
Abstract
Porphyromonas gingivalis (Pg) is involved in the link between periodontal diseases and atherosclerosis worsening. In periodontal cells, Pg modifies IL-1β expression via the NLRP3 inflammasome pathway activation. Our aim was to investigate NLRP3 inflammasome activation in endothelial cells (ECs) after Pg infection and Pg-LPS stimulation. In both situations, RT-PCR experiments demonstrated an increase of the NLRP3 mRNA level that can be potentiated by pre-treatment of ECs with 5 mM ATP. However, Western blotting analysis revealed that Pg infection induced a proteolysis of NLRP3 protein and a major decrease of the native protein. After ATP pre-treatment and/or Pg-LPS stimulation, this proteolysis was not observed, while NLRP3 protein levels were increased. Proteolysis of the NLRP3 protein was not observed with heat-killed Pg and inhibition of ECs protein synthesis with cycloheximide did not abolish the NLRP3 protein degradation induced by Pg infection in ATP pre-treated cells. Additionally, significant increases of secreted IL-1β were measured after ATP pre-treatment and/or Pg-LPS stimulation, but not after Pg infection. These data showed that Pg and Pg-LPS differentially controlled the NLRP3 inflammasome pathway in ECs, and suggested a novel potential mechanism developed by Pg to reduce IL-1β secretion and to escape host immune response.
Introduction
The infection of atheromatous plaques is one of the various pathways involved in the worsening of atherosclerosis. 1 Different periodontal pathogens, mainly Porphyromonas gingivalis (Pg), have been found in atherosclerotic lesions. 2 However, many aspects of the inflammatory response to infection remain unclear.
Periodontal diseases are inflammatory diseases caused by pathogenic bacteria in oral biofilms that result in the destruction of tissues surrounding teeth 3 and affect systemic diseases, including atherosclerosis, via the increase of circulating pro-inflammatory cytokines, such as IL-1β, and repeated bacteremia of virulent pathogens. 4 Pg and its LPS (Pg-LPS) stimulate different types of cells and inflammatory pathways involved in atheromatous plaque development, such as endothelial cells (ECs) 5 through activation of TLRs 2 and 4. 6
ECs are at the interface between blood and vessel, and play several roles, including the transport of plasma molecules, lipid homeostasis and immune recognition. 7 In response to injury or aggression, these cells could either produce cytokines 8 or enter apoptosis. 7
Inflammasomes are multi-protein complexes that contain NLRs, an adaptor protein (ASC) and pro-caspase-1. 9 NLRPs 3 and 1 activate caspase-1- and NF-κB-related pathways leading to the conversion of pro-IL-1β and pro-IL-18 to their respective mature forms resulting in their secretion.10,11 The control of inflammasome activation involves a large number of endogenous factors, including cholesterol, ATP and microbial components. 12 In mice deficient in low-density lipoprotein receptor and several components of the inflammasome, the size of the atheromatous plaque appears reduced, demonstrating a role for this complex in the development of atherosclerosis. 13 Recently, a clinical study in human showed that NLRP3 expression is correlated with the severity of coronary artery disease and cardiovascular risk factors. 14
Pg has been shown to deregulate the NLRP3 inflammasome complex in a cell-specific manner in various cells types, such as oral epithelial cells, 15 gingival fibroblasts 16 and macrophages. 17 NLRP3 inflammasome deregulation reduces IL-1β secretion 16 and endocytosis, 17 but no specific bacterial virulence factor has been directly related to these biological processes.
The aim of our study was to determine if Pg infection could activate the NLRP3 inflammasome pathway in ECs and to assess Pg-LPS involvement.
Materials and methods
Bacterial culture
The Pg strain (ATCC 33277) was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and was cultivated under strict anaerobic conditions at 37℃ in a brain–heart infusion medium supplemented with hemin (5 mg/ml) and menadione (1 mg/ml). All products were purchased from Sigma (St. Louis, MO, USA). On the day of infection, bacteria were centrifuged, washed twice with PBS and counted as previously described. 18 When using heat-killed Pg (HPg), bacteria were heated at 85℃ for 10 min before the beginning of the experiment.
The Pg-LPS used was a commercial ultrapure preparation purchased from InvivoGen (San Diego, CA, USA).
Cell culture
Human umbilical vein ECs were cultured in a complete EGM2 medium containing 100 000 U/l of penicillin and 100 mg/l of streptomycin at 37℃ in a humidified atmosphere with 5% CO2. All the media and antibiotics were obtained from PromoCell (Heildelberg, Germany).
