
Abstract
Neuroinflammation is becoming increasingly recognized as a critical factor in the pathology of both acute and chronic neurological conditions. Inflammasomes such as the one formed by NACHT, LRR, and PYD domains containing protein 3 (NLRP3) are key regulators of inflammation due to their ability to induce the processing and secretion of interleukin 1β (IL-1β). IL-1β has previously been identified as a potential therapeutic target in a variety of conditions due to its ability to promote neuronal damage under conditions of injury. Thus, inflammasome inhibition has the potential to curtail inflammatory signaling, which could prove beneficial in certain diseases. In this review, we discuss the evidence for inflammasome contributions to the pathology of neurodegenerative conditions such as Alzheimer’s disease and Parkinson’s disease, epilepsy, and acute degeneration following brain trauma or stroke. In addition, we review the current landscape of drug development targeting the NLRP3 inflammasome.
Introduction
Inflammation and Inflammasomes
Inflammation is a host response to injury and infection and is generally considered beneficial. However, inflammation is becoming recognized as a contributor to disease. This is well illustrated by the recent Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS), where patients with a history of myocardial infarction were treated with canakinumab, a monoclonal antibody targeting the pro-inflammatory cytokine interleukin-1β (IL-1β). 1 In the CANTOS trial, it was found that canakinumab treatment reduced the rate of recurrent cardiovascular events and cancer mortality, in addition to improving many other clinical outcomes. 1 IL-1β and other IL-1 family members are strongly implicated in inflammatory disease.2,3 IL-1β is produced as an inactive precursor (pro-IL-1β) by cells of the innate immune system, such as monocytes and macrophages, in response to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs). PAMPs are molecular motifs carried by pathogens (e.g., lipopolysaccharide [LPS] from Gram-negative bacteria). DAMPs are endogenous molecules released from dead cells or modified during disease (e.g., high-mobility group box 1 [HMGB1]). These danger signals are recognized by pattern recognition receptors (PRRs) (e.g., toll-like receptors [TLRs]) on the membranes of innate immune cells and induce signaling pathways (e.g., nuclear factor kappa-light-chain-enhancer of activated B cells [NF-κB]) that result in the upregulation of pro-IL-1β. A major mechanism by which pro-IL-1β is cleaved to an active secreted form of IL-1β is via the formation of large cytoplasmic multimeric protein complexes called inflammasomes. 4
Inflammasomes are typically composed of a sensor (a cytosolic PRR) and an adaptor protein called apoptosis-associated speck-like protein containing a caspase-recruitment domain (CARD) (ASC), and an effector such as the protease caspase-1. 4 The most commonly studied inflammasome is composed of the cytosolic PRR NACHT, LRR, and PYD domains containing protein 3 (NLRP3). In a PAMP or DAMP primed cell in which pro-IL-1β and NLRP3 have been expressed, an additional encounter with another PAMP or DAMP is typically required to induce formation of the NLRP3 inflammasome ( Fig. 1A ). 4 Upon activation, NLRP3 undergoes a conformational change and interacts with never in mitosis A-related kinase 7 (NEK7).5,6 The adaptor protein ASC is then recruited and undergoes oligomerization into large inflammasome specks upon which pro-caspase-1 is recruited and activated.7,8 Caspase-1 then cleaves pro-IL-1β to a mature active form, which is then released from the cell to promote inflammation. 9 Posttranslational modifications involving NLRP3 phosphorylation and deubiquitination are crucial for inflammasome assembly.10–13 K+ efflux is now widely accepted as an essential step in the activation of NLRP3 inflammasomes, 14 and recent studies from us and others suggest an involvement of Cl− channels.15–17 Caspase-1 also drives a rapid inflammatory form of programmed cell death called pyroptosis, in which cell lysis occurs and pro-inflammatory mediators are released. 18 Pyroptosis requires the caspase-1-dependent cleavage of gasdermin D (GSDMD) and the formation of plasma membrane pores by the cleaved GSDMD N-terminus.19,20 The secretion of IL-1β is poorly characterized, and we recently speculated that membrane pore formation may represent the secretory mechanism semianalogous to the mechanism used by the unconventionally secreted fibroblast growth factor 2 (FGF2). 9 The inflammasome specks can themselves also be released from pyroptotic cells and can further contribute to inflammation in the extracellular space.21,22
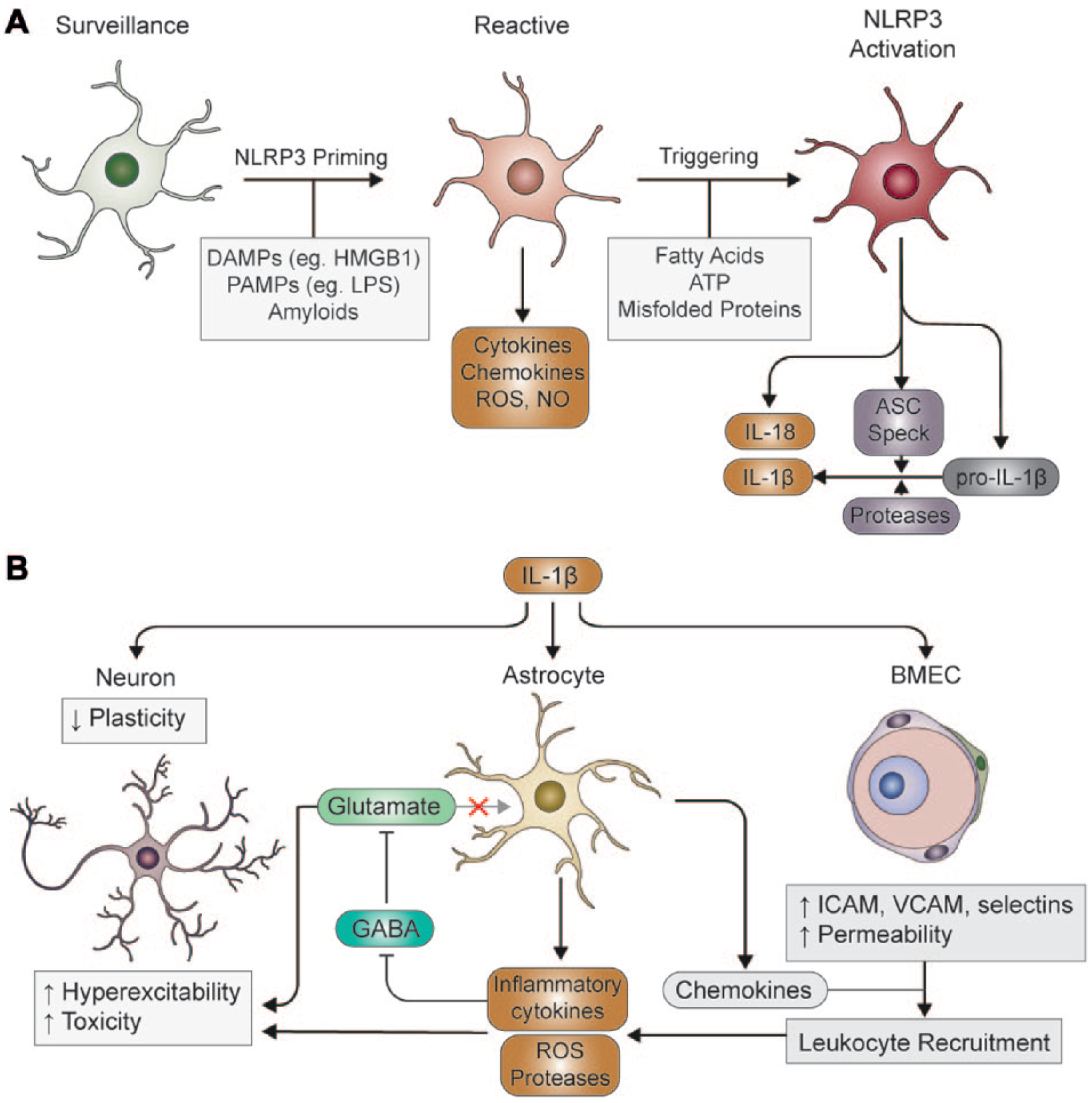
Production and action of IL-1β in neuroinflammation. (
Glial Cells and Neuroinflammation
The brain was long believed to be an immune-privileged organ, in which inflammation only occurred following infiltration of immune cells from the periphery due to blood–brain barrier (BBB) breakdown. 23 However, it is now understood that the brain possesses resident immune cells, known as microglia, which are able to initiate and sustain an inflammatory response. 24 In the healthy brain, microglia exist in a lattice-like organization; each microglial cell curates an individual section of the parenchyma, which it constantly monitors with long, motile processes. 25 In this state, microglia are said to appear “ramified,” which refers to the highly branched process morphology, and are now termed “surveillance microglia,” in an attempt to diverge away from the use of “resting,” which implies an inactive, nonfunctional state. 26 Microglia also facilitate synaptic development and plasticity by secreting brain-derived neurotrophic factor (BDNF) and by actively pruning synaptic terminals, making them critical for development and learning.27,28
As immune-competent cells microglia express a variety of PRRs that allow them to sense diverse PAMPs and DAMPs and evoke an innate immune response. TLRs 1–9, are present in microglia, along with numerous co-receptors, such as CD14, CD36, CD33, and triggering receptor expressed on myeloid cells 2 (TREM2), in addition to inflammasome components such as NLRP3. 29 Many proteins found to be misfolded or aggregated in neurodegenerative diseases, such as amyloid-β (Aβ), α-synuclein, and superoxide dismutase 1 (SOD1), as well as DAMPs released from dying cells, provoke innate immune responses through microglial PRRs.30–32 Moreover, certain aggregated protein species have been reported to activate the inflammasome in primed microglia.33–35 PRR ligands and other signaling factors activate a wide range of transcriptional programs in microglia, which promote a reactive phenotype that is distinct from that of surveillance microglia. 26 For example, in response to LPS, like peripheral macrophages, reactive microglia can evoke inflammatory, cytotoxic (M1-like) responses.36,37 Alternatively, challenges such as IL-4 or IL-13 can activate transcriptional profiles that enhance the phagocytic, anti-inflammatory activity of microglia (M2-like), thereby promoting tissue repair.36–40 However, recent profiling studies have highlighted considerable heterogeneity in microglial responses both in the context of disease and under homeostatic conditions, which do not readily conform to the M1/M2 model.41–44 As such, microglia are considered highly dynamic cells, with their transcriptional profile being regulated in a time- and context-dependent manner. 45 Nonetheless, many forms of neurodegeneration may be caused, perpetuated, or exacerbated by inappropriate skewing of the microglial response toward excessive release of inflammatory cytokines, such as tumor necrosis factor α (TNF-α) and IL-1β. Often, this is accompanied by production of reactive oxygen species (ROS) and neurotoxic enzymes that promote neuronal damage. 46 Microglia are therefore regarded as key players in determining disease progression, and thus modulating microglial behavior may present novel therapeutic avenues in many diseases.
