
Abstract
Human African trypanosomiasis, Chagas disease, and leishmaniasis are human infections caused by kinetoplastid parasites of the genera Trypanosoma and Leishmania. Besides their severity and global impact, treatments are still challenging. Currently available drugs have important limitations, highlighting the urgent need to develop new drugs. Phosphoglucose isomerase (PGI) is considered a promising target for the development of antiparasitic drugs, as it acts on two essential metabolic pathways, glycolysis and gluconeogenesis. Herein, we describe the identification of new nonphosphorylated inhibitors of Leishmania mexicana PGI (LmPGI), with the potential for the development of antiparasitic drugs. A fluorescence-based high-throughput screening (HTS) assay was developed by coupling the activities of recombinant LmPGI with glucose-6-phosphate dehydrogenase and diaphorase. This coupled assay was used to screen 42,720 compounds from ChemBridge and TimTec commercial libraries. After confirmatory assays, selected LmPGI inhibitors were tested against homologous Trypanosoma cruzi and humans. The PGI hits are effective against trypanosomatid PGIs, with IC50 values in the micromolar range, and also against the human homologous enzyme. A computational analysis of cavities present on PGI’s crystallographic structure suggests a potential binding site for the proposed mixed-type inhibition mechanism.
Introduction
Human African trypanosomiasis, Chagas disease, and leishmaniasis are neglected tropical diseases caused by the protozoan parasites Trypanosoma brucei, Trypanosoma cruzi, and Leishmania ssp., respectively, usually referred to as the Tritryps parasites. According to the World Health Organization, these three diseases combined affect more than 20 million people and cause more than 95,000 deaths per year. 1 The currently available chemotherapeutics are unsatisfactory, and consequently, more effective and safe drugs are urgently needed. In order to discover new drug candidates, it is necessary to improve our knowledge about the parasite biology and disease. Accordingly, studies on the metabolisms of these parasites may lead to the identification of new potential drug targets useful for drug discovery, which would fulfill key criteria such as essentiality, selectivity, assayability, and druggability. 2
In order to find new drug candidates for kinetoplastid diseases, the identification of inhibitors of the enzyme glucose-6-phosphate (G6P) isomerase, also known as phosphoglucose isomerase (PGI; EC 5.3.1.9), is considered a promising strategy. PGI is an aldose-ketose isomerase that catalyzes the reversible interconversion of G6P into fructose-6-phosphate (F6P). 3 The isomerization reaction proceeds with an intermolecular proton transfer, between C1 and C2, and the formation of a 1,2-cis-enediol(ate) intermediate.4–6 PGI participates in the glycolytic and gluconeogenesis pathways and can also be a shuttle to the oxidative branch of the pentose phosphate pathway (PPP) when G6P is used by the NADP-dependent enzyme, glucose-6-phosphate dehydrogenase (G6PDH). PPP represents an important source of NADPH, which acts as a reducing agent in various biosynthetic processes and for defense against oxidative stress.
Much experimental evidence indicates the relevance of glycolysis and gluconeogenesis for the viability and infectivity of the Tritryps parasites. RNAi-mediated PGI depletion causes the death of T. brucei procyclic forms growing in glucose-rich medium, probably as a consequence of glycolytic flux reduction. 7 In Leishmania major, the knockout fructose-1,6-biphosphatase, a key gluconeogenic enzyme, compromises the parasite proliferation inside the macrophage’s phagolysosomes and the development of skin lesions in infected mice. 8 For T. cruzi, metabolic profiling studies demonstrated that intracellular amastigotes stimulate the import of glucose by infected host cells, which is then directly taken up by the parasite and used for energy production through glycolysis. 9
Although PGI is essential in carbohydrate metabolism, its druggability and the identification of potent inhibitors that can be developed into effective drugs require further investigation. So far, the only available PGI inhibitors are phosphorylated compounds, analogous to the 1,2-cis-enediol reaction intermediate, 10 designed to assist in the elucidation of the enzyme catalytic mechanism.11,12 The presence of a phosphate group and the competitive inhibition mechanism compromise these compounds’ druglikeness. In the present work, we report the identification and characterization of the first nonphosphorylated Leishmania mexicana PGI (LmPGI) inhibitors, identified by chemical library screening. These inhibitors were also tested against human and T. cruzi homologous PGIs for the assessment of target selectivity, cellular-based viability assays for the evaluation of in vitro efficacy against parasites, and toxicity for mammalian cells.
Materials and Methods
Compound Libraries and Assay Reagents
The primary high-throughput screening (HTS) assay was performed against a total of 42,720 compounds that compose the DiverSet Library (29,680) purchased from ChemBridge (San Diego, CA) and the Diversity Screening Set (Diversity Set—10,000) and Natural Derivatives Library (NDL3000–3040) purchased from TimTec (Newark, DE). Libraries were received in 96-well (TimTec) or 384-well (ChemBridge) microplates, with compounds at a concentration of 10 mM in 100% DMSO. According to the library suppliers, DiverSet and Diversity Set are composed of chemically and structurally diverse compounds with druglike proprieties. Compounds in the NDL library are derivatives, analogues, or semisynthetics of natural products. Before screening, libraries were reformatted to 384 microplates and diluted to 1 mM in 100% DMSO. Columns 1, 2, 23, and 24 were filled with DMSO to be used as controls. For confirmatory assays, all hits were resupplied at 10 mM in 100% of DMSO as solvent. Leuconostoc mesenteroides G6PDH (EC 1.1.1.49), resazurin, NADP+, NADPH, G6P, F6P, and DMSO were purchased from Sigma-Aldrich (St. Louis, MO). Clostridium kluyveri diaphorase (EC 1.8.1.4) was obtained from Worthington Biochemical (Lakewood, NJ). Tris-HCl and NaCl were acquired from Merck (Whitehouse Station, NJ), Triton X-100 from Serva (Heidelberg, Germany), and the microplates from Greiner Bio-One (Monroe, NC).