Infection of ECs with Pg or stimulation by its purified Pg-LPS
Twenty-four hours before the beginning of the experiment, 2 × 105 cells were plated in 25 cm2 flasks. At the day of the experiment, ECs were washed twice with PBS and infected for 6 h 16 with Pg at a multiplicity of infection (MOI) of 200 bacteria/cell5,19 and stimulated for 24 h by Pg-LPS used at a final concentration of 1 µg/ml in the cell growth medium.5,8 Uninfected and non-stimulated cells served as controls. Two h before the start of the experiment, certain batches of cells were incubated for 2 h with 5 mM ATP, a known inducer of the NLRP3 inflammasome; when indicated, 25 µg/ml of cycloheximide (Sigma), a potent inhibitor of protein synthesis, was added in the culture medium 1 h before the addition of Pg.
RT-PCR experiments
To assess, mRNA expression of NLRP1, NLRP3 and IL-1β, RT-PCR was performed. Total RNA was extracted using the RNeasy kit (Qiagen, Les Ulis, France), according to the manufacturer’s instructions. RNA concentrations and purity were determined using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Rockland, DE, USA). Reverse transcription and PCR were performed as described previously. 18 Validated PCR primers pairs for the human genes NLRP1, NLRP3, IL-1β and GAPDH were purchased from Qiagen (Quantitect Primer Assay) and used at a final concentration recommended by the furnisher. Samples were processed in triplicate and for each sample the band intensity of the amplified fragment was expressed relative to the intensity obtained with GAPDH that was used as an internal control. 18
Western blotting
In order to detect the protein level of NLRP3, Western blot was performed. SDS-PAGE followed by immune-blotting were performed in conditions previously described.
18
