
Abstract
Kinase inhibitors have dramatically increased patient survival in a multitude of cancers, including hematological malignancies. However, kinase inhibitors have not yet been integrated into current clinical trials for patients with T-cell-lineage acute lymphoblastic leukemia (T-ALL). In this study, we used a high-throughput flow cytometry (HTFC) approach to test a collection of small-molecule inhibitors, including 26 FDA-approved tyrosine kinase inhibitors in a panel of T-ALL cell lines and patient-derived xenografts. Because hypoxia is known to cause resistance to chemotherapy, we developed a synthetic niche that mimics the low oxygen levels found in leukemic bone marrow to evaluate the effects of hypoxia on the tested inhibitors. Drug sensitivity screening was performed using the Agilent BioCel automated liquid handling system integrated with the HyperCyt HT flow cytometry platform, and the uptake of propidium iodide was used as an indication of cell viability. The HTFC dose–response testing identified several compounds that were efficacious in both normal and hypoxic conditions. This study shows that some clinically approved kinase inhibitors target T-ALL in the hypoxic niche of the bone marrow.
Introduction
T-cell-lineage acute lymphoblastic leukemia (T-ALL) is an aggressive hematopoietic malignancy that arises in the thymus from malignantly transformed T-cell progenitors. 1 It comprises ~15% of newly diagnosed children and young adults with ALL. Currently, patients with T-ALL require intensified therapies, for which risk-adjusted treatments are primarily determined by minimal residual disease (MRD) response. 2 Importantly, patients with high-risk T-ALL do not benefit from further therapy intensification and often experience additional adverse events. Although cure rates have shown dramatic improvement over time, poor outcomes are still observed in patients who failed induction or relapsed after conventional chemotherapy. Affected patients are typically adolescents or young adults for whom a second remission is often very short. Therefore, the development of novel treatment programs for pediatric T-ALL is essential. Novel therapies could cure patients for whom cure has been elusive or could provide bridging therapies to eradicate persistent MRD and facilitate stem cell transplantation.
Tyrosine kinase inhibitors (TKIs) have revolutionized modern pharmacology, leading to increased survival in a multitude of solid tumors, as well as precursor B-cell ALL (B-ALL) and chronic myelogenous leukemia (CML) with BCR-ABL1 fusion. 3 While B-ALL and CML have become an intensive area of study for kinase-directed therapies, TKIs have not yet been integrated into current therapies for patients with T-ALL. To date, a few studies have investigated the efficacy of kinase pathway inhibition in T-ALL aberrantly overexpressing FLT3, JAK-STAT, mTOR, and so forth.4–6 However, the development of targeted therapies is challenged by the high degree of heterogeneity in T-ALL, in which most cytogenetic or molecular driver mutations occur at very low frequencies. 1
Genome-wide studies have played a prominent role in drug response profiling, 5 but the repertoire of specific biomarkers for targeted therapies remains limited in T-ALL. Notably, kinase signaling pathways are critically involved in all aspects of cellular function. In cancer cells, they drive aberrant signaling, regulating cell proliferation, cell survival, and chemoresistance. Dysregulation of tyrosine kinase and other kinase signaling pathways is commonly seen in T-ALL, providing an opportunity for therapeutic intervention. 7 Such pathways might be targeted with FDA-approved small-molecule inhibitors, which are available for drug repurposing, and the development of novel targeted therapies. Because kinase inhibitors have relatively safe adverse event profiles, they have generally been able to safely integrate into dose-intensified therapies in leukemia treatment regimens. 3 We hypothesized that T-ALL may be sensitive to treatment with clinically approved kinase inhibitors. Therefore, we tested a library of FDA-approved inhibitors to identify the most potent drug candidates with antitumor activity against T-ALL.