Astrocytes are the most numerous cell type in the brain, and primarily support neuron function via regulation of the extracellular environment, secretion of trophic factors, and maintenance of the BBB. 47 Astrocytes express scavenger receptors, complement receptors, and are suggested to express TLRs 2–5 and 9. 48 However, recent studies utilizing PCR and RNA-sequencing have found that TLR3 is the only TLR significantly expressed by astrocytes, with the remainder predominantly found on microglia.49,50 Astrocytes are also suggested to express inflammasome sensors NLRP3,51–53 NLRP2, 54 and NLR family CARD domain containing protein 4 (NLRC4), 53 as well as ASC. 55 The role of astrocyte-mediated inflammation in brain disease is controversial, and many studies now indicate that the protective or toxic function of astrocytes is highly context and stimulus dependent.56–58 In response to brain injuries, astrocytes undergo a variety of responses, including extension of processes, hypertrophy, and in more severe settings, proliferation followed by assembly of a dense “glial scar.”59,60 This behavior, termed gliosis, protects adjacent neural networks by confining toxic factors and inflammatory cells to the injured region.59,61 Elimination of reactive astrocytes in acute injury is therefore generally counterproductive, increasing damage and impeding repair. 62 However, astrocytes also produce chemokines, particularly CCL2, CXCL1, CXCL2, and CXCL10, as well as certain cytokines, ROS, and proteases that may exacerbate neuronal damage ( Fig. 1 ).57,63 Building on these opposing observations, Liddelow et al. 58 propose an astrocyte activation model analogous to the M1/2 designation, termed A1 (toxic) and A2 (protective), based on transcriptional profiling. Interestingly, induction of the A1 phenotype is dependent on cytokines and complement factors released primarily by microglia, and elimination of microglia largely prevents astrocyte activation in response to LPS in vivo. 58 Furthermore, postmortem analysis of human brain tissue revealed that A1 astrocytes predominate in active amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), and Alzheimer’s disease (AD). Together with analyses of TLR localization, these results underscore the importance of microglia in orchestrating central nervous system (CNS) inflammation and astrocytes as critical effector cells.
IL-1β and Neuroinflammation
The production of IL-1β by microglia or infiltrating peripheral cells can have various effects depending on the cell type on which it acts ( Fig. 1 ). 64 In vitro studies have identified hundreds of IL-1β-responsive genes in astrocytes, including complement components, adhesion molecules, growth factors, and chemokines.58,63,65,66 While microglia are not considered primary targets of IL-1β, in vitro data indicate that IL-1β may nonetheless affect microglia indirectly via astrocyte-derived factors. 67 As such, both chronic and acute elevation of IL-1β in vivo result in pronounced reactive astrogliosis and microgliosis.68–70 Vascular endothelial cells also respond to IL-1β stimulation with upregulation of chemokines and adhesion molecules, which can affect the function of the BBB.67,71 It is well documented that IL-1β elevation can promote the influx of peripheral neutrophils and monocytes from the circulation across the BBB,68–70,72–75 which is dependent on both endothelial cell and astrocyte responses ( Fig. 1 ).64,72,73 Invading leukocytes have the potential to exacerbate inflammation by producing further cytokines, ROS, and proteases that increase tissue damage, as well as stimulating lymphocyte recruitment.76,77 As peripheral immune cells are suggested to contribute to the pathogenesis of AD,78,79 MS, 80 epilepsy, 81 stroke, and traumatic brain injury (TBI),82–85 it is feasible that IL-1β-dependent responses are detrimental in this regard.
Despite the potential neurotoxicity of reactive glia and invading leukocytes, IL-1β elevation alone is not normally sufficient to cause overt neurodegeneration.2,69 Instead, it has been suggested that IL-1β only promotes toxicity in the presence of additional cytokines, toxins, or existing CNS injury. 2 In support of this idea, co-administration of IL-1β with excitotoxins, or following ischemic or traumatic insults, exacerbates neuronal loss, while inhibition by neutralizing antibodies or IL-1 receptor antagonist (IL-1Ra) is protective. 86 While this would support a pathological role of IL-1β, it has been argued that IL-1β is also critical for CNS repair. In the cuprizone model of demyelination, mice lacking IL-1β exhibit defective remyelination, possibly due to the role of IL-1β in regulating the differentiation of oligodendrocyte precursors.87,88 Similarly, in a model of Parkinson’s disease (PD), dopaminergic neuron recovery following injury is reduced in IL-1β knockout animals, which may reflect lack of trophic support from reactive glia. 89
Inflammation and Neuronal Function
Inflammatory cytokines have direct effects on neuronal function and synaptic transmission, which may contribute to the neurocognitive symptoms associated with certain diseases. While physiological concentrations of IL-1β are involved in hippocampal long-term potentiation (LTP) and memory formation, aberrant elevation of IL-1β has the opposite effect, suppressing LTP.90–92 Accordingly, treatment with LPS, which induces activation of microglia and robust upregulation of IL-1β in the hippocampus, impairs performance in memory tasks, 93 which may occur partially through suppression of BDNF signaling. 94 Furthermore, it was recently demonstrated that both aging and prior exposure to IL-1β sensitizes hippocampal synapses to IL-1β-dependent LTP inhibition, 95 and that mice lacking NLRP3 are resistant to age-related cognitive impairment. 96 Rats lacking NLRP1 have also been demonstrated to be resistant to age-related cognitive deficits. 97 Together, these studies provide a compelling mechanism linking neuroinflammation to cognitive decline.
Inflammation also affects neuronal excitability (
Fig. 1
). First, various reports have indicated that inhibitory γ-aminobutyric acid (GABA) tone can be suppressed by a number of inflammatory cytokines, including IL-1β and IL-6.98–102 Second, IL-1β signaling increases the amplitude of N-methyl
Contribution of Inflammasomes to CNS Disease
Alzheimer’s Disease
AD is a neurodegenerative disease associated with progressive neuronal loss in areas of the brain associated with memory and higher cortical function, most notably the hippocampus and neocortex. 110 Consequently, AD patients show severe signs of dementia and cognitive decline, symptoms that worsen over time. Extracellular Aβ plaques and tau-containing neurofibrillary tangles are the pathological hallmarks of the disease found within the brain.110–112 Aβ plaques are largely composed of Aβ40 and Aβ42 peptides that are generated by amyloidogenic processing of amyloid precursor protein (APP). Among these species, Aβ42 has the highest propensity to aggregate and form oligomers and fibrils that are toxic to neurons.113–117
Neuroinflammation is now also gaining acceptance as a core component of AD pathology, and there is evidence to suggest a role for the NLRP3, NLRP1, and NLRC4 inflammasomes in AD ( Table 1 ).34,53,118–121 Perhaps the most direct evidence implicating inflammation in AD has come from genome-wide association studies (GWAS) identifying polymorphisms within genes of the innate immune system that are associated with an increased risk of developing AD. Such polymorphisms have been identified in the gene encoding complement receptor 1 (CR1), which plays an important regulatory role in the complement system and has been suggested to promote phagocytic clearance of Aβ by microglia.122,123 AD-associated polymorphisms have also been identified within the gene encoding TREM2, another receptor involved in microglial phagocytosis, and within human leukocyte antigen-D-related beta 1 (HLA-DR-B1) and 5 (HLA-DR-B5).124,125 HLA-DR-B1 and HLA-DR-B5 encode the major histocompatibility complex II (MHC II) beta chain paralogues β1 and β5, which are responsible for the presentation of foreign antigens to cells of the adaptive immune system. As such, HLA-DR expression is commonly used as a marker of activated immune cells. 126 HLA-DR-positive microglia are found in abundance within the AD brain, particularly around amyloid plaques, suggesting that Aβ is a source of immune activation in AD.127–129
Summary of Evidence Implicating Inflammasomes in Neurological and Neurodegenerative Conditions.

Only evidence supporting the role of inflammasomes in each disease is listed.