Briefly, ECs collected from infection with Pg and from stimulation by Pg-LPS were lysed for 5 min on ice in 200 µl of ice-cold RIPA buffer (65 m
ELISA assay
Dosage of IL-1β was done through ELISA analysis. A volume of 100 µl cell culture medium was used to quantify the level of secreted IL-1β using the BioLegend ELISA Max Deluxe set kit distributed by Ozyme (Saint Quentin en Yvelines, France), in accordance with the manufacturer’s specifications. Each sample was quantified in triplicate.
Statistical analysis
Statistical analyses were performed using the non-parametric Mann–Whitney rank sum test. A P-value of < 0.05 was considered statistically significant. The reported data are the means of at least three separate experiments performed under similar conditions.
Results
Pg infection and Pg-LPS stimulation induce the mRNA expression of NLRP3
During the time course infection of ECs by Pg, a significant increase in the NLRP3 mRNA levels were measured, with a maximum observed at 3 h (Figure 1). When ECs were pre-treated with 5 mM ATP, a significant increase of the NLRP3 mRNAs levels was also observed. Pg infection increased the NLRP3 mRNA level in ATP pre-treated cells modestly. No significant variations of the levels of the mRNA encoding for all others tested genes (NLRP1, IL-1β) were measured, even if ECs were pre-incubated with ATP.
Increasing expression of the NLRP3 mRNA in ECs infected with Pg. Human umbilical vein ECs were pre-incubated for 2 h in the presence or absence of 5 mM ATP and then infected with Pg at an MOI of 200 for 6 h. Total RNA was extracted and the expression of the mRNA encoding for the NLRP1, NLRP3 and IL-1β genes were quantified by RT-PCR. GAPDH was amplified as the housekeeping gene and relative gene expression was calculated. Assays were done in triplicate and data shown are the mean ± SD (n = 3; *P < 0.05 in comparison to the control group).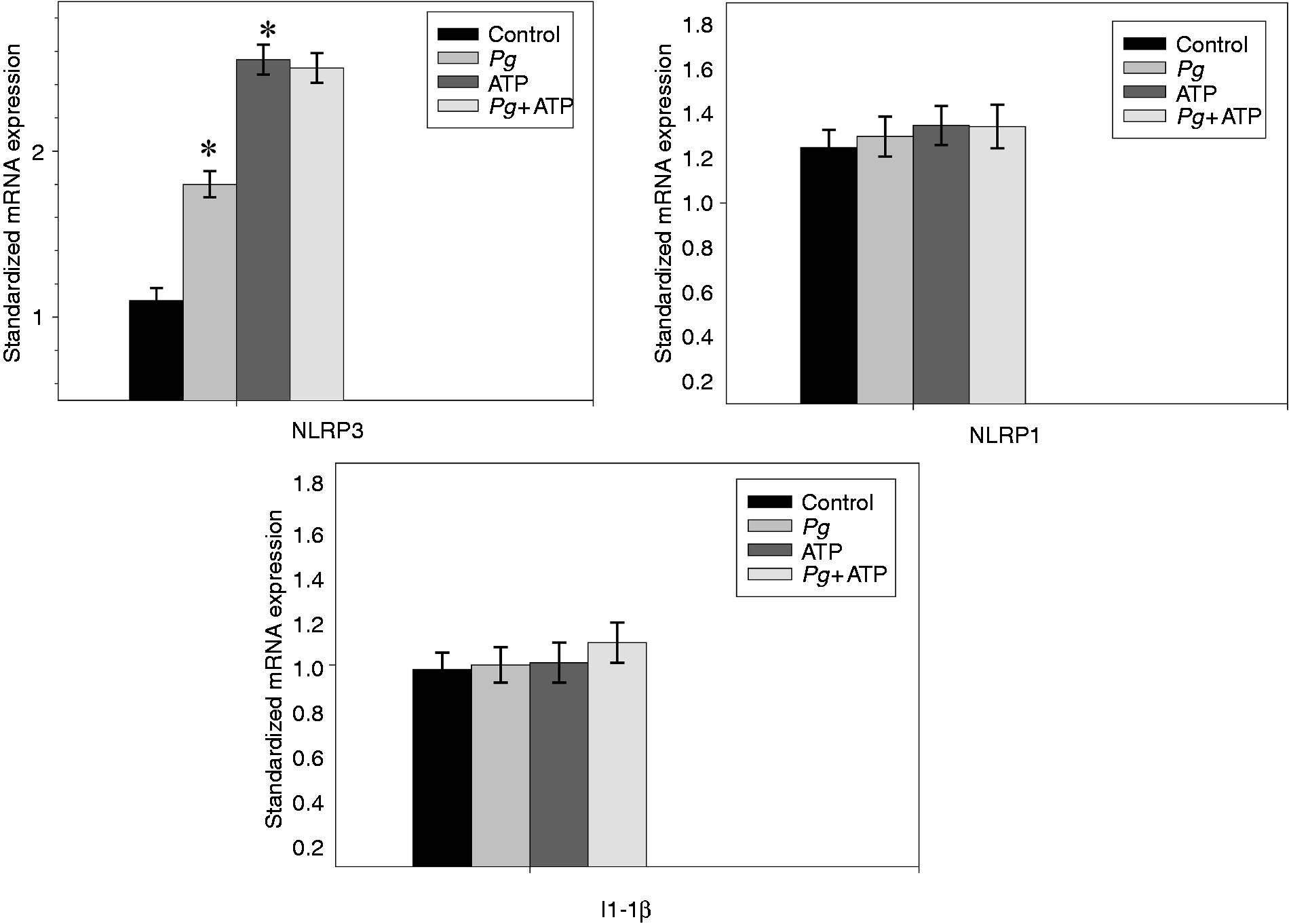
