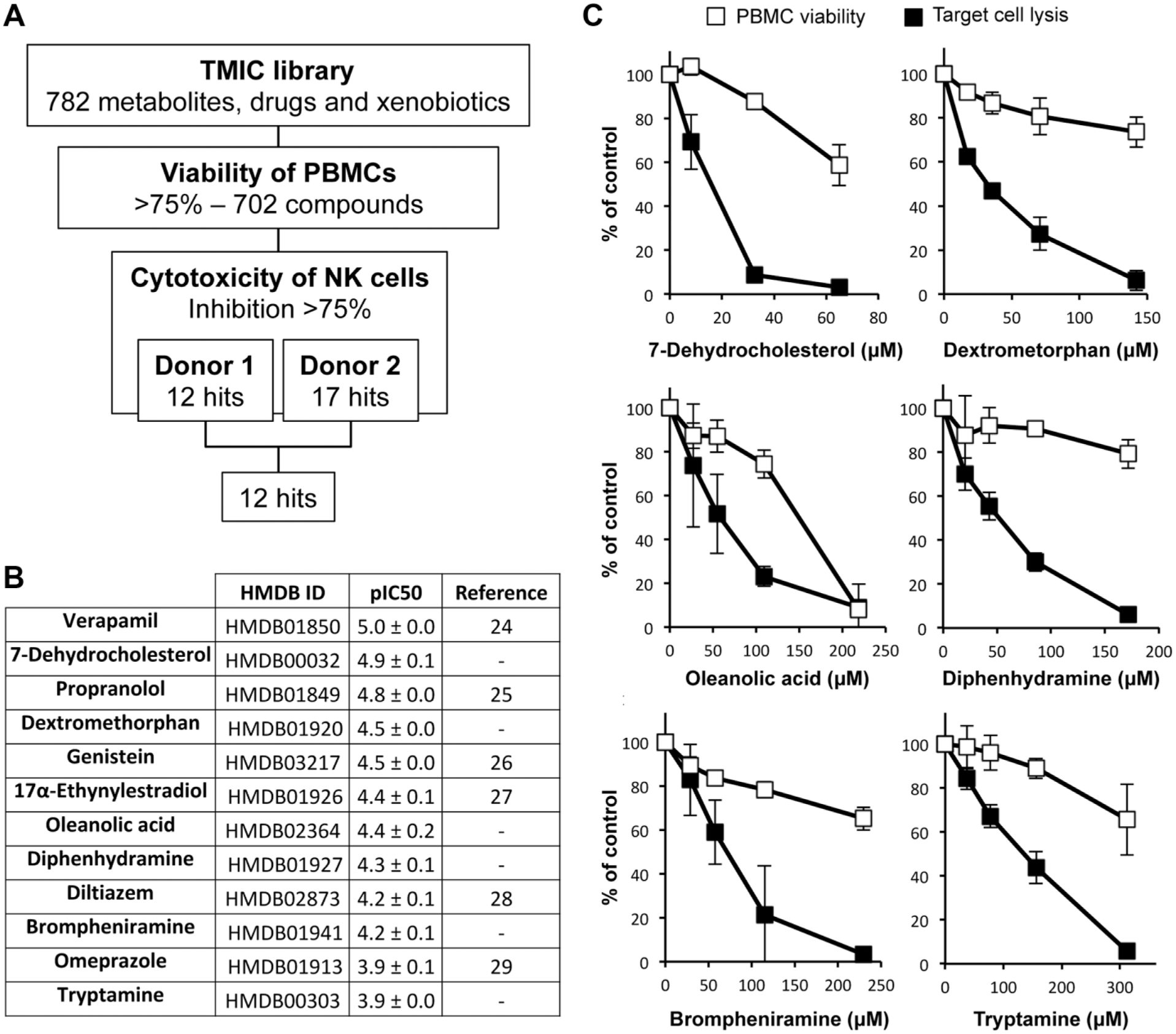
Screening a chemical library of metabolites, xenobiotics, and common drugs. PBMCs from two healthy donors were used to screen the TMIC Human Metabolome Library. (
Interestingly, six of these compounds were previously reported to inhibit NK cells ( Fig. 5B ), thus confirming the robustness of our screening pipeline.24–29 We also identified six compounds as new inhibitors, including three metabolites (7-dehydrocholesterol, oleanolic acid, tryptamine) and three medications (dextrometorphan, diphenhydramine, brompheniramine). These compounds were retested in dose–response experiments using PBMCs from at least three healthy donors to estimate the half-maximal inhibitory concentrations, which range from 10 to 126 µM ( Fig. 5C ). Dextrometorphan and diphenhydramine were particularly interesting because of the potent inhibition of target lysis, and a limited impact on the viability of PBMCs.