Expression and Purification of Recombinant PGIs
Cloning of LmPGI and its human homologue (HsPGI) genes in pET vectors has been reported previously.13,14 Escherichia coli BL21(DE3) competent cells were transformed with pET28-LmPGI and pET29-HsPGI constructs. Cells were cultivated in 500 mL of autoinduction ZYM5052 medium, 15 supplemented with kanamycin (100 µg/mL) for 48 h at 22 °C. Cells were harvested by centrifugation (10,000g for 20 min), resuspended in 50 mL of buffer A (50 mM triethanolamine, pH 8.0), and stored at −20 °C. Cell lysis, clarification, and purification proceeded as described previously.13,14 For production of T. cruzi PGI (TcPGI), the construct pET14b_TcPGI was kindly provided by Dr. Robin Stacy from the Seattle Structural Genomics Center for Infectious Diseases (Seattle, WA). E. coli BL21(DE3) competent cells were transformed with pET14b_TcPGI and grown in 500 mL of autoinduction ZYM5052 medium, supplemented with ampicillin (100 µg/mL) for 48 h at 22 °C. Cells were harvested by centrifugation (10,000g for 20 min), resuspended in 50 mL of buffer B (50 mM triethanolamine, pH 8.0, 300 mM NaCl, 10 mM imidazole, and 3 mM β-mercaptoethanol), and stored at −20 °C. The cells were lysed by sonication, in the presence of lysozyme at 0.5 mg/mL, and cell debris was removed by centrifugation at 40,000g for 20 min. Total soluble protein was further clarified by 35% ammonium sulfate precipitation. The clarified sample was equilibrated in buffer B, by overnight dialysis, and then passed through a column filled with 5 mL of Ni-NTA resin (Qiagen, Madison, WI). Contaminants were washed out with 30 mM imidazole, and the protein eluted with 300 mM imidazole, in buffer B. Thereafter, eluted fractions were pooled together and subjected to size exclusion chromatography, using a HiLoad Superdex 200 16/60 column equilibrated with buffer C (20 mM HEPES, pH 7.5, 300 mM NaCl, and 1 mM β-mercaptoethanol). Purified proteins were concentrated and stored at −80 °C with glycerol 25%.
Primary HTS Assay
An assay with fluorescence readout was established by coupling the PGI activity in the direction of F6P to G6P with the auxiliary enzymes G6PDH and diaphorase ( Fig. 1 ). In this coupled reaction system, G6P produced through PGI activity serves as substrate for G6PDH that oxidizes it to 6-phosphogluconolactone, with the concomitant reduction of NADP+ to NADPH. Diaphorase uses the NADPH to reduce resazurin to resorufin, which can be measured spectrophotometrically, by monitoring the change in fluorescence intensity with excitation at λ = 545–20 nm and emission at λ = 600–40 nm.
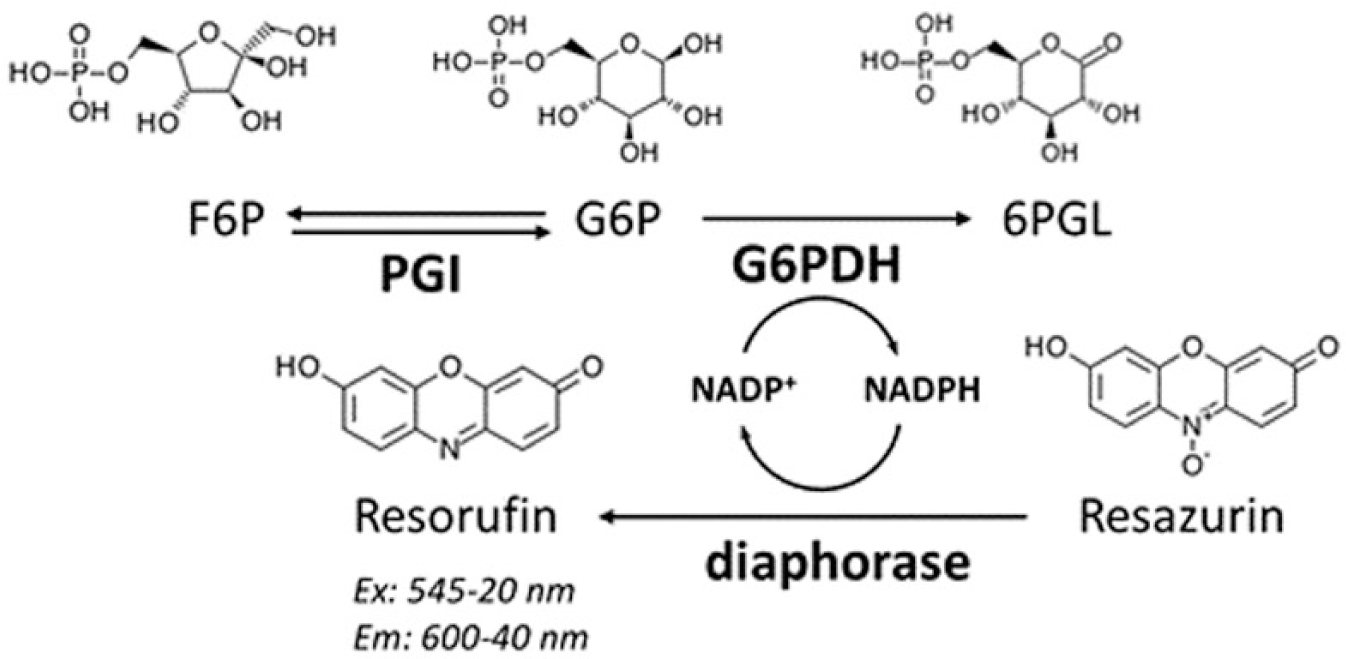
Coupled reaction system used to measure PGI activity and to establish the HTS assay. PGI catalyzes the conversion of F6P into G6P, which then is oxidized into 6-phosphogluconolactone (6PGL) by the NADP-dependent G6PDH. Finally, NADPH is used to reduce resazurin into resorufin, and the fluorescence intensity signal is detected using a microplate reader.