In addition to the previously discussed roles of IL-1β in neuronal injury, there are a number of specific mechanisms by which IL-1β is thought to be detrimental in AD. For example, IL-1β has been shown to promote the production of S100β, a cytokine that is overexpressed in the AD brain predominantly by astrocytes and is thought to contribute to neuritic dystrophy.130,131 In human endothelial cells, glial cells, and neurons, IL-1β can upregulate APP expression.132–134 IL-1β also enhances the activity of several protein kinases involved in the phosphorylation of tau, including mitogen-activated protein kinase p38 (MAPK-p38), glycogen synthase kinase 3β (GSK-3β) and cyclin-dependent kinase 5 (Cdk5), suggesting that IL-1β may promote the formation of neurofibrillary tangles.135–138
Upon interaction with the scavenger receptor CD36 on the surface of microglia, fibrillar Aβ activates NF-κB signaling by promoting the assembly of a CD36-TLR4-TLR6 heterotrimer. 31 Activation of this pathway by Aβ upregulates IL-1β mRNA expression and thus primes microglia for IL-1β production.31,139 Furthermore, fibrillar Aβ has also been shown to promote NLRP3-mediated IL-1β release in LPS-primed microglia and bone marrow-derived macrophages (BMDMs), an effect that is absent in LPS-primed BMDMs isolated from NLRP3- or ASC-deficient mice. 34 Thus, it is suggested that Aβ can provide both the first and second signals for activation of the NLRP3 inflammasome.
In vivo evidence further supports a role for the NLRP3 inflammasome in the pathology of AD. Total IL-1β and active caspase-1 expression is significantly upregulated in the brains of APP/PS1 mice and in postmortem brain tissue of AD patients.118,140 Furthermore, APP/PS1 mice deficient in either NLRP3 or caspase-1 show a reduction in cerebral Aβ40 and Aβ42 load and are protected from memory and learning deficits. Impaired activation of the NLRP3 inflammasome in APP/PS1 mice deficient in NLRP3 or caspase-1 is suggested to enhance the phagocytic capacity of microglia and subsequent clearance of Aβ, thus providing a potential explanation for the reduced amyloid burden seen in these mice. 118 Recently, an additional role for extracellular ASC specks in the seeding and spread of amyloid burden in AD was demonstrated. ASC specks released into the supernatant of LPS-primed, ATP-activated mouse microglia interact directly with Aβ42 peptides and promote their aggregation in vitro. 121 In support of this, APP/PS1 mice deficient in ASC display reduced amyloid burden and are protected from memory loss. Furthermore, intrahippocampal injection of brain lysates derived from 16-month-old APP/PS1 mice into 3-month-old APP/PS1 mice yet to develop severe Aβ pathology accelerates both the level and distribution of Aβ pathology within this hemisphere at 8 months. Notably, this effect is absent in APP/PS1 mice deficient in ASC. 121 In this same study, ASC specks were found within the core of Aβ plaques isolated from the hippocampus of patients with AD and from the brains of APP/PS1 mice, further supporting the notion that ASC promotes Aβ seeding. 121
The NLRP1 inflammasome is also implicated in AD pathology ( Table 1 ). Within the brain, NLRP1 is predominantly expressed by neurons, and its protein expression is significantly upregulated in the brains of APP/PS1 mice compared with those of wild-type mice.119,141 Cultured rat cortical neurons exhibit enhanced mRNA and protein expression of NLRP1 in response to oligomeric Aβ42, and siRNA-mediated knockdown of NLRP1 in these neurons significantly reduces Aβ-induced neuronal pyroptosis and IL-1β release. 119 Furthermore, knockdown of NLRP1 or caspase-1 within the brains of APP/PS1 mice is neuroprotective, with these mice showing greater neuronal survival in the hippocampus and cortex compared with APP/PS1 mice treated with control siRNA. These mice also demonstrate improvements in learning and memory. 119
Several lines of evidence implicate a high-fat diet and aberrant lipid metabolism as risk factors for AD. 142 The saturated fatty acid palmitate promotes the release of IL-1β in astrocytes. 120 Furthermore, culturing rat cortical neurons in palmitate-treated astrocyte-conditioned media enhances Aβ42 production in these neurons, an effect that is ablated through transient silencing of NLRC4 in astrocytes. 120 In further support of a role for the NLRC4 inflammasome in AD, NLRC4 and ASC expression is upregulated at both the mRNA and protein level within the neocortex of AD patients postmortem. 120 The phospholipid lysophosphatidylcholine (LPC) is also implicated in AD. LPC promotes glial recruitment and activation, pro-inflammatory cytokine production, Aβ42 oligomerization, demyelination, and neuronal death.143–147 LPC is produced as a metabolite of phospholipase A2 (PLA2)-mediated hydrolysis of phosphatidylcholine and is an important constituent of oxidized low-density lipoprotein (ox-LDL). 147 Elevated levels of PLA2 have been found within the hippocampus of APP mice and human AD patients postmortem; thus, LPC levels are also likely to be elevated.148–151 Furthermore, PLA2-deficient hAPP mice are protected against learning and memory deficits. 151 LPC was recently shown to activate NLRP3 and NLRC4 inflammasome pathways in LPS-primed microglia and astrocytes. 53 This suggests that the potential role of LPC in AD may be mediated, at least in part, by co-activation of NLRC4 and NLRP3.
Parkinson’s Disease
PD is a neurodegenerative disease that is associated with severe atrophy of dopaminergic neurons within the substantia nigra pars compacta, an area of the brain associated with motor function. 152 As a result, PD patients experience tremors, muscle stiffness, and impaired voluntary movement, symptoms that are often accompanied by dementia and cognitive dysfunction. 153 Pathologically, PD is characterized by the buildup of α-synuclein aggregates within the brain. α-Synuclein aggregates accumulate within Lewy bodies that disrupt neuronal function and ultimately cause neuronal death.154–156 As such, specific mutations within the gene encoding α-synuclein are causal of familial PD. 157
Several lines of evidence suggest that inflammation also plays an important role in the pathology of PD. GWAS have identified polymorphisms within several inflammatory genes that are associated with PD risk, including human leukocyte antigen-D-related alpha (HLA-DR-A) and HLA-DR-B5, which encode specific MHC II molecule alpha and beta chains, respectively.158,159 HLA-DR-positive microglia are found in abundance within the substantia nigra of PD patients postmortem,127,160 accompanied by high levels of IL-1β, IL-6, and TNF-α.127,161 Polymorphisms in IL-1α, IL-1β, and TNF-α are also associated with an increased risk of developing PD; however, the significance of these polymorphisms is yet to be established through GWAS.162,163
Given their close association with HLA-DR-positive microglia within the PD brain, α-synuclein aggregates are considered key drivers of neuroinflammation in the disease. 127 In human monocytes, monomeric and fibrillar species of α-synuclein activate NF-κB signaling upon interaction with TLR2, upregulating mRNA and protein expression of pro-IL-1β. Furthermore, α-synuclein can also induce the processing and secretion of mature IL-1β in human monocytes, though this property is exclusive to fibrillar species.35,164
A commonly used experimental model of PD involves the administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which is directly toxic to nigral dopaminergic neurons. Mice deficient in NLRP3 are protected from MPTP-mediated dopaminergic neuronal loss within the substantia nigra, suggesting that the NLRP3 inflammasome might play an important role in PD-associated neurodegeneration. 165 Interestingly, dopamine has been shown to negatively regulate NLRP3 inflammasome activation in primary mouse BMDMs and astrocytes by promoting cyclic AMP (cAMP)-mediated ubiquitination and degradation of NLRP3. 165 Dopamine is thought to do this via activation of dopamine receptor D1 (DRD1), which is abundantly expressed by immune cells.165,166 Furthermore, mice deficient in DRD1 show enhanced levels of active caspase-1 within the brain following MPTP administration, accompanied by increased levels of total IL-1β within serum. The extent of nigral dopaminergic neuronal loss is also exacerbated by DRD1 deficiency in mice administered MPTP compared with wild-type mice. However, mice deficient in both DRD1 and NLRP3 show dopaminergic neuronal loss comparable to that of wild-type mice in response to MPTP treatment. 165 Dopamine-mediated regulation of the NLRP3 inflammasome is thus likely to be an important mechanism by which inflammation is controlled within the CNS. 165 However, given the deficit in dopaminergic signaling within the PD brain, this endogenous control over the NLRP3 inflammasome may be impaired in PD.152,165
An alternative role for the inflammasome, but more specifically caspase-1, in promoting the aggregation of α-synuclein has also been suggested. 167 In response to inflammasome-activating stimuli, caspase-1 cleaves α-synuclein at residue 121 within its C-terminus, resulting in the production of a highly aggregation-prone species of α-synuclein that are neurotoxic in vitro. 167 C-terminally truncated species of α-synuclein have a higher propensity to aggregate and are found in abundance within Lewy bodies. Such truncated species are more toxic to dopaminergic neurons.168–170 Furthermore, the caspase-1 inhibitor VX-765 is neuroprotective in a transgenic mouse model of multiple system atrophy (MSA), another neurodegenerative movement disorder that is characterized by the buildup of α-synuclein aggregates within the brain. In this mouse model of MSA, VX-765 treatment significantly reduces striatal α-synuclein load, protects nigral dopaminergic neurons from α-synuclein-mediated neurotoxicity, and improves motor function. 171 However, it is important to note that PD and MSA are distinct diseases and caution should be taken when applying these findings to PD.