Development of a Dual-Reporter Assay to Measure Both Target Cell Lysis and IFN Secretion
NK cell activation is associated with the production of type II interferon (IFN-γ), which is released through vesicles and secretory pathways that are distinct from cytotoxic granules. 30 In order to further improve our screening assay, we developed new target cells for monitoring both the cytotoxic activity and the production of interferons by NK cells. We previously developed a HEK-293 cell line stably transfected with a firefly luciferase reporter gene controlled by ISREs in its promoter (STING-37 cell line). 20 These reporter cells not only respond to type I IFNs but also to IFN-γ, which efficiently induced the expression of firefly luciferase ( Fig. 6A ), making them suitable to detect the secretion of this cytokine by activated NK cells. Thus, STING-37 cells were engineered to express NanoLuc, so that NK cell cytotoxicity can be concomitantly monitored in the same culture wells. Best clones were amplified and carefully selected to establish a reporter cell line that we called twINNE because of its dual-reporter system based on firefly and NanoLuc luciferases. When these cells were co-cultured with PBMCs for 7 h at relatively low E/T ratios ranging from 0.5:1 to 4:1, NanoLuc expression in culture supernatants increased as a consequence of target cell lysis ( Fig. 6B ). In parallel, the cellular expression of firefly luciferase increased as the ISRE-luciferase reporter was induced in co-cultures ( Fig. 6B ). When the same experiment was performed at higher E/T ratios, the expression of firefly luciferase did not increase (data not shown), probably because target cells were lysed before the induction of the ISRE-luciferase reporter. Therefore, this dual-reporter system is allowing the quantification of both target cell lysis and interferon signaling from a single well, provided that experiments are performed at low E/T ratios. Finally, we tested Torin2, diphenhydramine, and dextromethorphan in this new assay. As expected, all three compounds were found to inhibit the lysis of target cells, but interestingly ( Fig. 6C , upper panel), only Torin2 completely blocked the expression of the ISRE-luciferase reporter ( Fig. 6C , lower panel). The situation was intermediate with dextromethorphan, while the ISRE-luciferase reporter was barely inhibited by diphenhydramine ( Fig. 6C , lower panel). These profiles demonstrate the inhibitory properties of these drugs, but also support distinct modes of action for Torin2, dextromethorphan, and diphenhydramine and different consequences on NK cell functions. Altogether, this validates our assay and highlights the interest of such a dual-reporter system.