One critical factor that influences cancer cell sensitivity to drug treatment is the microenvironment, with an emphasis on hypoxia.8,9 Many promising drug candidates with demonstrated antitumor potential in in vitro assays paradoxically fail in preclinical trials in vivo because the microenvironment niche serves as a site for resistance to chemotherapy. On the other hand, some inhibitors show increased antitumor activity toward hypoxic cells, suggesting that conventional drug sensitivity testing under normoxic conditions may result in overlooking promising drug candidates. 8
Recent works suggest Src activation as an important mechanism by which cancer cells maintain chemoresistance under hypoxia to promote cell survival, progression, and metastasis of a variety of human cancers. 9 In T-ALL, the hypoxic bone marrow niche is commonly infiltrated with rapidly proliferating T-cell lymphoblasts. A growing body of evidence indicates that the hypoxia present in the bone marrow microenvironment alters the activity of multiple clinically approved TKIs contributing to drug resistance. 9 Therefore, novel approaches that will identify drug candidates that effectively target leukemic cells in the hypoxic microenvironment are warranted.
We utilized the unique drug screening capabilities of the University of New Mexico Center for Molecular Discovery 10 to identify small-molecule kinase inhibitors for drug repurposing in T-ALL. In addition to T-ALL cell lines, we performed drug sensitivity profiling using T-ALL primary samples and patient-derived xenografts (PDXs), which were maintained as monoculture in serum-rich media. We established a high-throughput flow cytometry (HTFC) synthetic niche that mimics the low oxygen levels found in leukemic human bone marrow to assess leukemic cells for sensitivity against clinically approved inhibitors. We successfully identified inhibitors that were efficacious in normoxia as well as niche-mimicking conditions and which have not been used in modern T-ALL regimens. Our HTFC screening platform provides a rapid and convenient tool for the implementation of new therapeutic options and drug repositioning opportunities for suspension target cells such as leukemic blasts.
Material and Methods
Reagents
All reagents were purchased from Thermo Fisher Scientific (Waltham, MA) unless specified otherwise.
T-ALL Cell Lines
Human T-ALL cell lines (Loucy, ALL-SIL, Jurkat, and CCRF-CEM) were purchased from DSZM-German Collection of Microorganisms and Cell Cultures. The CUTLL1 cell line was a generous gift from Dr. Ferrando at Columbia University. The cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM
T-ALL Patient Samples and Patient-Derived Xenografts
Cryopreserved primary samples were obtained from patients enrolled in Children’s Oncology Group T-ALL trial AALL0434 and/or the University of New Mexico Health Sciences Center. All patients or their parent(s) or guardian(s) provided written, informed consent for future research in accordance with the Declaration of Helsinki and local institutional human research guidelines. The Institutional Animal Care and Use Committee approved the animal studies. Characteristics of PDXs are listed in
Supplemental Table S1
. PDXs were established by injecting 1–2 × 106 cells via the tail vein into nonobese diabetic and severe combined immunodeficiency NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ mice. Leukemia engraftment was assessed by flow cytometry analyses of peripheral blood with fluorescent-labeled anti-human APC-CD45+ and anti-mouse BV21-CD45+ antibody (BD Biosciences, San Jose, CA). Leukemic cells were purified via centrifugation in Ficoll-Paque/Percoll density gradient (GE Healthcare, Piscataway, NJ). The cells were preserved with freezing medium (90% FBS, 10% DMSO) and stored in liquid nitrogen. PDX samples were used at low passage up to three to limit the effect of mouse positive selection on PDX divergence from the original tumor sample. The cells were thawed and rested for ~4 h at 37 °C in complete RPMI-1460 medium supplemented with 2 mM
Compounds
We investigated several drug classes, including TKIs (n = 26), cyclin-dependent kinase inhibitors (n = 2; alvocidib and palbociclib), Hedgehog signaling inhibitor (n = 1; vismodegib), and proteasome inhibitors (n = 2; bortezomib and carfilzomib), all purchased from LC Laboratories (Woburn, MA) ( Suppl. Table S2 ). The compounds were solubilized in DMSO (250 µM for alvocidib, bortezomib, and carfilzomib; 20 mM for vandetanib; 25 mM for all others) and stored frozen at −80 °C.