The primary HTS assay was performed at a single point, in 384-well, black, v-bottom microplates (Greiner Bio-One, cat. 781289). Except for tested compounds, other reagents were prepared in reaction buffer: 50 mM Tris-HCl (pH 7.5), 25 mM NaCl, and 0.01% Triton X-100. The HTS assay was assembled in three sequential steps as follows: dispense of the reaction mix (4.7 pM PGI, 1.1 U/mL G6PDH, 1.1 U/mL diaphorase, 0.022 mM resazurin, and 0.11 mM NADP+) in columns 1–24, transfer of compounds from 384-well library plates (1 mM in 100% DMSO) to columns 3–22, and dispense of substrate (6.25 mM F6P) in columns 2–23. Columns 1 and 24, which received reaction buffer but not substrate, were used as positive controls (100% inhibition). Columns 2 and 23, which received substrate but not compounds, were used as negative controls (0% inhibition). Steps involving dispensing were performed with the Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific, Waltham, MA). For the screening involving DiverSet Library, the final assay volume was 25 µL, transfer steps were performed with the JANUS MDT Automated Workstation (PerkinElmer, Waltham, MA), and fluorescence intensities were measured using the EnVision microplate reader (PerkinElmer). For screening involving the Diversity Set and NDL, the final assay volume was 50 µL, transfer steps were performed with the Versette Liquid Handling System (Thermo Fisher Scientific), and fluorescence intensities were measured using the ClarioStar microplate reader (BMG LABTECH, Ortenberg, Germany). To reduce the amount of reagent used, the screening of the largest library, DiverSet, was performed in a lower final reaction volume of 25 µL, without compromising assay quality. In this screening, the JANUS MDT Automated Workstation was used for the pipetting steps because it was able to pipet volumes lower than 1 µL, in an accurate manner.
Final assay concentrations of reagents and enzymes were as follows: 4.3 pM PGI, 1 U/mL G6PDH, 1 U/mL diaphorase, 0.02 mM resazurin, 0.1 mM NADP+, 50 µM F6P, 20 µM compounds, and 1% (v/v) DMSO. Fluorescence intensities were measured 1 h after the substrate dispense step, and the percentage of inhibition for each sample was calculated considering the mean values of positive (100% inhibition) and negative (0% inhibition) controls in each plate. Compounds that inhibited LmPGI activity greater than 30% were considered hit candidates. A reference compound was not present among the controls because specific PGI inhibitors known to date are not commercially available. The HTS assay was evaluated by the Z factor calculated for each plate screened. A Z factor higher than 0.5 is considered indicative of acceptable quality for HTS assays. 16
Confirmatory Assays
To remove false positives and define a resupply list, hit candidates were cherry-picked from reformatted library plates and retested against LmPGI and auxiliary enzymes of the coupled reaction in independent assays. Confirmatory assays were assembled in three pipetting steps, similar to the primary HTS assay, using the Multidrop Combi Reagent Dispenser and the Versette Liquid Handling System (Thermo Fisher Scientific), in the dispense and transfer steps, respectively. In summary, an enzyme mix was dispensed, and then compounds were transferred and the reaction was started by substrate dispensing. Final concentrations of reagents in the LmPGI inhibition confirmatory assay were as follows: 20 pM LmPGI, 1 U/mL G6PDH, 1 U/mL diaphorase, 0.02 mM resazurin, 0.1 mM NADP+, 20 µM compounds, 1% DMSO, and 50 µM F6P (as substrate). For G6PDH inhibition assays, final concentrations of reagents were 0.01 U/mL G6PDH, 1 U/mL diaphorase, 0.02 mM resazurin, 0.1 mM NADP+, 20 µM compounds, 1% DMSO, and 0.5 mM G6P (substrate). For the diaphorase inhibition assay, final concentrations of reagents were 0.04 U/mL diaphorase, 0.1 mM NADPH, 20 µM compounds, 1% DMSO, and 0.02 mM resazurin (substrate).
Confirmatory assays were performed in the kinetic mode with fluorescence intensities followed for 30 min, using the ClarioStar microplate reader (BMG LABTECH). Resorufin formation rates were calculated at an interval of 5 min, in MARS Data Analysis Software (BMG LABTECH), and normalized considering the mean values of positive and negative controls in each plate. Compounds that inhibited diaphorase activity greater than 20% were excluded for further confirmatory assays. For the remaining compounds, LmPGI inhibition greater than 50% and a difference between G6PDH and LmPGI inhibition greater than 30% (that corresponded to the average minus 3 standard deviations) were used to defy the resupply list.
IC50 Calculation
Confirmed hits were resupplied from supplier’s available stocks, at 10 mM in 100% DMSO, following the same purity criteria (minimum of 90%) as compounds in the library, confirmed by nuclear magnetic resonance and mass spectrometry. For IC50 experiments, compounds were serially diluted (1:2 factor) in 96-well, v-bottom microplates, from column 1 to 11. Column 12 received only DMSO to be used as positive control (100% enzyme activity). The negative control was defined as 0% enzyme activity. The serial dilution procedure was performed within the Versette Liquid Handling System (Thermo Fisher Scientific).
IC50 values of the resupplied hits were determined against LmPGI, HsPGI, and TcPGI homologue enzymes. For LmPGI and HsPGI, the reagent concentrations were the same as used in the confirmatory assay. For TcPGI, the target enzyme concentration was 30 pM. IC50 assays were performed in quadruplicate, in 384-well, black, v-bottom microplates, with a final volume of 50 µL/well. Final concentrations of reagents were 20 pM (LmPGI and HsPGI) or 30 pM (TcPGI) PGI, 1 U/mL G6PDH, 1 U/mL diaphorase, 0.02 mM resazurin, 0.1 mM NADP+, serial diluted compounds from 100 to 0.09 µM, 1% DMSO, and 50 µM F6P (as substrate). Positive and negative controls received 1% of DMSO instead of compound and reagent buffer instead of substrate, respectively. Pipetting procedures were similar to the procedures performed for the confirmatory assays. Resorufin formation rates were calculated at an interval of 5 min, in MARS Data Analysis Software (BMG LABTECH). Data were normalized and fitted to a sigmoidal curve in GraphPad Prism v6.0 (GraphPad, San Diego, CA).