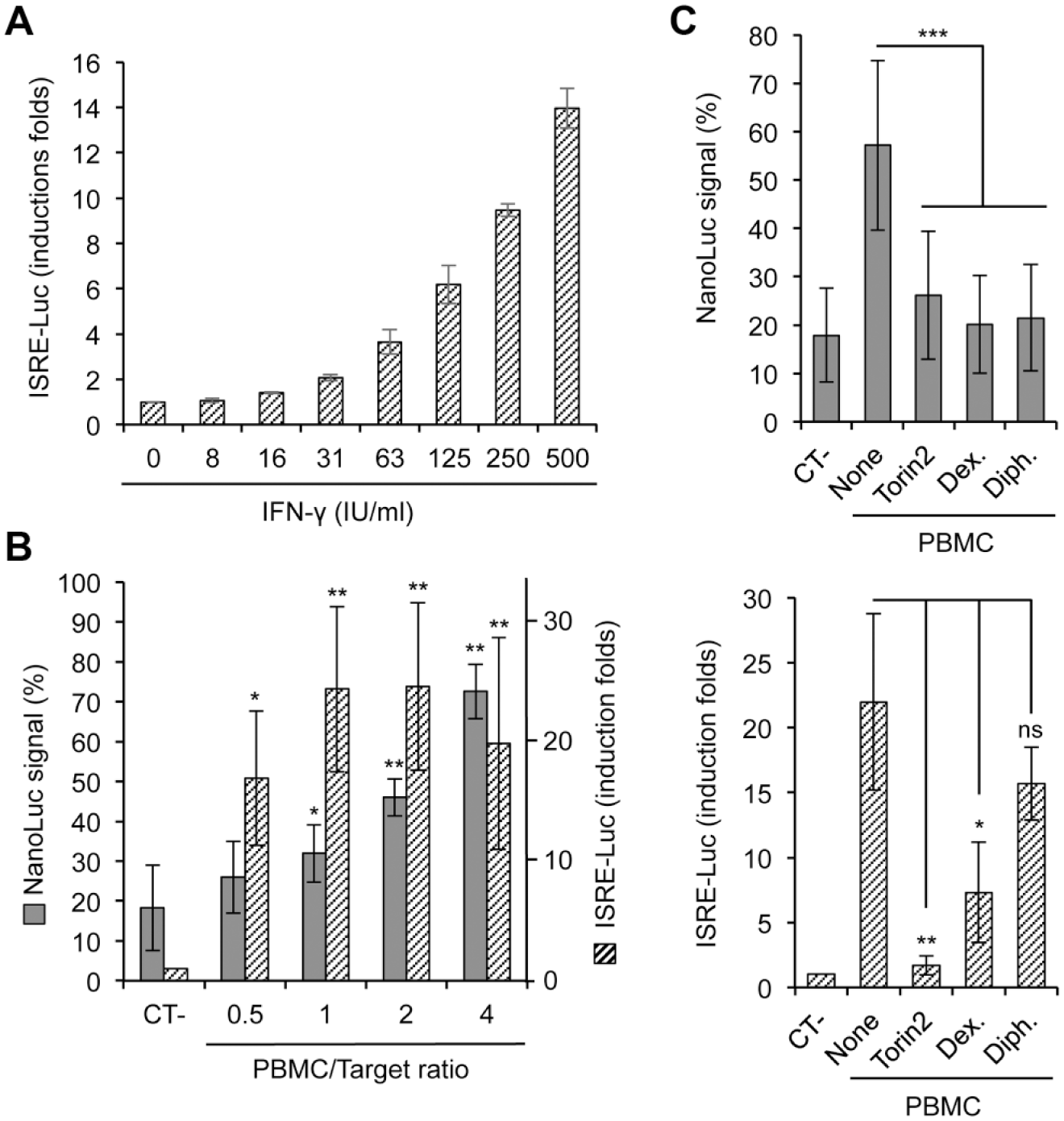
Measuring NK cell cytotoxicity and IFN secretion with dual-reporter cells. (
Discussion
NK cells play a central role in the innate immune response against intracellular pathogens and tumor cells and are considered prime targets for innovative immunotherapies, and functional defects are associated with pathological conditions. Both natural and artificial molecules, including cytokines, drugs, and natural products, have been defined as modulators of NK cell activity, and some of them are developed for medical purposes. The identification of novel modulators requires both target-based approaches and functional screenings that are complementary. But the latter is complicated since current NK cell assays are not easily adapted to the HTS of large libraries of compounds. Besides, quick functional assays, ideally usable at the patient’s bedside, are needed for the quick diagnosis of pathological conditions such as FHL where NK cell cytotoxicity is altered.