Multiple Sclerosis
MS is an autoimmune disease characterized by CD4+ T-cell-mediated autoreactivity toward myelin, resulting in severe demyelination, axonal degeneration, and chronic inflammation within the CNS. 172 These changes result in the clinical manifestation of a variety of neurological complications, including fatigue and problems with movement and vision. 173 Although inflammation is thought to be important in the pathology of MS, its role in the disease is not completely understood given that demyelination has also been shown to occur independently of inflammation. 172 For example, peripheral immune infiltrates are generally found dispersed throughout the CNS rather than being specifically localized to MS lesions. Thus, it has been argued that these infiltrates may instead be involved in regeneration and repair.172,174
Experimental autoimmune encephalitis (EAE) is a commonly used animal model for MS that is usually achieved through immunization with myelin peptides such as myelin oligodendrocyte glycoprotein (MOG), which evoke strong CD4+ T-cell-mediated immune responses that promote demyelination and inflammation within the CNS. 175 Th1 and Th17 cell responses are thought to be important in the pathology of EAE. 176 In EAE, following differentiation within the periphery, Th1 and Th17 cells are recruited into the CNS and produce interferon gamma (IFN-γ) and IL-17, respectively, in response to antigen-presenting cells (APCs). 177 IFN-γ and IL-17 enhance the permeability of the BBB and promote the recruitment of peripheral immune cells into the CNS, which initiate immune responses toward myelin and bring about demyelination and axonal degeneration. 176
Evidence suggests that the NLRP3 inflammasome plays an important role in the recruitment of CD4+ Th1 and Th17 cells into the CNS and in the orchestration of Th1 and Th17 responses in EAE ( Table 1 ). IL-1β and another pro-inflammatory cytokine that is secreted following inflammasome activation, IL-18, are important for the differentiation of Th1 and Th17 cells.178–181 IL-1β is expressed by macrophages and microglia in MS lesions isolated from the CNS tissue of MS patients postmortem and EAE rats.182,183 Furthermore, NLRP3 mRNA expression is upregulated within the spinal cord of EAE mice in response to MOG peptide and increases with disease progression. 184 EAE mice also show similar increases in caspase-1, IL-1β, IL-6, TNF-α, and IFN-γ mRNA within blood. 185 In NLRP3-, ASC-, or caspase-1-deficient mice, the development of EAE in response to MOG peptide is impaired and there is a marked reduction in the recruitment of peripheral immune cells into the CNS.184–186 MOG-immunized mice deficient in NLRP3 also demonstrate a marked reduction in the production of IFN-γ and IL-17 within the spinal cord. These findings suggest that CD4+ Th1 and Th17 cell responses are dampened by NLRP3 deficiency in these mice, providing a potential explanation for the impaired development of EAE. 184 Similar findings have been demonstrated in MOG-treated EAE mice deficient in IL-1α and IL-1β, whereas the development of EAE in MOG-treated mice lacking the endogenous IL-1Ra is augmented. 187
CD4+ Th1 and Th17 cells isolated from the peripheral lymphoid organs of MOG-immunized EAE mice deficient in NLRP3 or ASC show downregulated mRNA levels of various genes involved in chemotaxis, including those encoding C-C chemokine receptor type 2 (CCR2) and C-X-C motif chemokine receptor 6 (CXCR6). mRNA levels of the corresponding chemotactic molecules (e.g., CCL7 and CXCL16) are also downregulated in APCs concomitantly in EAE mice deficient in NLRP3 or ASC. Thus, it is suggested that in EAE, activation of the NLRP3 inflammasome within the periphery promotes the infiltration of CD4+ T cells and APCs into the CNS by enhancing the expression of chemokine molecule and receptor pairs in these cells. 188
The development of EAE in rodents can occur both independently of NLRP3 and in a NLRP3-dependent manner, depending on the method and intensity of experimental immunization.189–191 Aggressive immunization methods used to achieve EAE do not require activation of the NLRP3 inflammasome, a subtype of EAE referred to as type B EAE. 189 Interestingly, IFN-β, which is currently approved for the treatment of MS, is ineffective in type B EAE and has therefore been suggested to mediate its effects through the NLRP3 inflammasome.189,191 In line with this, IFN-β treatment has been shown to inhibit the production of total IL-1β and active caspase-1 in LPS-primed splenocytes isolated from nonaggressively immunized EAE mice, and lower levels of total IL-1β are also observed in the spleens of these mice. 189 As some MS patients are nonresponsive to IFN-β treatment, it is feasible that patient responsiveness to IFN-β treatment may be determined, at least in part, by the extent of NLRP3 inflammasome activation in patients. To investigate this, a patient cohort study was carried out to compare the expression of NLRP3 and IL-1β in peripheral blood mononuclear cells (PBMCs) from IFN-β-responsive and nonresponsive MS patients. 192 At baseline, the expression of NLRP3 and IL-1β mRNA was significantly higher in the PMBCs of IFN-β-resistant MS patients. However, following IFN-β treatment, NLRP3 and IL-1β mRNA expression significantly increased from baseline in the IFN-β-responsive group and remained unchanged in the IFN-β-resistant group. The authors suggest that the upregulated expression of NLRP3 and IL-1β mRNA that was seen in the responsive group following IFN-β treatment was required for their response to treatment. In this same study, an association was found between a gain-of-function NLRP3 polymorphism (rs35929419) and IFN-β response, with the polymorphism being found at a higher prevalence in IFN-β-responsive patients. 192 However, the significance of NLRP3 polymorphisms in the response to IFN-β treatment in MS patients is yet to be confirmed through GWASs.
A role for the inflammasome has also been demonstrated in the cuprizone model of MS. Cuprizone is a copper chelator that is toxic to oligodendrocytes; 4-week treatment of cuprizone causes severe gliosis, neuroinflammation, and demyelination in C57BL/6 mice. 193 NLRC4 protein expression is abundant within astrocytes and, to a lesser degree, microglia in corpus callosum tissue sections of mice after 4 weeks of cuprizone treatment. In line with this, astrocytic NLRC4 protein expression is also elevated within MS lesions from postmortem brain tissue of patients with chronic MS. 53 Furthermore, in the cuprizone model of MS, mice deficient in both NLRP3 and NLRC4 demonstrate a significant reduction in gliosis and extent of demyelination, whereas disease phenotype is more severe in mice deficient in just NLRP3 or NLRC4. 53 As previously discussed, the phospholipid LPC, which promotes demyelination and is thought to be elevated in MS due to enhanced PLA2 activity,143,194 promotes inflammasome activation in LPS-primed microglia and astrocytes by engaging both NLRP3 and NLRC4. 53 Together these findings suggest that both NLRP3 and NLRC4 are important for MS-associated demyelination and that LPC may be an important mediator of this process. 53
Amyotrophic Lateral Sclerosis
ALS is a devastating neurodegenerative condition characterized by a steady loss of spinal motor neurons, leading to progressive motor impairment followed by death. The average survival is 3–5 years following diagnosis, and there are currently very few treatments. Familial ALS (fALS) is linked to variants in more than 30 separate genes, including in SOD1, fused in sarcoma (FUS), TAR DNA-binding protein 43 kDa (TDP-43), and the noncoding chromosome 9 open reading frame 72 (C9orf72) region.195–199 The cause of sporadic ALS (sALS) is not fully understood but is thought to be a multihit process arising through both genetic and environmental risk factors. 200 Various environmental factors have been suggested to increase ALS risk, including excessive physical activity, military deployment, smoking, and exposure to neurotoxins. 201 However no definitive, reproducible associations have yet been established, which may reflect either genuine population differences, differences in study design, or low study power. 201 Due to the extreme variety in causative factors, ALS pathology is heterogeneous. 199 However, it is notable that aggregated protein inclusions in motor neurons are prevalent even in sALS cases and are commonly composed of TDP-43, as well as SOD1, FUS, and other products of ALS risk genes. 202
ALS patients display evidence of an increased inflammatory profile in both the periphery and cerebrospinal fluid (CSF), and radiological studies have highlighted microglial reactivity along spinal motor tracts and in the motor cortex, which colocalize with structural abnormalities.203–205 However, microglial reactivity may be a response to motor neuron injury rather than a cause, and may be either beneficial or detrimental. Both rats and mice bearing the disease-causing human SOD1G93A mutation are widely used as ALS models, and several studies using SOD1G93A mice indicate that the microglial phenotype in the early stages of ALS is protective and promotes neuronal survival.206,207 Accordingly, when microglial reactivity is attenuated via P2X7 receptor inhibition, early-stage SOD1G93A mice deteriorate faster than controls, whereas late presymptomatic mice experience prolonged survival. 208 These observations highlight a switch in microglial behavior to a neurotoxic phenotype as the disease progresses, yet the molecular basis of this transition is poorly understood. Sequencing and protein studies have demonstrated upregulated protein expression of NLRP3, in addition to upregulated IL-1β mRNA, in symptomatic but not presymptomatic mice. 207 Deep RNA sequencing of SOD1G93A microglia by Chiu et al. 41 found evidence of a progressive alteration in microglial phenotype, but one which deviated significantly from both the classical M1 and M2 states. Thus, a unique neurodegeneration-specific signature appears to predominate in SOD1-related ALS.