Mechanism of Inhibition Determination
The mechanism of inhibition was determined against LmPGI, in which the effects of different inhibitor concentrations were evaluated on Lineweaver–Burk plots for variable F6P concentrations. For these experiments, compound and substrate (F6P) were serially diluted (1:2 factor) in 96-well, v-bottom microplates. Assays were performed in quadruplicate, in 384-well, black, v-bottom microplates, in a final volume of 25 µL/well. Resorufin formation rates were calculated at an interval of 5 min, in MARS Data Analysis Software (BMG LABTECH). Inhibition constant calculations and Lineweaver–Burk plots were carried out using GraphPad Prism v6.0.
Molecular Docking Studies
Cavities in the homodimeric LmPGI protein structure (PDB ID: 1T10) were generated by KVFinder software. 17 The atomic structure of compound ST082230 was obtained from the ZINC database 18 (ZINC32117634) and protonated in aqueous solution using LeadIt software (BioSolveIT, Inc., Bellevue, WA) before docking. Docking was performed in the cavity identified by KVFinder software, using the FlexX method, 19 with a maximum number of solutions per interaction and per fragmentation set to 2000. After the docking procedure, the complex structure was minimized in solvent by steepest descent, and simulated annealing, using Yasara software, 20 with force field AMBER14. Figures were prepared using PyMOL v1.9 software (Schrödinger, Inc., New York, NY).
In Vitro Parasiticidal Effect and Cellular Toxicity Assay
The LmPGI inhibitors resupplied were tested against the Leishmania amazonensis amastigote form infecting the J774 monocyte/macrophage lineage (ATCC TIB-67), T. cruzi (Tulahuen strain) infecting NIH/3T3 cells (ATCC CRL-1658), and NIH/3T3 cells alone. Strains of L. amazonensis and T. cruzi used in these assays were genetically modified to constitutively express β-lactamase and β-galactosidase, respectively.21,22 These enzymes catalyze the conversion of commercially available substrates (CENTA β-Lactamase Substrate or Chlorophenol Red-β-D-galactopyranose-CPRG, respectively) into products suitable for spectrophotometric light absorbance measurements.
J774 macrophages were cultivated in RPMI medium (Corning, Troy, MI), supplemented with 10% fetal bovine serum (FBS; Seradigm, Radnor, PA), and 1% pentamycin-streptomycin-glutamine (PSG; Corning) for 96 h, at 37 °C and 5% CO2. The L. amazonensis promastigotes were grown in the same medium, at 26 °C, until cultures reached thestationary phase. T. cruzi trypomastigotes were cultivated with LLC-MK2 cells growing in Dulbecco’s modified Eagle’s medium (DMEM) without phenol red (Corning), supplemented with 2% FBS and 1% penicillin-streptomycin (PS; Corning), at 37 °C and 5% CO2. To recover active trypomastigotes, the supernatant of the LLC-MK2-infected culture flask was harvested between the fifth and seventh day after infection (at 1000g for 7 min) in Falcon tubes. After centrifugation, the tubes were incubated for 3 h, at 37 °C, to allow the active trypomastigotes to migrate from the pellet to the supernatant, which was used to infect NIH/3T3 cells, growing separately in DMEM with phenol red, supplemented with 10% FBS and 1% PS, at 37 °C and 5% CO2.
The L. amazonensis amastigote inhibition assay was adapted from the colorimetric assay described previously by Buckner and Wilson. 22 Briefly, the compounds were tested in 96-well, clear, sterile plates (Corning, cat. 353072), using RPMI supplemented with 10% FBS and 1% PSG as the assay medium. J774 cells (5 × 104 cells/well) and the promastigote parasites (125 × 104 parasites/well) were concomitantly plated to a final volume of 200 µL/well. After 24 h of incubation at 37 °C and 5% CO2, the extracellular parasites that did not infect the host cells were washed out and compounds previously diluted in medium (50 µM, 1% DMSO) were transferred to the assay plates. The infected cells were incubated with compounds for 96 h and parasite viability was evaluated by light absorbance at 405 nm using the CENTA substrate at 100 µM (Calbiochem, Billerica, MA) and 0.1% Nonidet P-40 (Roche, Mannhein, Germany).
The T. cruzi intracellular inhibition assay was developed as previously described by Bettiol and coworkers. 23 The compounds were tested in 96-well, clear, sterile plates (Corning, cat. 353072), using DMEM without phenol red, supplemented with 2% FBS and 1% PS as the assay medium. The NIH/3T3 (5 × 104 cells/well) and active T. cruzi trypomastigotes (5 × 104 parasites/well) were concomitantly plated to a final volume of 200 µL. The extracellular parasites that did not infect the host cells were washed out after 24 h of incubation at 37 °C and 5% CO2, and compounds previously diluted in medium (50 µM, 2% DMSO) were transferred to the assay plates. The infected cells were further incubated with compounds for 72 h, and parasite viability evaluated by light absorbance was measured at 570–595 nm using 500 µM CPRG and 0.5% of Nonidet P-40.
The inhibitor’s cytotoxicity was evaluated against NIH/3T3 fibroblastic cells line. The assay was performed in white, 96-well, clear-bottom, sterile plates (Greiner Bio-One, cat. 655098), using DMEM with 2% FBS and 1% PSG, and without phenol red. The NIH/3T3 (5 × 104 cells/well) cells and compounds (100 µM, 2% DMSO) were plated in a final volume of 200 µL and incubated for 3 days at 37 °C and 5% CO2. The cell viability was evaluated by fluorescence measured at λ 485 nm excitation and λ 530 nm emission, using the Alamar Blue reagent (Thermo Scientific) as a mitochondrial activity indicator.
Absorbance or fluorescence readouts were measured in a VICTOR X3 microplate reader (PerkinElmer). The corresponding relative parasite or host cell viability inhibition was calculated using negative (cells or infected host cells) and positive (cells treated with 100 µM ionomicyn and infected cells with 4 µM amphotericin) controls. The samples and controls were present in triplicate and quintuplicate per plate, respectively. The calculation of EC50 and TC50 values was performed with GraphPad Prism v6.0 by a nonlinear fit.