Luciferase-based functional assays have become increasingly popular because of the exceptional performances of this family of bioluminescent enzymes that emit a strong signal and are fully compatible with biological systems. Some attempts have been made to develop cell cytotoxicity assays based on firefly luciferase, but the poor stability of this enzyme after release in the extracellular medium greatly limited their performances.15,16,31 This analytical issue was solved with the development of brighter, smaller, and much more stable luciferases of marine origins, such as NanoLuc, that use coelenterazine or its chemical analog furimazine as a substrate.17–19 As we were developing our system, two research groups recently developed with success their own cytotoxicity assays by expressing these enzymes in target cells. They were used to measure antibody-dependent cellular cytotoxicity (ADCC), target cell lysis by chimeric antigen receptor (CAR) expressing T cells, and complement-dependent cytotoxicity.32,33 Performances were found to be at least similar to 51Cr release, which is considered the gold standard. Here, we established our own assay to measure the natural cytotoxicity of primary NK cells using appropriate targets, that is, K562 or HEK-293. Using PBMCs as a source of NK cells, this assay can be used to measure the inhibitory or stimulatory properties of small compounds or cytokines, but also to detect functional defects in PBMCs from patients ( Fig. 4C ). In the future, it will be interesting to further miniaturize this assay using 384-well plates as blood samples from patients, especially when testing babies, are limited in volume. Furthermore, we demonstrate the scalability of this assay as assessed by excellent statistical parameters. As proof of concept, we used the following settings to identify inhibitors of NK cells: 0.4 × 106 PBMCs/well at a 25/1 E/T ratio in a 96-well format. If we consider that 1 × 109 PBMCs are usually purified from one standard blood bag (500 mL), cells from a single donor are sufficient to screen 25 collection plates, including 80 compounds and control wells, which corresponds to 2000 different molecules. This makes possible the screening of medium-scale libraries on primary effector cells, a key point when looking for relevant cytotoxicity modulators.
From the screen of the TMIC Human Metabolome Library, we identified six molecules that were previously described in literature for their inhibitory properties on NK cells24–29 and six molecules that were not reported for this activity before. Interestingly, several of these molecules fall into pharmacological classes of drugs previously identified by Theorell et al. 34 as modulators of antibody-dependent NK cytotoxicity, including adrenergic receptor ligands (propranolol), Ca2+ channel blocker (verapamil, diltiazem), H1R-antagonist (diphenhydramine, brompheniramine), and serotonergic, dopaminergic, and muscarinergic antagonists (diphenhydramine, brompheniramine, dextrometorphan)—sometimes with opposite effects (17α-ethynylestradiol). However, all these drugs have multiple side targets, and mechanisms responsible for their biological activity toward PBMCs, and NK cells in particular, remain elusive without further investigations. Among the new NK cell inhibitors identified in the screen, dextromethorphan and diphenhydramine were the most interesting, showing some potent effects with a limited cytotoxicity on PBMCs. Dextrometorphan (d-3-methoxy-17-methylmorphinan) belongs to the morphinan family and is widely prescribed as a cough suppressant but is also used in the treatment of specific neurological disorders. It shows no affinity for the opioid receptor but was characterized as a nanomolar to low-micromolar-range antagonist of the NMDA receptor, an agonist of the σ1 receptor (σ1R), a serotonin–norepinephrine reuptake inhibitor, an antagonist of nicotinic receptors, and an inhibitor of voltage-gated calcium and potassium channels. 35 Interestingly, it was previously reported to have immunosuppressive properties at higher concentrations, for example, inhibiting the maturation of conventional dendritic cells, 36 but also disease progression in experimental mouse models of autoimmune disorders,37,38 and inflammation and oxidative stress in heavy smokers. 