Various reports suggest that inflammasomes may contribute to ALS pathology ( Table 1 ). In mice, aggregated SOD1G93A is released by severely injured neurons in the later stages of progression and activates ASC-dependent mature IL-1β release in microglia. 33 TDP-43, which is often misfolded in ALS and frontotemporal dementia (FTD), also activates NLRP3-dependent IL-1β secretion in microglia, as well as increasing secretion of TNF-α.209,210 In vivo, several groups have reported cleaved caspase-1 in spinal homogenates of mice bearing SOD1 mutations.33,51,211 Furthermore, SOD1G93A mice lacking either IL-1β or caspase-1 display slower loss of motor neurons and extended survival. 33 More recently, ASC and NLRP3 staining was observed in spinal cord tissue from sALS patients, which primarily localized to astrocytes. 51 Thus, inflammasome activation may occur in both human cases and animal models of ALS. Nonetheless, the significance of individual inflammasome components in ALS pathogenesis remains to be demonstrated. While ALS mice lacking caspase-1 experience longer survival, it is unclear which inflammasome is responsible for caspase-1 activity in the SOD1G93A model. 33 It is also important from a therapeutic angle that no studies have so far tested the efficacy of specific inflammasome inhibitors in ALS models. However Heitzer et al. 212 administered 17β-estradiol, which is known to suppress expression of inflammasome components, 213 to symptomatic SOD1G93A mice. 17β-Estradiol treatment improves motor performance and extended survival, as well as reducing the expression of NLRP3, ASC, mature IL-1β, and active caspase-1 in the spinal cord. 212
Overall, studies to date suggest a potential involvement of the inflammasome in the course of ALS ( Table 1 ). However, there are several prominent gaps in our understanding of the precise consequences of each inflammasome for disease progression. A recent clinical trial assessing IL-1Ra treatment in ALS patients failed to demonstrate any improvement in motor scores, though the study’s primary outcome was tolerability. 214 Due to the complex and heterogeneous pathology of ALS, it seems likely that disease stage and individual mutations will radically influence the benefits of inflammasome inhibition. Thus, more studies are required to investigate the efficacy of direct inflammasome modulation in ALS models.
Stroke
Stroke is a neurovascular disease that is caused by an interruption in blood flow to the brain, resulting in cerebral hypoxia. Given the high metabolic demand of the brain, these hypoxic conditions bring about rapid cell death, irreversible brain damage, and subsequent loss of brain function. Ischemic stroke is the most common form of stroke and is caused by either thrombotic (e.g., an atherosclerotic plaque) or embolic blockage of a cerebral blood vessel. In hemorrhagic stroke, a cerebral blood vessel becomes ruptured, resulting in the leakage of blood either into the brain, in the case of intracerebral hemorrhage (ICH), or into the subarachnoid space, in the case of subarachnoid hemorrhage (SAH). In addition to interrupted cerebral blood flow, hemorrhagic stroke is also characterized by the formation of a neurotoxic hematoma. 215
Rapid inflammatory responses are seen during the acute phase of stroke. DAMPs, including ATP, HMGB1, and heat shock protein (HSP), are released from necrotic tissue poststroke and promote the recruitment of microglia, neutrophils, lymphocytes, and macrophages to the site of ischemic injury.216–218 Enhanced immune cell activation and pro-inflammatory cytokine production are seen within both the ischemic core and the peri-infarct region, which is most commonly referred to as the “inflammatory penumbra.”219–221 Of these pro-inflammatory cytokines, evidence suggests that IL-1 is a particularly important mediator of poststroke neuronal injury. 2 In the commonly used middle cerebral artery occlusion (MCAo) model of ischemic stroke, mice deficient in both IL-1α and IL-1β or the IL-1 receptor (IL-1R1) are protected from neuronal injury.222,223 Furthermore, administration of recombinant IL-1β into the brains of rats undergoing transient MCAo promotes postischemic neuronal injury, 224 whereas administration of IL-1Ra is neuroprotective in the MCAo model and in rodent models of SAH.225,226 Such findings have led to the testing of IL-1Ra in phase II clinical trials for ischemic stroke and SAH. IL-1Ra was not associated with significant toxicity issues in these studies, and as such, the efficacy of IL-1Ra in stroke patients is now being tested in phase III clinical trials.227–229
The evidence implicating IL-1 in poststroke brain injury suggests a role for the inflammasomes. NLRP3 mRNA and protein expression within the brain of rodents is upregulated following experimental induction of ischemic and hemorrhagic stroke.230–236 The mRNA expression of the inflammasome sensors NLRP1, absent in melanoma 2 (AIM2), and NLRC4, as well as the protein expression of NLRP1, also appears to be upregulated within the brain in rodent MCAo models of ischemic stroke.231,236 NLRP3-deficient mice are protected from ischemic brain damage, BBB breakdown, and neurological deficits following MCAo. 233 However, work by Denes et al. 237 suggests that the NLRP3 inflammasome is not important for the acute injury after MCAo in mice; rather, the injury is regulated by AIM2 and NLRC4 inflammasomes. In this study, NLRP3-deficient mice that underwent MCAo were not protected from ischemic brain injury or neurological deficits, whereas ASC-, NLRC4-, or AIM2-deficient mice were. 237 Further studies are required to explain the discrepancies between these studies and to elucidate the mechanisms by which the AIM2 and NLRC4 inflammasomes may be activated in stroke. Antibody-mediated neutralization of NLRP1 has been shown to reduce the expression of IL-1β and active caspase-1 within the brain following thromboembolic stroke in mice. However, NLRP1 neutralization does not reduce infarct volume, suggesting that other inflammasomes may be more important in this model. 238
Free heme, a toxic metabolite of hemoglobin, is released into the brain parenchyma following cerebral vessel rupture and is thought to have detrimental effects in hemorrhagic stroke. 226 Heme induces oxidative stress in brain tissue, which in turn promotes neuronal and endothelial cell death, with the latter resulting in the breakdown of the BBB and infiltration of peripheral immune cells into the CNS. 239 In line with this, heme oxygenase 1 (HO-1), an enzyme responsible for the breakdown of heme, is abundantly expressed around the bleed site following SAH and is found in close proximity to IL-1α-expressing microglia. 226 Furthermore, treatment of LPS-primed mixed glial cultures with hemin, the oxidation product of heme, induces the processing and release of mature IL-1α but not IL-1β. In hippocampal organotypic slices, hemin treatment induces neuronal death concomitantly with IL-1α expression, an effect that is ablated with IL-1Ra treatment, suggesting that the neurotoxic effect of hemin may in part be mediated through IL-1α. 226 Conversely, free heme has been shown to activate the NLRP3 inflammasome in LPS-primed BMDMs and induce the release of mature IL-1β. Furthermore, the production of total IL-1β within the peritoneal cavity of mice is also enhanced in response to intraperitoneal injection of heme and mice deficient in either NLRP3, ASC, or caspase-1 are protected from hemolysis-induced lethality. 240 Thus, in addition to DAMPs released from necrotic tissue, heme may also serve as an important DAMP for the release of IL-1α and IL-1β following hemorrhagic stroke.
Acute Brain Trauma
TBI and SCI are neuropathological events resulting from direct physical trauma to the head or vertebrae, respectively. Following the primary trauma, a series of cascades initiate the “secondary” injury, whereby inflammatory, excitotoxic, and oxidative stress drives neuronal loss and neurological impairment.241,242 DAMPs released by necrotic cells following the initial hemorrhage drive an inflammatory response that is likely protective in the initial phase. 242 mRNA and protein expression of IL-1β and IL-18 is upregulated within the brain and CSF of TBI and SCI patients, and is accompanied by the recruitment of leukocytes to the site of injury, which may transform the initial protective response to aggravate damage.243–247 Exogenous administration of IL-1β following experimental TBI increases neuronal apoptosis, whereas neutralization of IL-1β decreases neutrophil infiltration, lesion volume, and cognitive deficits in controlled cortical impact (CCI) models.248,249 Furthermore, neutralization of the inflammasome component ASC is protective in rodent models of TBI and SCI.250,251
NLRP1 expressed by both neurons and microglia may be a contributor to neuronal pyroptosis following acute injury. 252 In patients with moderate to severe TBI, increases in NLRP1, ASC, and caspase-1 protein expression in the CSF have been reported, with higher levels correlating with unfavorable outcomes. 253 However, only one study has directly and specifically addressed the role of NLRP1 in TBI. Brickler et al. 254 reported that mice lacking NLRP1 or ASC are not protected against motor impairment following CCI, despite decreases in IL-1β and IL-18 production. However, two previous reports indicate that IL-1β contributes primarily to cognitive deficits following TBI, which were not measured by the former study, and that motor deficits are unaffected by IL-1β neutralization.248,249 Thus, motor deficit alone may not be fully indicative of the extent of brain trauma and does not rule out a role of inflammasomes in cognitive outcome.
NLRP1 is also present in exosomes isolated from the CSF of patients following SCI, and rodent models demonstrate NLRP1 protein expression and activation in spinal cord motor neurons following experimental SCI.255,250,256 Antibody-mediated ASC neutralization results in reduced IL-1β production, tissue damage, and motor deficits, supporting a role of inflammasomes in SCI. 250 However, ASC neutralization is a rather broad approach and does not specifically implicate NLRP1 in SCI pathology. HO-1 is expressed in rat spinal cord neurons after SCI and downregulates NLRP1 by modulating endoplasmic reticulum stress signaling. 256 When overexpressed in spinal cord motor neurons prior to SCI, HO-1 reduces NLRP1 expression, which correlates with decreased neuronal loss and motor recovery. 256 Thus, while NLRP1 has a potential role in CNS trauma, direct evidence for its involvement is lacking and will require further study.