Results and Discussion
Three commercial libraries, totaling 42,720 compounds, were screened for LmPGI inhibition using a coupled reaction assay with G6PDH and diaphorase as auxiliary enzymes ( Fig. 1 ). The mean Z-factor values for DiverSet (ChemBridge), Diversity Set (TimTec), and NDL3000 (TimTec) plates were 0.72, 0.89, and 0.89, respectively. The complete HTS process consisted of four consecutive biochemical assay steps, starting with the primary HTS, for the selection of hit candidates, and then proceeding with three confirmatory assays to remove false-positive compounds acting on the auxiliary enzymes ( Fig. 2A ). In the first step, 291 compounds were selected as hit candidates, with LmPGI inhibition above 30% ( Fig. 2B ). Those compounds were cherry-picked from library stocks to be used in the confirmatory assays.
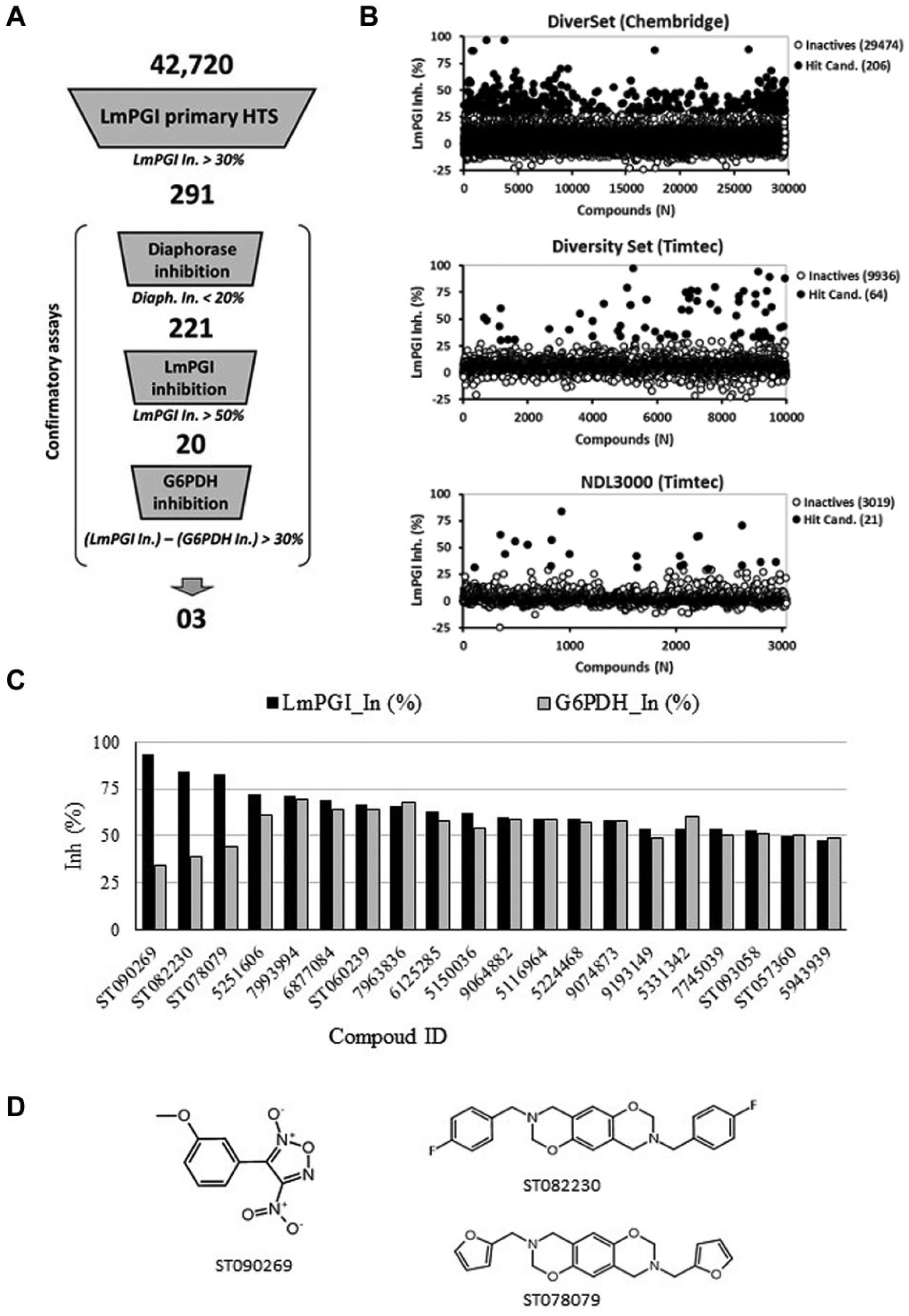
(
In the first confirmatory assay, 70 compounds exceeded the diaphorase inhibition limit of 20% and were excluded from further assays. The remaining 221 compounds were retested against LmPGI, and among them, only 20 compounds had confirmed LmPGI inhibition greater than 50%. The final confirmatory assay consisted of measuring G6PDH inhibition to exclude compounds that inhibited both enzymes. The 20 compounds selected as LmPGI inhibitors were also active to some extent against G6PDH (Supplemental Sections S1 and S2). To select the best hits, compounds were ranked by the difference between LmPGI and G6PDH inhibition values ( Fig. 2C ). Three compounds, ST090269, ST082230, and ST078079 ( Fig. 2D ), presented the largest difference between LmPGI and G6PDH inhibition (greater than 30%) and so were selected for further characterization.
An unusual kinetics inhibition pattern was observed for compound ST090269 from the dose–response experiment, with increasing IC50 values calculated at different time intervals, over the time course of the experiment. This pattern suggested that the compound lost affinity for the target during the experiment, probably by modification of its chemical structure. For this reason, this compound was excluded from further assays. IC50 values were calculated for compounds ST082230 and ST078079 against LmPGI and homologous enzymes from T. cruzi and humans ( Table 1 ). There were no significant differences in the IC50 values observed from the tested enzymes. This lack of selectivity suggested conserved binding sites for those compounds in the homologous enzymes. Indeed, target selectivity is highly desired when considering drug development for infectious diseases. Although the hits described here were not selective to parasite enzymes, they are the first nonphosphorylated and druglike PGI inhibitors described to date. In this way, they can be useful tools to investigate PGI function in biochemical and cell-based assays. Additionally, understanding how these compounds interact with PGI might also suggest chemical modification for gain of target affinity and selectivity.