39 Mechanisms involved are still poorly understood, but could also control the activation of NK cells. One hypothesis is that dextrometorphan, through interactions with σ1R or voltage-gated channels, perturbs Ca2+ mobilization that is essential for NK cell activation and degranulation. A second inhibitor of interest identified in the screen was diphenhydramine, a common first-generation H1-antihistamine, which is closely related to brompheniramine, which was also among selected hits. The IC50 of diphenhydramine for the H1 receptor is in the nanomolar range, which is incompatible with the higher concentrations required for inhibiting NK cells ( Fig. 5C ). Diphenhydramine has pharmacological interactions with multiple secondary targets, including muscarinic and α-adrenergic receptors, but also organic cation, serotonin, dopamine, and norepinephrine transporters. 40 Several of these receptors are expressed by immune cells and are potentially involved in the inhibitory effects of diphenhydramine on NK cells. This is supported by a report showing that diphenhydramine suppresses the innate immune response in a mouse model of septic peritonitis, and these effects are not mediated by H1R. 41 Altogether, our data suggest that diphenhydramine, dextrometorphan, and related drugs that are widely used to treat the symptoms of common cold may have unwanted effects on the innate immune response. This adds elements to the current debate on the low benefit/risk ratio of these molecules. Finally, it should be noted that we did not identify NK cell activators in the screen, probably because immunostimulatory molecules, which potentially induce inflammation, are relatively rare among prevalent metabolites or frequently used drugs.
Finally, we further improved our screening system by developing an original dual-reporter cell line to monitor both the secretion of IFN-γ by NK cells and their cytotoxic activity. Results obtained showed a good correlation between NK cell cytotoxicity and IFN-γ expression, as expected ( Fig. 6B ). Interestingly, when testing NK cell inhibitors in this dual-reporter system, we found that Torin2 efficiently blocked target cell lysis and the IFN response, whereas the effects of dextromethorphan and diphenhydramine were skewed for the inhibition of NK cell cytotoxicity ( Fig. 6C ). This suggests that these drugs have different modes of action. Cytotoxic granules with perforin/granzymes and vesicles containing IFN-γ are separate entities, which are delivered to the surface of NK cells through different secretory pathways. 30 While the secretion of IFN-γ vesicles is not polarized, cytotoxic granules are released at the contact interface with target cells, and this pathway could be selectively inhibited, in particular by diphenhydramine. Further experiments using purified NK cells and cellular imaging should be performed to test this hypothesis. Finally, it should be stressed that the dual-reporter assay we developed should be performed at a relatively low E/T ratio to let the expression of IFN-γ induce the ISRE-luc reporter before target cells are killed. Provided that this rule is properly followed, this dual-reporter system will provide much more complete functional data on the activation of NK cells when screening libraries of compounds or biologics in high-throughput settings. In the future, this should help identify modulators of NK cells of interest for developing innovative therapies.
Footnotes
Acknowledgements
We acknowledge the technical support of Stéphanie Dupuy from the Flow Cytometry Platform at Université Paris Descartes. Vincent Hervin, Eloi Coutant, and Yves L. Janin are acknowledged for a generous gift of furimazine. This work was supported by the Agence National de la Recherche (ChemInnate program to P.O.V. and S.N.), the Agence Nationale de Recherche sur le SIDA et les hepatitis virales (ANRS), Campus France (Programme CEDRE), SantImmune from the Fondation Paris Descartes, the Centre National de la Recherche Scientifique (CNRS; www.cnrs.fr), and the Institut National de la Santé et de la Recherche Médicale (INSERM; ![]() ). S.H. was supported by the National Council for Scientific Research (Lebanon) and the Université Saint-Esprit de Kaslik (USEK).
). S.H. was supported by the National Council for Scientific Research (Lebanon) and the Université Saint-Esprit de Kaslik (USEK).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.