Pg-LPS stimulation also induced at 12 h a significant increase of the NLRP3 mRNA levels (Figure 2). The Pg-LPS stimulation did not significantly affect the levels of the mRNA encoding all others tested genes (NLRP1, IL-1β), even when EC stimulation was done in the presence of ATP.
Increasing expression of the NLRP3 mRNA in ECs stimulated by Pg-LPS. Human umbilical vein ECs were pre-incubated for 2 h in the presence or absence of 5 mM ATP and then stimulated by Pg-LPS at a final concentration of 1 µg/ml for 12 h. Total RNA was extracted and the expression of the mRNA encoding for the NLRP1, NLRP3 and IL-1β genes were quantified by RT-PCR. GAPDH was amplified as the housekeeping gene and relative gene expression was calculated. Assays were done in triplicate and data shown are the mean ± SD (n = 3; *P < 0.05 in comparison to the control group).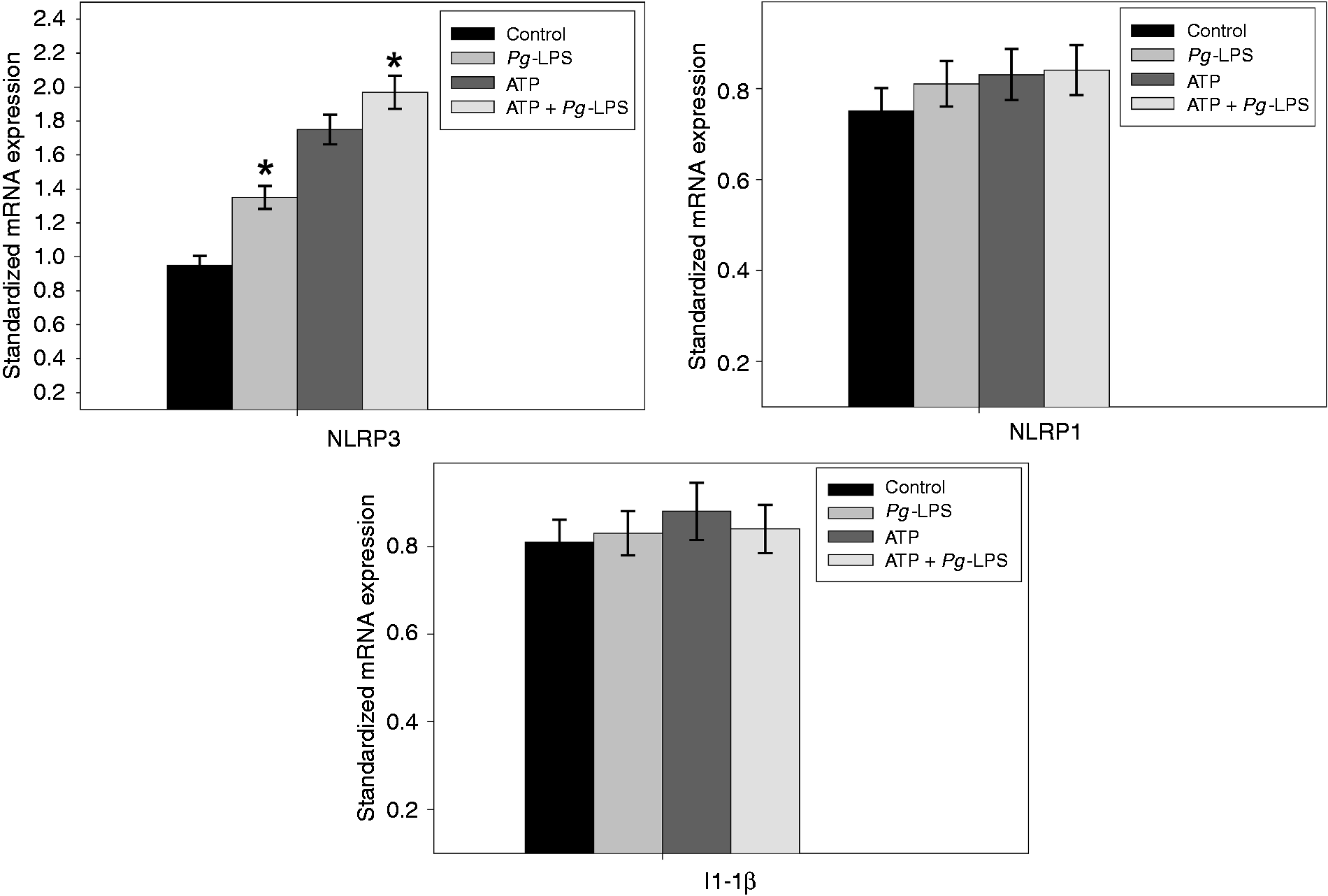
Infection with Pg induces proteolysis of NLRP3 protein
After 3 h, the NLRP3 protein was not detected in the uninfected control, but pre-incubation of ECs with 5 mM ATP led to a marked increase of the NLRP3 protein band (MM 109 ku), confirming ATP as an activator of the NLRP3 expression (Figure 3). However, in infected extracts obtained with or without ATP pre-treatment, we did not observe the NLRP3 protein except for a faint band at 109 ku, and several lower MW bands persisting in all the ATP pre-treated cells suggesting a proteolysis of the NLRP3 protein resulting from Pg infection (Figure 4). Interestingly, this process appeared to be specific, while NLRP1 protein was not affected by Pg infection (Figure 3).