High-Throughput Flow Cytometry Viability Testing
An Echo 555 Liquid Handler (Labcyte, San Jose, CA) was used both to create intermediate source plates and to add inhibitors to assay plates. The HTFC assays were conducted in 384-well Greiner Bio-One 784201 polypropylene plates (Monroe, NC). The 31 drugs were added to the plates to generate 10-point dose responses, with an assay final DMSO concentration of <1%. Each compound was tested at a clinically relevant concentration range (0.005–100 µM except for bortezomib, alvocidib, and carfilzomib, which were tested at 0.05–1000 nM) ( Suppl. Table S1 ). The cells were resuspended in fresh media and 15 µL was added to wells with a Biotek Multiflo system (Winooski, VT). The cell lines were tested at 5000 cells per well, and the primary samples were tested at 4500–6000 cells per well. The assay plates were incubated in a humidified atmosphere at 37 °C and 5% CO2. The hypoxic incubation, 1% O2, was achieved in a Cytomat24 C 10 (Thermo Fisher). The assay plates were covered with polystyrene lids for the first 24 h of incubation and sealed with a PlateLoc Thermal Microplate Sealer (Agilent, Santa Clara, CA) for the remainder of the incubation. Plates from normoxic conditions were flushed with ambient air for 1.2 s prior to sealing, and those from hypoxic conditions were flushed with 100% N2. Three replicate experiments were performed for the cell lines, which were incubated for 72 h. Due to the limited availability of primary samples, those experiments were done in single replicates, but with multiple plates to allow for 48 and 72 h incubations. On average, eight 384-well plates (3072 wells) were analyzed per day. With continuous round-the-clock automation, we have the potential to analyze ~100 × 384- or 1536-well plates per day, but our throughput was based on the availability of cells and primary samples.
The cell viability assay was performed as previously described. 11 Briefly, 5 µL of propidium iodide (PI; Sigma-Aldrich) was added to all wells of the assay plates to a final concentration of 0.75 µg/mL. The plates were then incubated on rotators at 4 °C for 20–45 min. The HTFC data were collected with a HyperCyt platform (IntelliCyt, Albuquerque, NM) configured to an Accuri C6 Plus flow cytometer (BD Biosciences, Franklin Lakes, NJ). The data were analyzed with HyperView software (IntelliCyt). The samples were gated on forward and side scatter to isolate the cell populations, and a binary gate was used on the histograms for fluorescence in the FL-3 channel to distinguish PI-positive and -negative populations.
Data Analysis
The data were normalized to the mean viability from negative control wells = 100% viability for inhibitor-treated samples. GraphPad Prism 7 software (La Jolla, CA) was used to plot and fit the data. Half-maximal effective concentration (EC50) values were calculated based on least-squares fit of the data to a four-parameter sigmoidal dose–response curve. EC50 values are only reported for compounds that yielded a maximum response value of 20% or greater. For EC50 values calculated to be beyond the concentration range tested and for those samples that yielded maximal responses <20%, they are reported as >100 µM. The hypoxia cytotoxicity ratio (HCR) was determined for each drug and cell line as EC50normoxia/EC50hypoxia. For each cell line and compound, the HCRs from independent experiments were compared with those from a two-tailed paired t-test.
Results and Discussion
Drug Sensitivity Screening in T-ALL Cell Lines
To determine the sensitivity of T-ALL cells to tyrosine kinase pathway inhibition in T-ALL, we compiled a library of 26 FDA-approved TKIs with clinically proven efficacy against multiple tyrosine kinases, including PDGFR, VEGFR, EGFR, and ALK ( Suppl. Table S2 ). Due to growing interest in kinases that regulate cell cycle, we also added two cyclin-dependent kinase inhibitors: alvocidib, targeting CDK7/CDK9 (under clinical development), and the FDA-approved CDK4/6 inhibitor, palbociclib. Our drug panel also included vismodegib, a Hedgehog pathway inhibitor, which was recently reported as a novel therapeutic option for T-ALL. 12 Because bortezomib, a reversible inhibitor of 26S proteasome, has been introduced into clinical trials in relapsed/refractory T-ALL (NCT02112916), we used bortezomib as a reference and also added a second-generation, nonreversible proteasome inhibitor, carfilzomib, to our screening library.