Several lines of evidence now also support a role of the NLRP3 inflammasome in the pathogenesis of TBI. A recent study in pediatric TBI patients reports elevated protein expression of NLRP3 in the CSF up to 72 h postinjury, which correlates with worse outcome. 257 NLRP3, active caspase-1, and ASC mRNA and protein expression is also increased following experimental CCI in rats, accompanied by enhanced production of IL-1β and IL-18. 258 Furthermore, Irrera et al. 259 demonstrate that NLRP3-deficient mice exhibit reduced tissue damage after TBI and score better in cognitive tests than wild-type controls. Accordingly, lack of NLRP3 reduces expression of the pro-apoptotic protein BAX in the hippocampus at 7 days postinjury, indicating that NLRP3 may contribute to the spread of neuronal death and to neurological deficit. 259 The selective NLRP3 inhibitor MCC950 ( Fig. 2C ) partially ameliorates neurological deficit at 72 h in mice following experimental CCI, and reduces activation of caspase-3, which may reflect protection against pro-apoptotic signaling. 260
Rodent models of SCI have also demonstrated robust upregulation of NLRP3, ASC, and caspase-1 protein within spinal cord tissue, though these findings have yet to be verified in human patients.212,261,262 Jiang et al. 262 found that BAY 11-7082, which inhibits NF-κB signaling, and A438079, an inhibitor of P2X7 receptor-dependent NLRP3 inflammasome activation, improve motor recovery in mice following spinal compression injury. Faster recovery in these mice was accompanied by reduced neutrophil recruitment; reduced spinal cord levels of IL-1β, IL-18, and active caspase-1; and reduced neuronal apoptosis. However, no studies have directly addressed NLRP3 involvement using either genetic models or specific inhibitors such as MCC950. Therefore, a definitive role for NLRP3 in experimental SCI remains to be demonstrated.
Epilepsy
Epilepsy is a neurological disorder associated with episodic seizures that range in severity and frequency. 263 Generally speaking, epileptic seizures are thought to occur due to an imbalance between excitatory and inhibitory signaling in the brain. The etiology of epilepsy is largely unclear, but genetics and comorbidities such as stroke, brain trauma, and infection have all been associated with an increased risk of epilepsy. 264 Evidence suggests that neuroinflammation may also be a core component of epilepsy, and in light of what has been previously discussed, it is feasible that neuroinflammation links epilepsy to these comorbidities. 2 IL-1β and IL-6 immunoreactivity is seen in rodent brains following the induction of an experimental seizure, alongside concomitant upregulated mRNA expression of these cytokines and their receptors on neurons.265–269 Of these cytokines, IL-1β is thought to be of particular importance in epilepsy. Recombinant IL-1Ra treatment inhibits the onset of experimentally induced seizures in rodents, an effect that is absent in mice deficient in IL-1R1. 270 In rodent models of febrile seizures, IL-1β protein levels are elevated within the brain and are associated with increased seizure risk. 271 The role of IL-1β and other pro-inflammatory mediators in epilepsy is yet to be fully established. However, these cytokines are thought to enhance the excitability of neurons, lower the threshold for the onset of seizures, and contribute to synaptic remodeling to allow for the generation of hyperexcitable neuronal circuits. 272
A role for the NLRP3 inflammasome in epilepsy was first demonstrated by Meng et al. 273 Status epilepticus (SE) is a condition associated with prolonged or frequently occurring seizures of short duration. 274 Experimental rat models of SE involving electrical stimulation of the amygdala show elevated hippocampal levels of NLRP3, active caspase-1, and IL-1β compared with sham controls. 273 siRNA-mediated knockdown of NLRP3 within the brain of SE rats enhances neuronal survival within the hippocampus and reduces the onset, duration, and severity of seizures, also implying a role for NLRP3 in epileptogenesis. 273 Furthermore, the caspase-1 inhibitor VX-765 has anticonvulsant activity in rodent experimental partial seizure models by reducing the production of IL-1β, and as such, its use for the treatment of drug-resistant partial epilepsy has been tested in clinical trials. 275 Evidence also suggests an involvement for the NLRP1 inflammasome in epilepsy ( Table 1 ). Elevated protein levels of NLRP1 and caspase-1 are seen within the hippocampus of patients with temporal lobe epilepsy (TLE), a form of epilepsy associated with hippocampal neuronal atrophy. 276 Furthermore, genetically silencing NLRP1 within the brain of rodent TLE models reduces the severity and occurrence of seizures and enhances neuronal survival within the hippocampus. 276
Injury Phases and Potential Points of Therapeutic Intervention in CNS Disease
It is critical to the development of inflammasome-targeting therapies to understand at what stage in disease inflammasome activation is most detrimental. In stroke models, administration of IL-1Ra and caspase-1 inhibitors is protective only up to 3 h poststroke.277,278 Therefore, any benefits of inflammasome inhibition are likely to be limited to this acute time window. It appears feasible, given prior preclinical data, that if inflammasome inhibition could be accomplished, the reduction in IL-1β levels might favor reduced neutrophil recruitment and attenuated neuronal damage. However, preclinical validation of this strategy in stroke, TBI, and SCI models will first be required.
In neurodegenerative conditions such as AD, PD, and ALS, in which there is marked proteotoxic involvement, the picture is less clear. These diseases all involve a period of clinical latency, followed by gradual onset of symptoms as neurons are progressively lost.201,279,280 Inflammasomes could act during the latent phase to drive the underlying pathological changes that occur prior to overt neurodegeneration, or alternatively could act as downstream effectors exacerbating neuronal demise in response to the primary pathology. This is a critical distinction in determining whether inflammasome inhibition might be prophylactic or therapeutic in these conditions. In AD, there is evidence that NLRP3 could act at all disease stages, via ASC-mediated nucleation of Aβ plaques, as well as IL-1β-driven tau aggregation and cognitive decline.121,135–138 Similarly, in PD, caspase-1 can promote α-synuclein aggregation, 167 and in a reciprocal manner, fibrillar α-synuclein can also promote activation of the NLRP3 inflammasome.35,164 Pharmacological targeting of the inflammasome pathway could therefore potentially delay PD and AD onset if given sufficiently early in the disease course, or slow the disease progression by ameliorating neuronal death. In ALS, there is relatively little evidence specifically addressing the temporal involvement of inflammasomes. In the SOD1G93A model, caspase-1 deficiency lengthens survival but does not delay disease onset. 33 This suggests that, at least in SOD1-related pathologies, inflammasome activation is an accompanying rather than an initiating event and might therefore be targeted to slow ALS progression. However, as ALS comprises a particularly heterogeneous set of pathologies, more research is required to determine whether other forms of familial and sporadic ALS might be similarly tractable.
MS is still more complex, with some patients experiencing distinct attacks separated by quiescent periods (relapsing-remitting), while others present with progressive disease that does not remit (primary progressive). 281 Inflammasome deficiency can reduce the severity of demyelination in EAE and delay disease progression, suggesting an involvement in the early stages of EAE.184–186 There is also some evidence that NLRP3 inhibitors can attenuate the severity of EAE. 282 Given the role of IL-1β in the development of Th1 and Th17 responses,178–181 it is possible that inflammasome inhibition might help reduce the infiltration of inflammatory immune cells and ameliorate demyelination in MS. However, due to inherent differences between EAE and clinical MS, the translational value of these results remains to be established.
Given that there are several different types of epilepsy disorders and that our current knowledge of the pathological processes involved in epilepsy is lacking, it is hard to conclude at this stage whether inflammasome inhibition would be beneficial in epilepsy. Inflammasomes have been implicated both in the onset of seizures and in epileptogenesis; 273 thus, further research is warranted in order to understand when therapeutic intervention might prove useful.
Inflammasome Inhibitors
Given the role of the NLRP3 inflammasome in the discussed diseases, there is great potential in developing NLRP3 inflammasome inhibitors as therapeutics for such conditions. We recently published a comprehensive review of small-molecule inhibitors of the NLRP3 pathway, and their potential for therapeutic intervention in neurological disorders. 283 A few biological inhibitors of IL-1β are already utilized clinically, including rilonacept (Arcalyst), canakinumab (Ilaris), and anakinra (Kineret). However, these inhibitors target solely IL-1 and, as a result, have no effect on other inflammasome-mediated processes, such as pyroptosis and IL-18 release. Moreover, the BBB penetration of these therapeutics is slow; thus, their utility may be limited to peripheral inflammatory diseases. On the other hand, small-molecule inhibitors of the NLRP3 inflammasome will likely be more cost-effective, and of greater use in CNS indications due to greater BBB permeability. Some promising small molecules are discussed below, illustrating the various approaches taken to inhibiting the NLRP3 inflammasome.
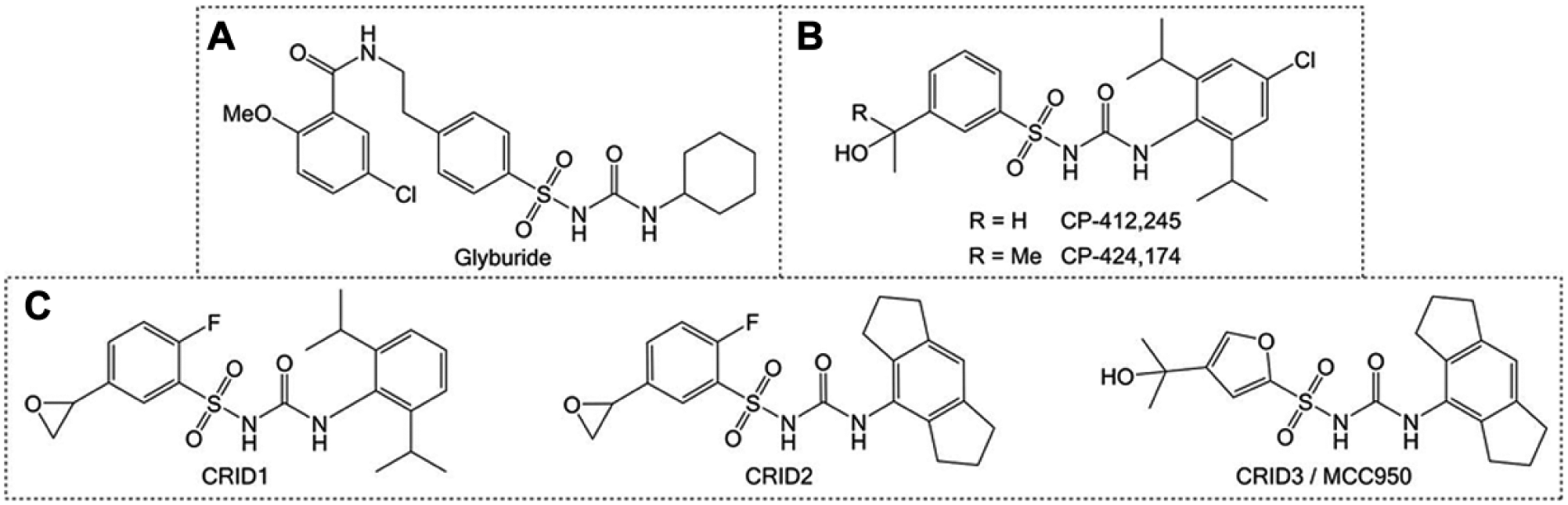
From Glyburide to MCC950: The CRID Story
A promising series of inhibitors was inspired by the discovery of the antidiabetic sulfonylurea glyburide ( Fig. 2A ) as an inhibitor of IL-1β production. 284 Following the discovery of the inflammasome, 285 glyburide was found to act via inhibition of NLRP3 inflammasome assembly. 286 Early compounds CP-412,245 and CP-424,174 took inspiration from the glyburide structure, with a diarylsulfonylurea motif incorporated ( Fig. 2B ). The CP compounds inhibit NLRP3 inflammasome-dependent posttranslational IL-1β processing with IC50 values of around 250 nM. 284 Subsequent mechanism of action studies suggested glutathione S-transferase omega 1 (GSTO1) as a possible target of two diarylsulfonylurea-containing cytokine release inhibitory drugs (CRIDs), CRID1 and CRID2, which also inhibit the release of IL-1β ( Fig. 2C ).287,288 The related analogue CRID3 was also reported in this study, with structural features taken from CP-424,174 and CRID2.