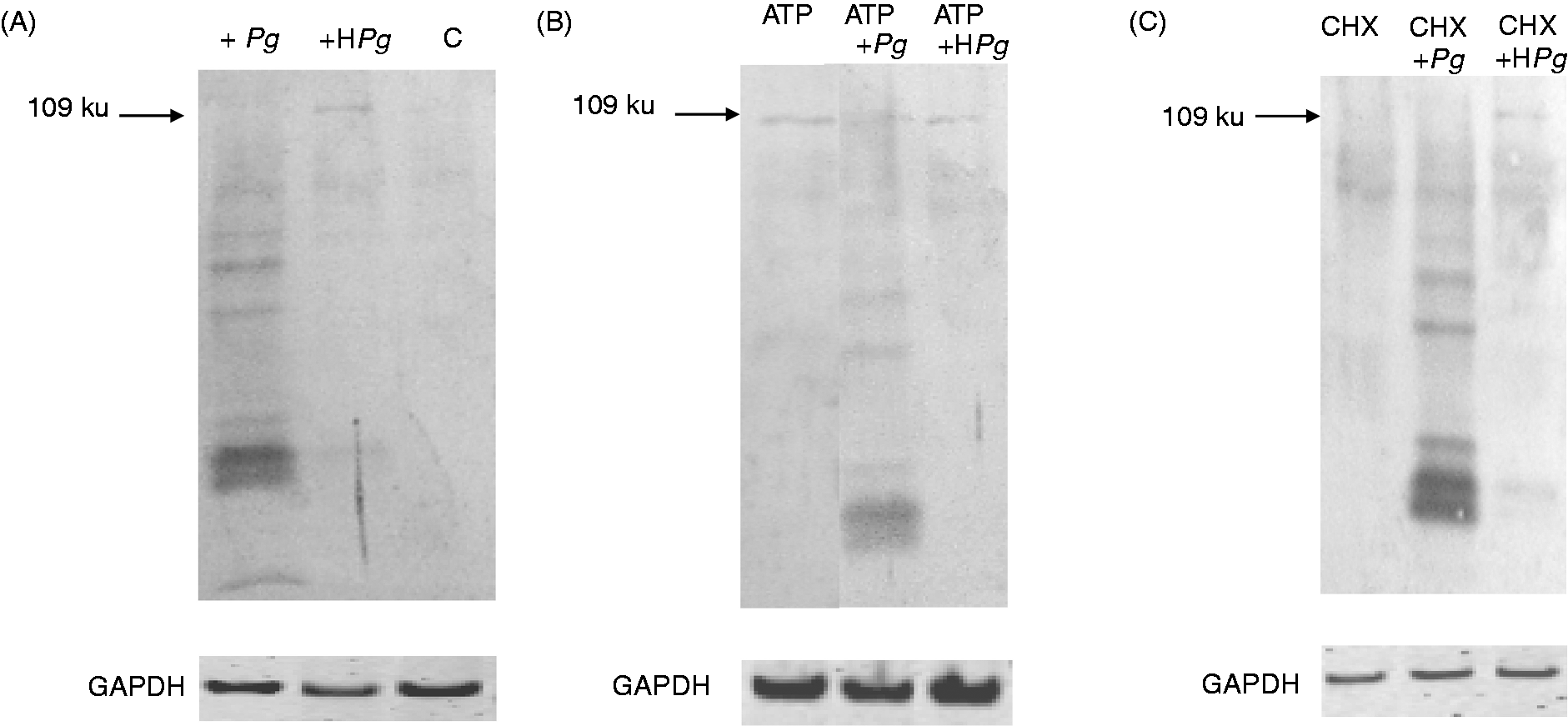
Infection with Pg induces proteolysis of the NLRP3 protein. ECs were pre-incubated for 2 h in the presence or absence of 5 mM ATP and then infected with Pg for 3 h or stimulated by Pg-LPS for 24 h. Cell extracts were prepared after infection (A) or stimulation (B) for Western blot analysis using Abs for NLRP3, NLRP1 and for GAPDH used as an internal control to check the equal loading of proteins in all wells. Assays were done in triplicate and data shown are the mean ± SD (n = 3; *P < 0.05 in comparison to the control group). Effect of HPg and of cycloheximide (CHX) on the proteolysis of NLRP3. ECs were pre-incubated for 2 h in the absence (A) or presence (B) of 5 mM ATP before the addition of Pg or HPg. To test the effect of CHX, an additional hour of incubation with CHX at 25 µg/ml was included before the addition of Pg or HPg for 3 h (C). Cell extracts were prepared for Western blot analysis using Abs for NLRP3 or for GAPDH used as an internal control to check the equal loading of proteins in all wells.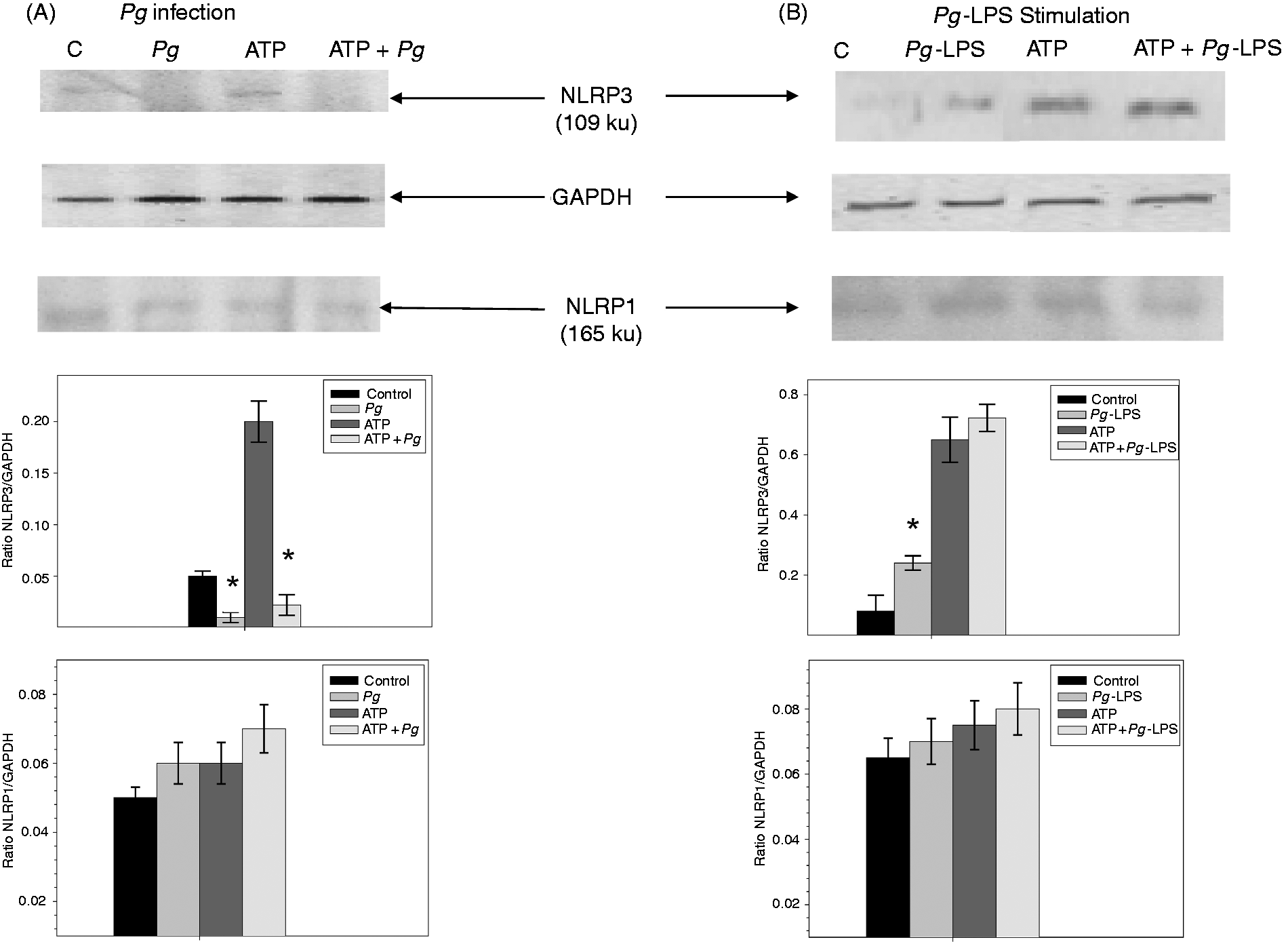
In ECs stimulated by Pg-LPS, we observed at 24 h an increase of the intensity of the NLRP3 band in extracts of activated cells, without any sign of NLRP3 proteolysis compared with the control cells (Figure 3). A more marked increase of the NLRP3 band intensity was observed in EC pre-treated with 5 mM ATP and was amplified after Pg-LPS stimulation. Taken together, these results showed that Pg infection induced a proteolysis of the NLRP3 protein that does not involve Pg-LPS.
Effects of HPg and of cycloheximide on the NLRP3 proteolysis
When ECs were infected for 3 h with HPg, we observed, using Western blotting, a considerable decrease of the proteolysis of NLRP3 band compared with infection performed with the viable bacterium (Figure 4), suggesting that NLRP3 proteolysis may be initiated by degrading enzyme(s) of bacterial origin.
Additionally, when the NLRP3 production was initially induced by ATP pre-treatment followed by an infection in the presence of cycloheximide, a potent inhibitor of protein synthesis added 1 h before the addition of Pg or HPg, we still observe the proteolysis of NLRP3 with viable Pg, but a reduced proteolysis when HPg was used (Figure 4). These data suggest that the degrading enzyme(s) did not originate from a de novo synthesis, but can be either constitutive of the live bacteria or be cellular enzyme(s) activated as a consequence of the Pg infection.
Secretion of IL-1β by ECs infected with Pg or stimulated by Pg-LPS
At any point of the time course infection, we did not measure any significant variation of the amounts of secreted IL-1β compared with uninfected controls. The presence of 5 mM ATP increased significantly the secretion of IL-1β only in the uninfected controls cells but not in infected ECs (Figure 5).