Drug sensitivity screening of five T-ALL cell lines revealed a differential response to the compounds tested under normoxia conditions ( Fig. 1 ). Despite the heterogeneity of the inhibitor responses, all the cell lines demonstrated high sensitivity to six kinase inhibitors: afatinib, axitinib, crizotinib, ponatinib, and sunitinib, which induced cytotoxic responses of EC50 ≤ 10 µM, and alvocidib (EC50 ≤ 1 µM) after 72 h of incubation ( Fig. 1A , C , Suppl. Table S3 ). Interestingly, alvocidib was the most efficacious among all the tested kinase inhibitors, with demonstrated cytotoxic activity at EC50 ≤ 1 µM across all the cell lines. In addition, the proteasome inhibitors bortezomib and carfilzomib were also effective against all T-ALL cells at a nanomolar concentration range (EC50 ≤ 1 µM). Another six inhibitors (bosutinib, cabozantinib, ceritinib, dasatinib, erlotinib, and lapatinib) were efficacious in at least three out of five cell lines tested (EC50 ≤ 10 µM) ( Fig. 1A , Suppl. Table S3 ). The remaining compounds were less potent (EC50 > 10 µM), showing selectivity against a single T-ALL cell line (e.g., vandetanib, EC50 = 3.4 µM in Loucy cells), or did not induce a significant response in the tested cells.
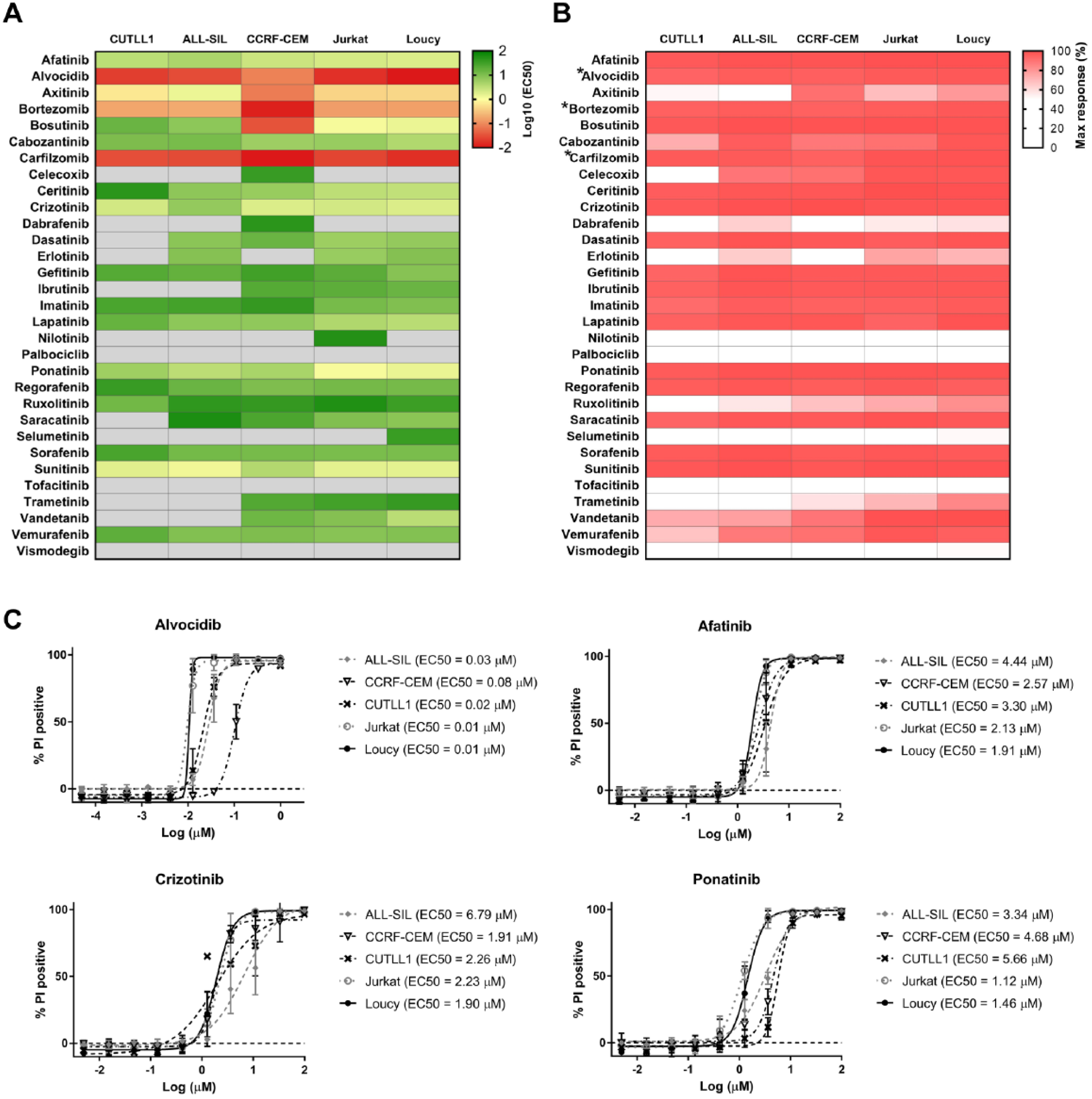
Drug response profiles of the T-ALL cell lines. (
To further delineate the cytotoxic activity of kinase inhibitors, we determined the maximum response of each drug at the highest tested dose (100 µM for each inhibitor except for alvocidib, bortezomib, and carfilzomib, for which the highest tested dose was 1 µM). As seen in Figure 1B , most of the tested agents effectively eradicated leukemic cells at the highest tested concentration, indicating that their cytotoxic activity increases substantially as the maximum tested dose was approached.
Since first described by Coustan-Smith et al., patients with early T-cell precursor phenotype (ETP-ALL) have received much attention for their increased risk for relapse. 13 In our study, Loucy cells, which show a transcriptional program related to ETP-ALL, were highly sensitive to the tested drugs, similar to the other four more differentiated T-ALL cell lines ( Fig. 1 , Suppl. Table S3 ). Our results identify attractive kinase inhibitor candidates and suggest that kinase inhibitors might be promising targeted therapies and are worthy of further exploration as potential treatment options in T-ALL.