In Vitro Characterization of Confirmed Hits against PGI Enzymes and Cell Viability Assay.
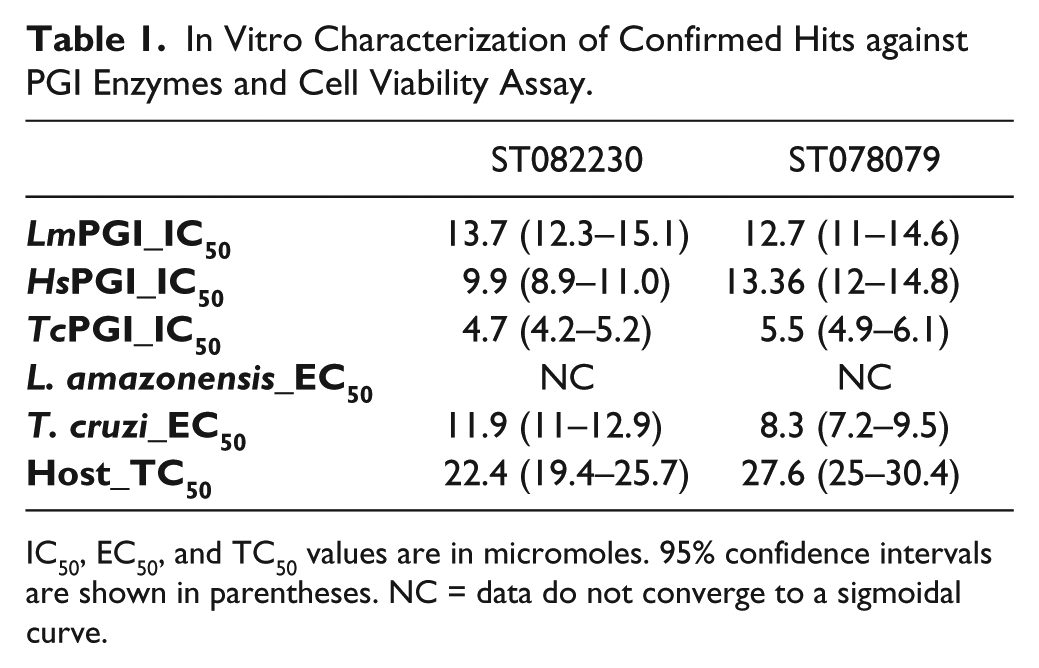
IC50, EC50, and TC50 values are in micromoles. 95% confidence intervals are shown in parentheses. NC = data do not converge to a sigmoidal curve.
Compounds ST082230 and ST078079 share a common benzo-oxaza-perhydroine ring system with a symmetrical chemical structure. These compounds are not structurally related to PGI substrates, and therefore may not bind directly in the enzyme catalytic site. The inspection of cavities on parasite and human PGI crystallographic structures points to a potential binding site to these compounds localized close to the substrate binding site, in the dimeric interface ( Fig. 3A ). Docking of ST082230 on this site suggests that there are two molecules of the inhibitor occupying symmetric sites of the cavity ( Fig. 3B ). The binding of the compound could perturb residues near the catalytic sites and compromise the affinity of the enzyme for its substrates. If this is the case, these compounds would not compete with the natural substrates for the catalytic site, and they should affect both Vmax and Km values in a mixed-model inhibition mechanism. The inspection of the ST082230 inhibition mechanism confirmed the mixed type with a 10-fold increase in Km values for F6P and a 2-fold reduction on Vmax ( Fig. 4 ).