Secretion of IL-1β in the culture mediums from ECs infected with Pg or activated by Pg-LPS. ECs were pre-incubated for 2 h in the presence or absence of 5 mM ATP and then infected with Pg for 6 h (A) or stimulated by Pg-LPS for 24 h (B). These different time points were chosen because they corresponded to the maximum biological effect observed in each case. At each indicated time point of the experiment, cell culture mediums were analyzed by ELISA to quantify the levels of secreted IL-1β. Assays were done in triplicate and data shown are the mean ± SD (n = 3; *P < 0.05 in comparison to the control group).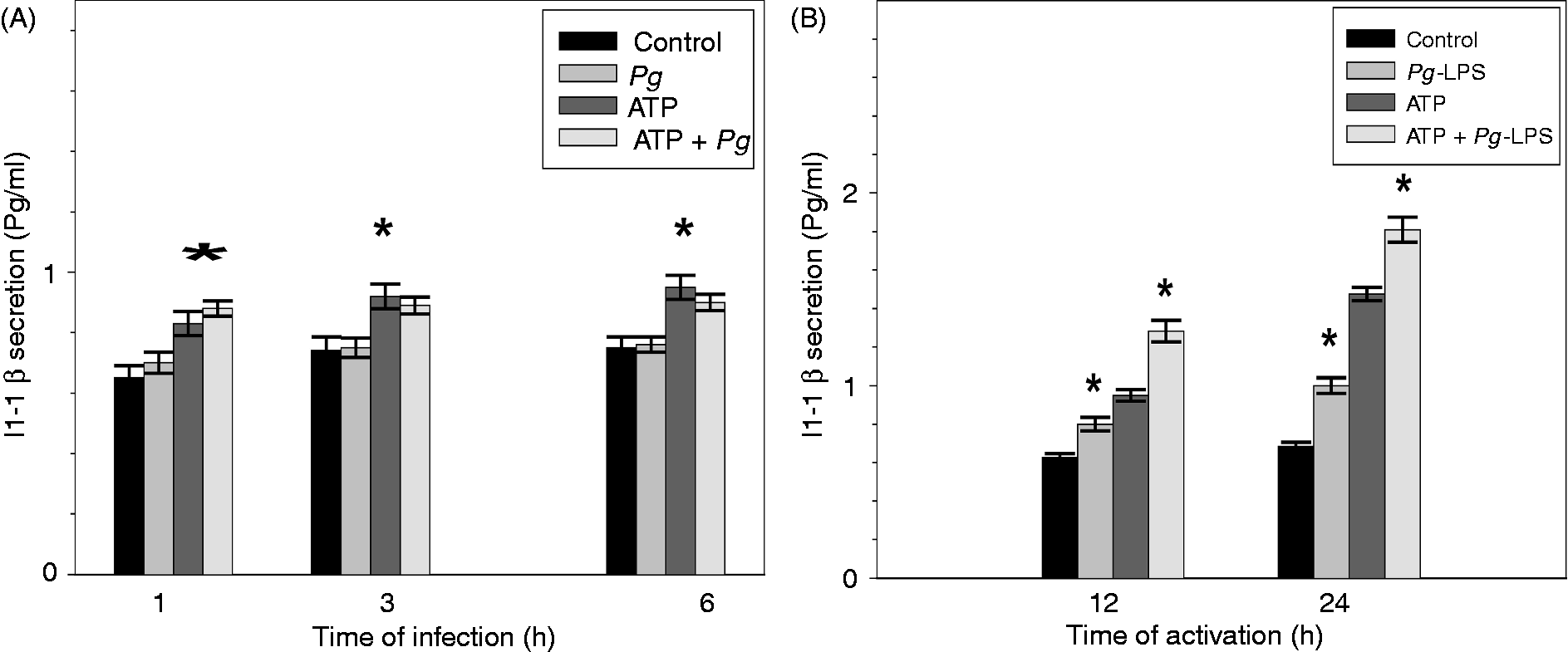
The stimulation of ECs by Pg-LPS induced the increase of IL-1β secretion at all time points, and this effect was more significant in ATP pre-treated cells (Figure 5). Our results showed that Pg infection did not induce IL-1β secretion, while Pg-LPS stimulation led to an increase of IL-1β production potentiated by ATP.