CRID3, also known as MCC950, is a selective small-molecule inhibitor of NLRP3 with an IC50 of 7.5 nM against NLRP3 inflammasome activation in BMDMs ( Fig. 2C ).282,287,289 In vivo, MCC950 reduces IL-1β production and attenuates symptoms of EAE. 282 MCC950 also abrogates neonatal lethality in a mouse model of cryopyrin-associated periodic syndrome (CAPS), an autoinflammatory disease that is caused by gain-of-function mutations in NLRP3. 282 MCC950 also inhibits inflammasome activation and microglial activation in the APP/PS1 mouse model of AD. 290 Thus, MCC950 is an interesting potential therapeutic for NLRP3-associated diseases.
The precise biological target for MCC950 is yet to be elucidated. Unlike glyburide, MCC950 does not target ATP-sensitive K+ channels, 282 with K+ efflux an event common to all NLRP3 activators. 14 Ca2+ signaling is also implicated in NLRP3 activation; 291 however, MCC950 has little effect on the ATP-induced increase of intracellular Ca2+. 282 MCC950 inhibits the NLRP3-induced oligomerization of ASC, yet does not interfere with either the NLRP3/NLRP3 or NLRP3/ASC interaction. 282
Michael Acceptors in NLRP3 Inflammasome Inhibitors
Compounds containing α,β-unsaturated carbonyl groups are called Michael acceptors (named after Arthur Michael), which react with nucleophiles at the β-carbon of the alkene ( Fig. 3A ). Michael acceptors are present in a number of drugs that are covalent modifiers of their target protein (suicide substrates). Michael acceptors are present in a number of reported NLRP3 inflammasome inhibitors. 292 3,4-methylenedioxy-β-nitrostyrene (MNS) and BAY 11-7082 ( Fig. 3A ) selectively inhibit the NLRP3 inflammasome independently of any effect on the NF-κB pathway in the priming stage by binding the ATPase region of the NLRP3 NACHT domain, with the Michael acceptor moieties being indispensable.293,294 Parthenolide ( Fig. 3B ), which has reported inhibitory activity against NLRP3, NLRP1, and NLRC4 inflammasome-dependent IL-1β release, is another example of an inhibitor with a Michael acceptor. The broad activity of parthenolide was found to be due to alkylation of both the caspase-1 p20 subunit and NLRP3. 294
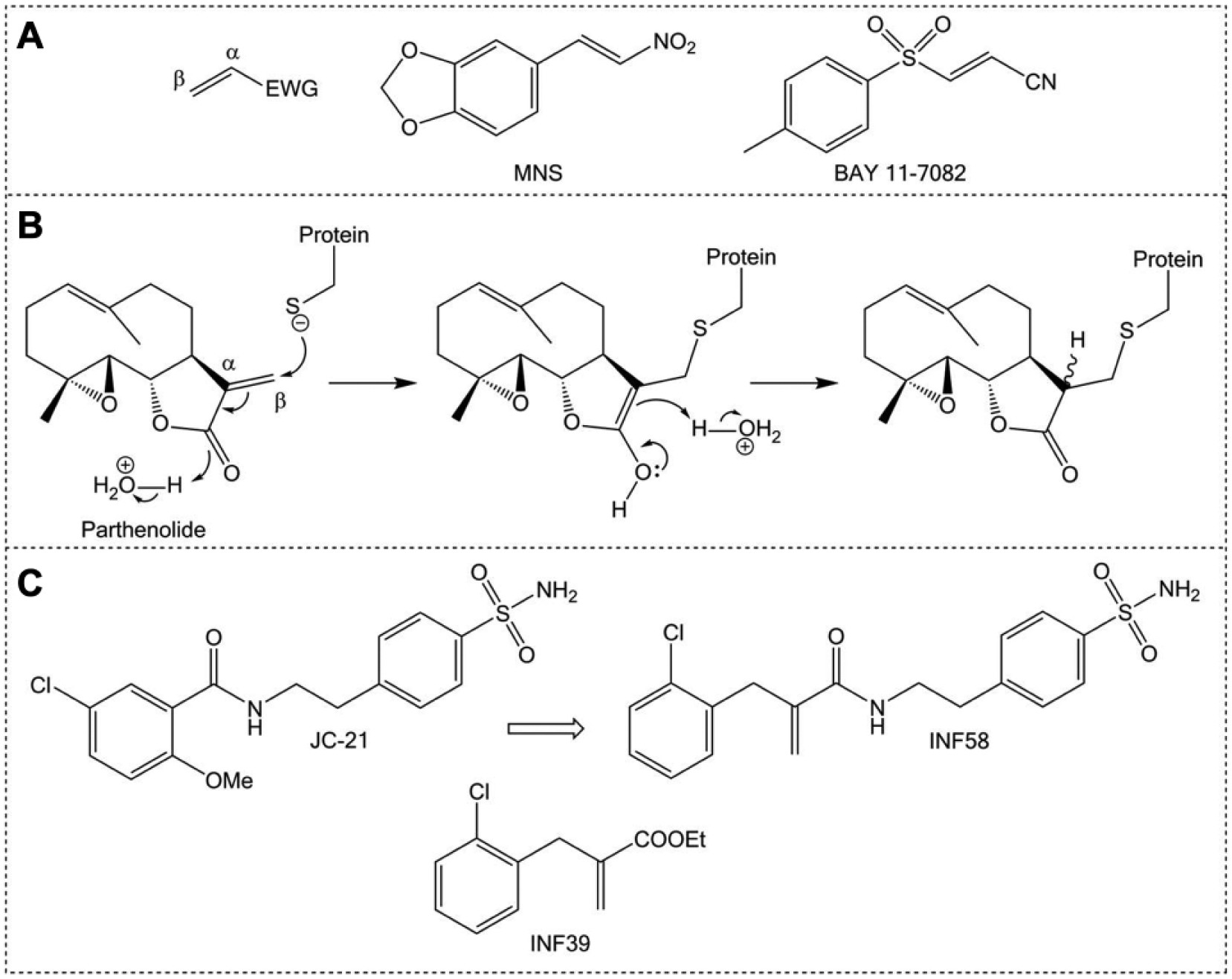
Michael acceptors in inflammasome inhibitors. (
A series of electrophilic Michael acceptor warheads inhibit ATPase activity of NLRP3 as well as caspase-1 activity. 292 Despite fine tuning of reactivity to avoid cytotoxic effects, the lead compounds of this initial study do retain some cytotoxicity, with a follow-up exploring less reactive acrylamide-based warheads. 295 In an interesting merging strategy, the electrophilic warhead was combined with structural motifs from the reported NLRP3 inflammasome inhibitor JC-21, itself an analogue of an intermediate in the synthesis of glyburide ( Fig. 3C ). 296 The resulting compound, INF58, is able to inhibit the ATPase activity of NLRP3, though with a relatively modest IC50 of 74 μM. 295 INF39 is the latest leading compound of this series ( Fig. 3C ), with an IC50 of around 10 μM, representing an irreversible, nontoxic inhibitor of the NLRP3 inflammasome that has undergone preliminary in vitro absorption, distribution, metabolism, excretion (ADME) studies. 297
NBC Compounds
Since the discovery of MCC950, the landscape for NLRP3 inhibition has broadened. Boron is an unusual element in drugs, with only one medicine in the clinic that contains boron, Velcade (or bortezomib), which is used to treat multiple myeloma. 298 We recently published our novel boron compound (NBC) series of inflammasome inhibitors based on an oxazaborine ring scaffold (exhibited by NBC-19; Fig. 4A ).299,300 2-Aminoethoxy diphenylborinate (2APB) is a small-molecule inhibitor of Ca2+ signaling used to probe the involvement of Ca2+ in inflammasome activation.301–304 Though 2APB likely affects the NLRP3 inflammasome independently of Ca2+, 305 the potential to develop 2APB as an NLRP3 inflammasome inhibitor is limited by its influence on Ca2+ homeostasis. A screen of a library of 2APB analogues suggests that boron is essential for inhibitory activity against IL-1β release, with the most active compounds consisting of a diarylborinic acid moiety and oxazaborine ring. Subsequently, several analogues were synthesized and screened, with the lead compounds also incorporating the trichloromethyl (CCl3) group, as well as an amide functionality as present in NBC-19 ( Fig. 4A ). This series of molecules effectively inhibit ASC speck formation and IL-1β release both in vitro and in vivo in a mouse model of peritonitis. 299 Importantly, these inhibitors appear to have no effect on Ca2+ signaling, with their inhibitory activity against IL-1β release independent of Ca2+. Thus, the NBC series represents a unique chemical scaffold for the development of NLRP3-targeting therapeutics.