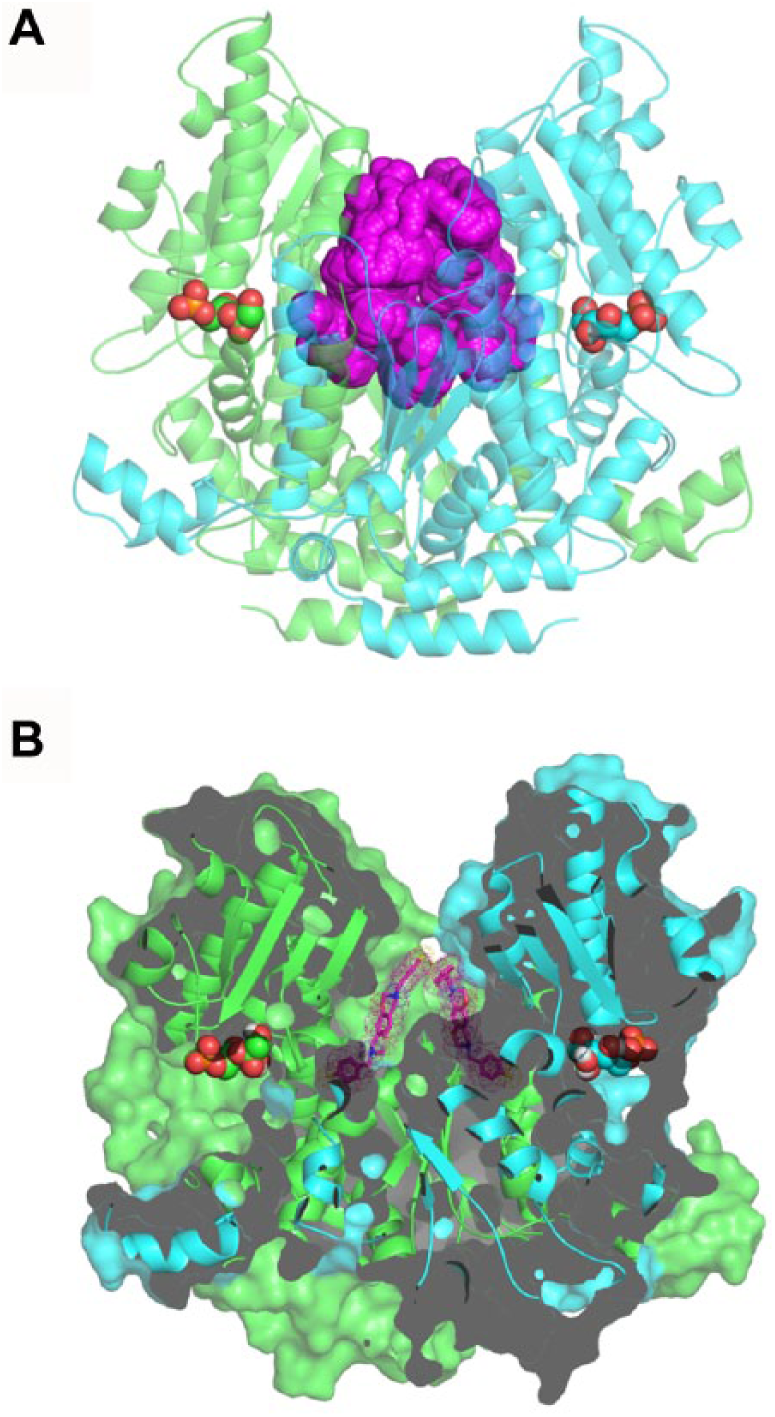
(
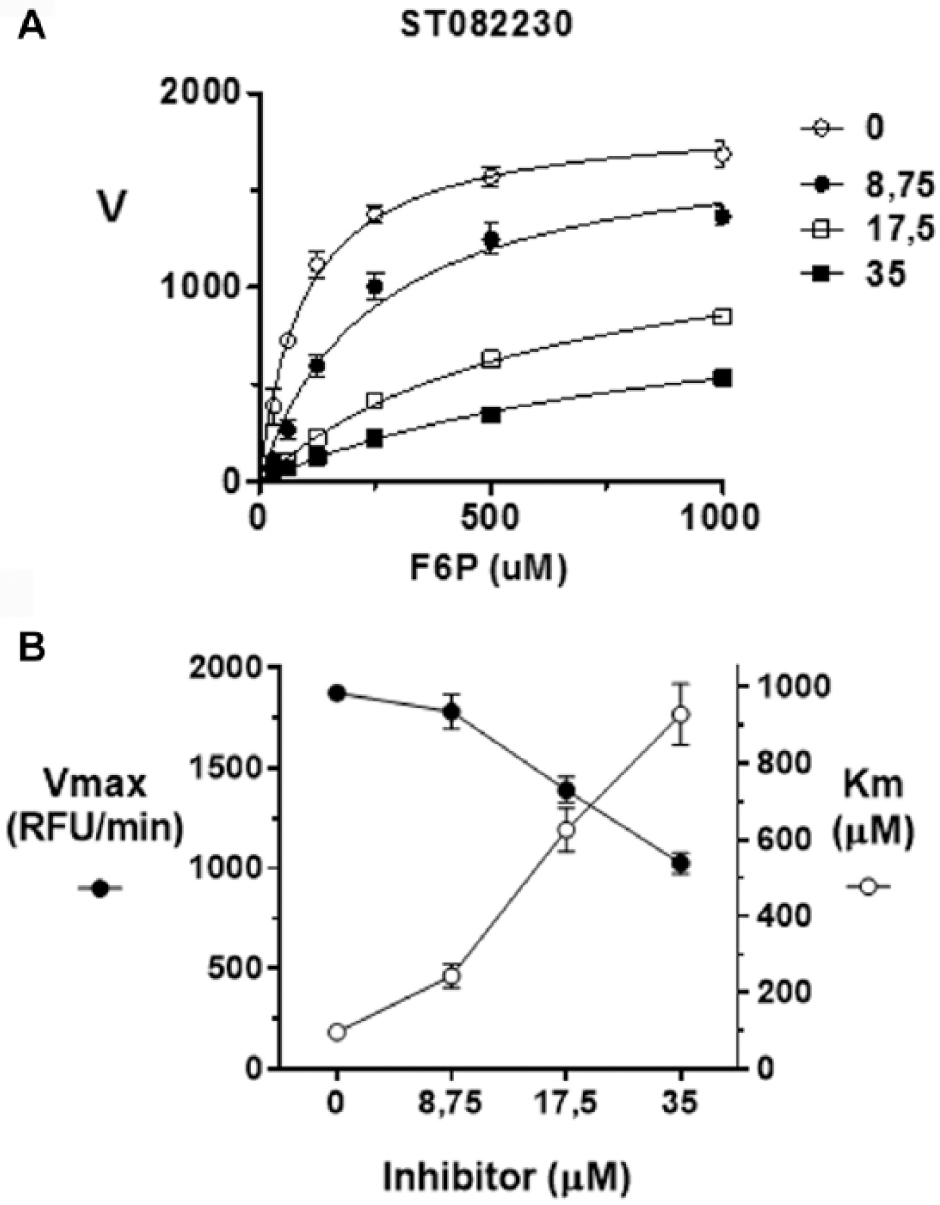
ST082230 mixed-model inhibition mechanism. (
In the cell-based viability assays, compounds ST082230 and ST078079 were not effective against Leishmania amastigote infection of J744 macrophages. For both compounds, the viability of infected macrophages exposed to the highest assayed concentration was not different from that of untreated cells. The absence of activity can be associated with multiple factors, like reduced drug access to macrophage phagolysosomes, where amastigotes reside, or even chemical instability due to the low pH of this cellular compartment. In experiments against the T. cruzi amastigote form and noninfected NIH/3T3 host cells, compounds ST082230 and ST078079 were effective against the parasite, with EC50 values below 10 µM, but they were also cytotoxic toward noninfected NIH/3T3 fibroblast host cells, with TC50 between 20 and 30 µM ( Table 1 ). Despite the narrow safety windows (TC50/EC50) of both PGI hits ( Fig. 5 ), in values close to the EC50, it is possible to observe the effectiveness of compounds on parasite viability without interference on host cells.