Discussion
Our study demonstrates, for the first time, that the NLRP3 inflammasome pathway could be a molecular target for Pg in ECs. In our model, Pg increased the mRNA expression of NLRP3 with or without pre-treatment with ATP, but not the mRNA expression of NLRP1 and IL-1β. EC stimulation by Pg-LPS also significantly increased mRNA expression of NLRP3. These data show that this Pg virulence factor was able to alone induce the NLRP3 inflammasome response. In some cell types, Pg infection increases NLRP3 mRNA expression, as shown in Mono-Mac-6 cells 21 and macrophages. 17 However, a decrease or a non-significant modification of NLRP3 mRNA expression has been observed in other cell types, such as gingival fibroblasts 16 and epithelial cells, 15 respectively, suggesting a cell/tissue specificity of inflammasome response to Pg infection. Other periodontal bacteria such as Aggregatibacter actinomycetemcomitans could induce an up-regulation of the mRNA expression related to NLRP3 in human mononuclear cells without any effect on the expression of mRNA encoding for caspase-1. 22 Interestingly, ATP, a compound known to activate the NLRP3 inflammasome, 13 potentiated this induction in our cellular model, as described in mouse macrophages. 17
At the protein level, infection with Pg specifically induced a major decrease and a proteolysis of NLRP3 protein, while NLRP1 protein was not affected by the infection. Furthermore, this specificity was confirmed by the observed absence of cathepsin B proteolysis in a study on Pg infection using the same cell and infection model. 5 Few studies have analyzed the bacterial effect on NLPR3 expression at the protein level and, to our knowledge, this could be the first demonstration of a proteolytic mechanism related to this particular inflammasome. In the same manner, recent data have shown that Pg is able to induce proteolysis of RIPK1 and RIPK2, cellular components involved in EC apoptosis and host defense, probably through the action of a lysine-specific gingipain. The proteolysis of key molecular compounds may be considered as a potential strategy for bacterial persistence within ECs. 23 The use of HPg considerably reduced NLRP3 proteolysis suggesting that the involved enzyme(s) may be of bacterial origin. On the contrary, however, Pg-LPS stimulation increased NLRP3 protein expression, showing that the proteolysis observed during the infection was Pg-LPS-independent. However, we could not exclude the notion that one or more cellular degrading enzymes may be activated in response to the Pg infection. Additionally, proteolysis of NLRP3 still occurred during infection with Pg in the presence of cycloheximide, an inhibitor of protein synthesis, supporting the hypothesis that the degrading enzymes may be constitutive of the bacterium and are not synthetized during infection.
This decrease in NLPR3 protein via proteolysis could be related to a NLRP3 inflammasome pathway inhibition in ECs by Pg infection. NLRP3 pathway impairment has also been observed in other cell types after Pg infection via different mechanisms such as mRNA down-regulation 16 and ubiquitination. 24
In ECs pre-treated or not with ATP, Pg infection did not change the secretion level of IL-1β, while an increase of IL-1β secretion was observed after ATP and/or Pg-LPS stimulation. These data suggest that the observed proteolysis of NLRP3 protein could specifically affect EC response to Pg infection. In gingival fibroblasts, the decrease of the NLRP3 mRNA level is also associated with a decrease in the mRNA encoding IL-1β. 16 Furthermore, the increase of IL-1β after Pg-LPS stimulation showed that NLRP3 could be involved in the control of ECs’ biological response to a vascular dissemination of Pg periodontal infection. Overall, the relatively weak increase in IL-1β secretion observed in response to ATP stimulation, Pg infection and Pg-LPS stimulation was relatively weak and could have been due to the relatively short experimental period, as shown here by the progressive increase of IL-1β secretion with the culture duration of ECs after ATP stimulation. However, in another cell type and in another model of stimulated ECs, the measured levels of IL-1β secretion after Pg infection were similar.15,25
In conclusion, our results obtained in human ECs show that Pg infection and Pg-LPS stimulation differentially activate NLRP3 inflammasome pathway. This differential effect of Pg infection and Pg-LPS stimulation on ECs has been previously observed for cathepsin B activation. 5 The whole bacteria can escape the immediate local immune inflammatory response via the proteolysis of the induced NLRP3 inflammasome, while Pg-LPS initiates a delayed pro-inflammatory IL-1β response via NLRP3 inflammasome activation. NLRP3 proteolysis could be considered as a new tool in the bacterial arsenal to escape the host immune response in ECs. Indeed, invasion of ECs by Pg has already been described as one of the mechanisms contributing to the worsening of atherosclerosis. 26 As NLRP3 is involved in atherosclerosis, 27 we could not rule out the possibility that the longstanding presence of Pg in ECs or of one of its virulence factors, such as Pg-LPS, worsened atherosclerosis via NLRP3 inflammasome pathway control. The longstanding presence of Pg led to low-grade chronic inflammation, a hallmark of pathogen-mediated chronic inflammatory disorders. However, further investigations are needed to clarify the precise role of Pg in EC responses, notably the identification of one or more possible bacterial or cellular degrading enzymes acting on NLRP3 or other inflammasome components induced by Pg infection.
Footnotes
Funding
The authors would like to thank their institutions for the financial support.