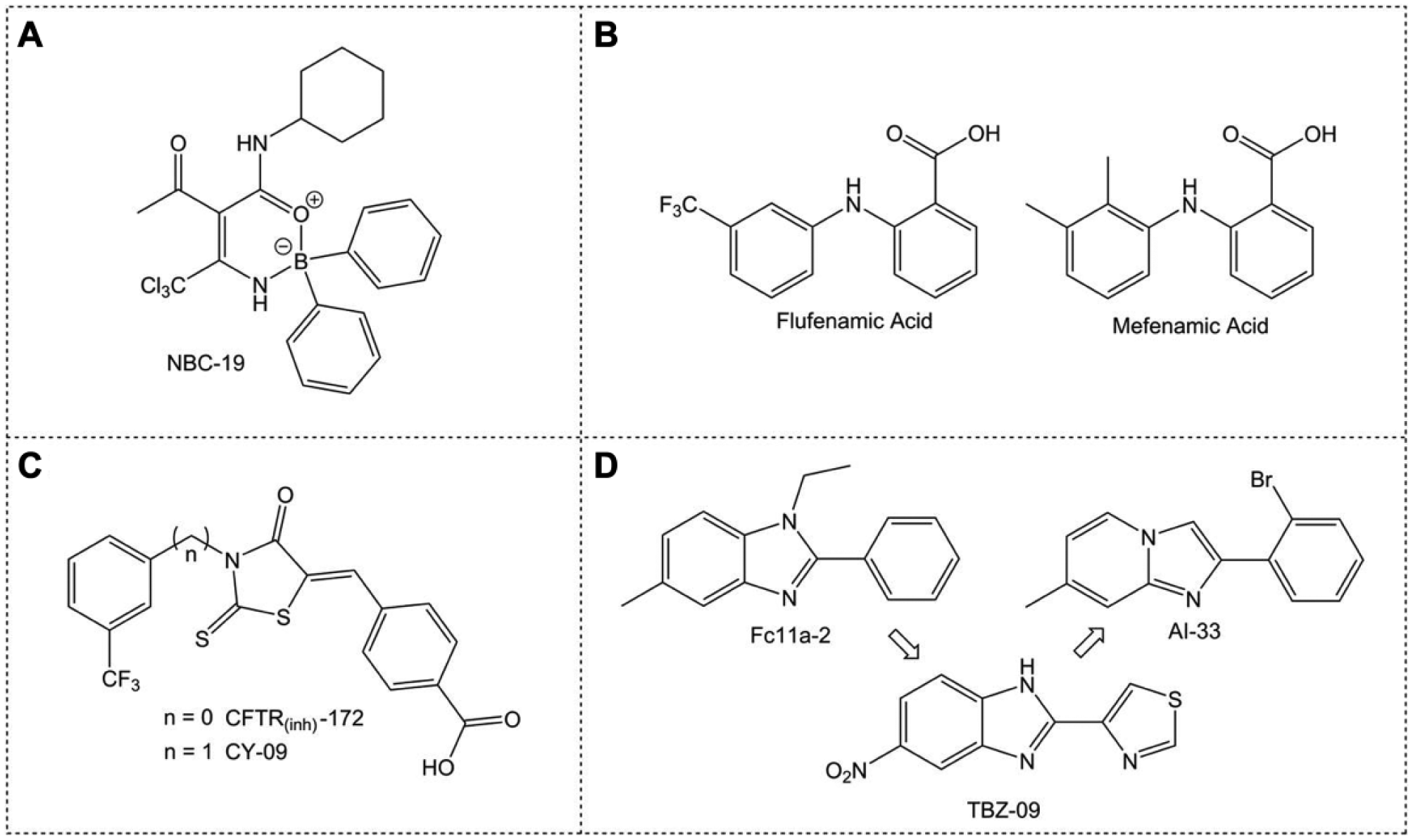
Structures of NLRP3 inflammasome inhibitors. (
Chloride Channels: A Potential Target?
Recent work has suggested that Cl− channel inhibition could be a good therapeutic angle for NLRP3 inhibition.15–17 We demonstrated that nonsteroidal anti-inflammatory drugs (NSAIDs) based on a fenamate scaffold could be repurposed as NLRP3 inhibitors ( Fig. 4B ). 15 The fenamate scaffold, exemplified by mefenamic and flufenamic acid, consists of two aromatic rings separated by an amine linker, with a carboxylic acid located in the position next (ortho) to the amine linker of one of the rings. The fenamates appear to inhibit the NLRP3 inflammasome through blocking Cl− channels, with pharmacology suggesting the volume-regulated anion channel (VRAC). 15 Further work is required to explore this approach of NLRP3 inflammasome inhibition.
Another Cl− channel inhibitor, the cystic fibrosis transmembrane conductance regulator inhibitor CFTR(inh)-172, specifically inhibited NLRP3 activation in LPS/ATP-treated BMDMs independently of any effect on CFTR ( Fig. 4C ). 306 Given the CFTR-independent nature of this effect, several CFTR(inh)-172 analogues were screened, which had no CFTR inhibitory activity. 306 One analogue, CY-09, exhibited NLRP3-specific inhibition of a magnitude similar to CFTR(inh)-172 ( Fig. 4C ). 306 CY-09 has no effect on LPS-induced priming, nigericin-induced K+ efflux, or VRAC-dependent Cl− efflux, but does inhibit NLRP3 oligomerization and subsequently inflammasome assembly. The proposed mechanism for CY-09 against NLRP3 is via inhibition of the ATP binding region of the NLRP3 NACHT domain, which prevents the ATPase activity necessary for NLRP3 self-association.306,307 In vivo, CY-09 is protective against NLRP3-dependent pathologies, both abrogating neonatal lethality in a mouse model of CAPS and reversing metabolic disorders in diabetic mice. 306 Interestingly CY-09 also inhibits caspase-1 activation and IL-1β production in synovial fluid cells from patients with gout, suggesting that CY-09 or its analogues may represent a potential therapeutic in NLRP3-dependent diseases. 306
Inhibiting NLRP3 Inflammasome Dissociation
In a search for small molecules with anti-inflammatory activity, a series of compounds were screened, yielding Fc11a-2 as an inhibitor of ATP-induced IL-1β release at 10 μM ( Fig. 4D ). 308 Fc11a-2 also dose dependently reduces the symptoms of dextran sodium sulfate (DSS)-induced experimental colitis in mice. 308 Inhibition of inflammasome activation by Fc11a-2 is through suppression of caspase-1 release from the ASC/NLRP3 complex. 308 Structurally similar thiabendazole analogues 309 can also act as anti-inflammatory agents. 310 An early lead, TBZ-09, inhibits the release of IL-1β and exhibits the structural feature of an electron withdrawing group (EWG) at position 5 of the benzimidazole core ( Fig. 4D ). A broader series was synthesized consisting of analogues retaining an EWG in this position, as well as alternatives to the benzimidazole scaffold. 310 The most potent compound of this series, AI-33 ( Fig. 4D ), has an IC50 of only around 10 μM; however, this series does represent a potential new approach to inhibiting NLRP3-dependent IL-1β release.
Summary
Inflammasomes have emerging roles in a variety of neurodegenerative conditions, and their inhibition may be an effective strategy for future therapeutics. Given the recent acceleration in advances surrounding the regulation, mechanism of action, and inhibition of inflammasomes, clinical manipulation of the pathway is becoming increasingly feasible. This is particularly evident in the discovery of existing drugs that target NLRP3. However, due to the multiphasic progression of many conditions, care must be taken to identify the disease stages at which inflammasome-directed therapies will be effective. It is also important to note from a translational standpoint that preclinical data are generally produced in laboratory mice under specified pathogen-free conditions. Therefore, the physiological importance of proteins with immunological functions such as inflammasomes may be masked in such studies. As humans exist in a far “dirtier”, more immunologically challenging environment, careful assessment of the potential impact of inflammasome inhibition on host–microbe interactions and pathogen resistance is required. At present, there are minimal data regarding side effects from inflammasome-targeting treatments specifically, though the CANTOS trial does note a slightly increased incidence of fatal infection in canakinumab recipients. 1 However, canakinumab targets all IL-1β irrespective of its origin. Immunosuppression might therefore be minimized by targeting specific inflammasomes, such as NLRP3, while leaving others, such as NLRC4 and AIM2, functional, thus preserving some aspects of the inflammasome response.
Work continues on a number of series in order to improve potency and pharmacokinetic properties, while attempting to limit off-target and cytotoxic effects. These small-molecule inhibitors of the NLRP3 inflammasome offer some promise in the development of potential anti-inflammatory therapeutics in the treatment of diseases involving the NLRP3-dependent release of IL-1β.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: T.S. is funded by the Alzheimer’s society. J.C. and J.A.B. are funded by MRC DTP studentships.