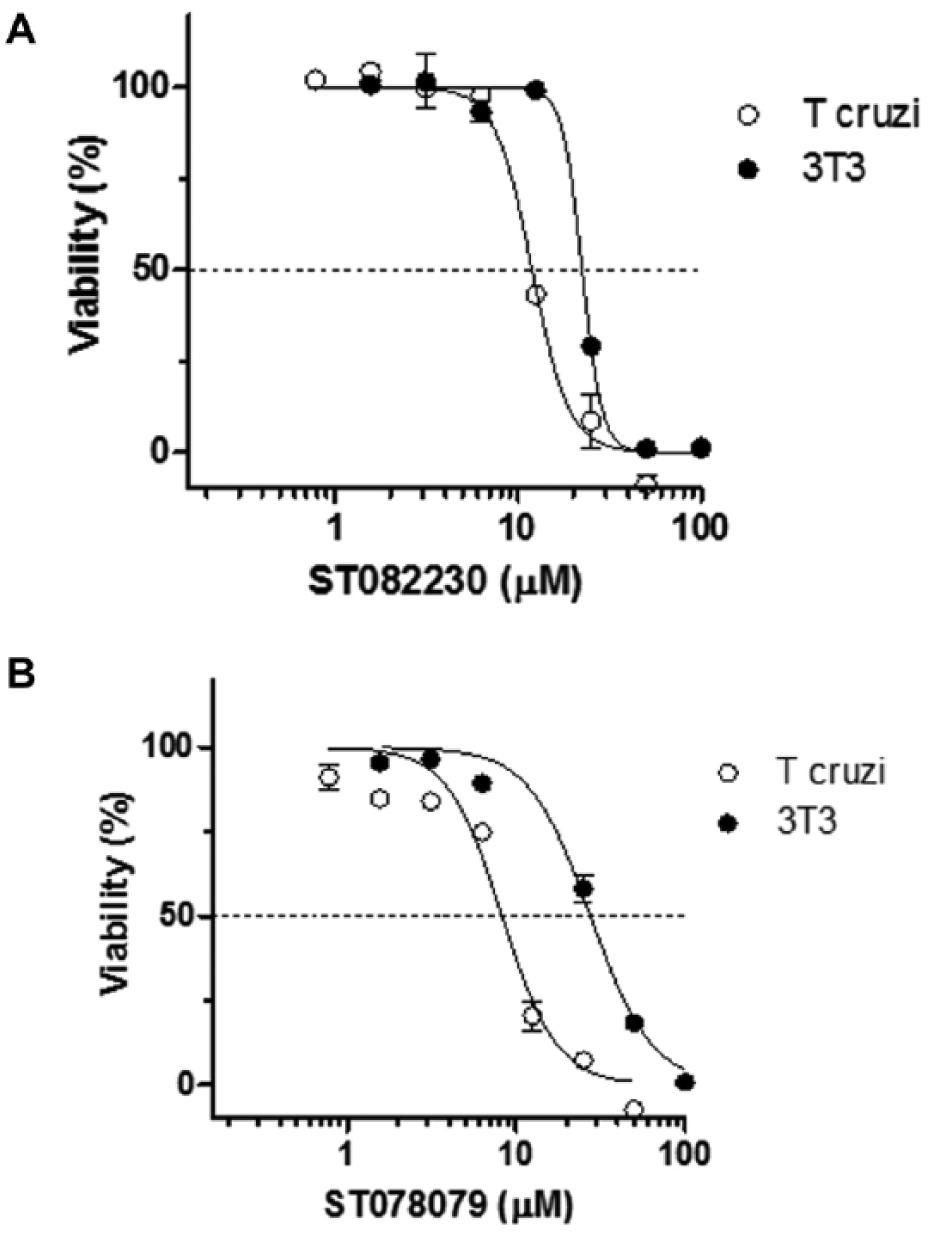
In vitro parasiticidal effect and cellular toxicity assay of PGI inhibitors identified: (
In conclusion, the few confirmed hits identified in the HTS assay targeting LmPGI suggest that this enzyme is hardly druggable. An alternative strategy for identifying initial hits for poorly druggable targets would be the screening of natural product libraries, which explore a diversity chemical space not necessarily encompassed within commercial libraries of synthetic molecules. In fact, HTS of microbial natural product extracts against kinetoplastid parasites has shown success in the identification of promising drug candidates. 24 Nevertheless, the hits discovered in the present work are the first nonphosphorylated PGI inhibitors with druglike characteristics and may be considered a starting point toward the development of lead compounds that might feed the antiparasitic drug discovery pipelines. Since compounds ST082230 and ST078079 were also active against human PGI, they can also be used to investigate the biological role of mammalian PGI in in vitro assays. Finally, those compounds active on both PGI and G6PDH can be considered for the development of multitarget inhibitors of glycolytic and the PPP.
Supplemental Material
Supp_Material_for_Cordeiro_et_al – Supplemental material for First Nonphosphorylated Inhibitors of Phosphoglucose Isomerase Identified by Chemical Library Screening
Supplemental material, Supp_Material_for_Cordeiro_et_al for First Nonphosphorylated Inhibitors of Phosphoglucose Isomerase Identified by Chemical Library Screening by Sabrina G. R. Mota, Gustavo F. Mercaldi, José G. C. Pereira, Paulo S. L. Oliveira, Ana Rodriguez and Artur T. Cordeiro in SLAS Discovery
Footnotes
Acknowledgements
We thank Dr. Robin Stacy (Structural Genomics Center for Infectious Disease, Seattle, WA) for supplying the TcPGI plasmid, the New York University School of Medicine for support with the development of the cell-based viability assays, the Brazilian National Laboratory of Biosciences (LNBio) from the Brazilian National Center for Research in Energy and Materials (CNPEM) for the infrastructure and technical assistance, and Dr. Edward L. D’Antonio from the University of South Carolina Beaufort for critical review of the manuscript.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: São Paulo Research Foundation (FAPESP) research grant for A.T.C. (2016/14271-4), PhD fellowship for S.G.R.M. (2014/15590-0; 2016/19141-1), and postdoctoral fellowship for G.F.M. (2016/03151-8).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.