Hypoxia Cytotoxicity Ratio in T-ALL Cell Lines
To test whether hypoxia modulates the activity of the tested compounds, we performed drug sensitivity profiling in T-ALL cell lines under hypoxia (1% O2). The results were integrated with the data obtained for the cells incubated in normoxic conditions (20% O2) as described above. The HCRs for the T-ALL cell lines (n = 5) treated with inhibitors for 72 h are presented in Figures 2 and 3 and Supplemental Figure S1 . Here we demonstrate that hypoxia can induce differential responses in T-ALL cells exposed to clinically approved kinase inhibitors. Unexpectedly, most of the tested inhibitors were generally equipotent under hypoxic and normoxic conditions (HCR 1; arbitrarily set to 0.8–1.2). However, specific patterns of drug response indicative of increased sensitivity under hypoxia (HCR >1.2) or decreased sensitivity under hypoxia (HCR <0.8) were also observed ( Figs. 2 and 3 , Suppl. Fig. S1 ). Interestingly, imatinib was preferentially more efficacious under hypoxia in all the tested cell lines (HCR 1.33–1.63). Sensitivity to ruxolitinib and sunitinib was also elevated under hypoxic conditions in at least three out of five T-ALL cell lines. Nevertheless, several of the tested compounds, including afatinib and crizotinib, were more active in normoxia (three cell lines or more), suggesting decreased sensitivity under hypoxia ( Figs. 2 and 3 , Suppl. Fig. S1 ). We have not observed any specific pattern of response among the tested drugs and cell lines since all three patterns of drug response could be observed in many cases. For example, axitinib was less potent under hypoxia in CUTLL1 (HCR 0.46) and Loucy (HCR 0.62) cells, but its antitumor activity was increased under hypoxia in CCRF-CEM (HCR 2.11) and Jurkat (HCR 1.30) cells ( Fig. 2 , Suppl. Fig. S1 ). Our EC50 results indicate that despite distinct HCR values for individual drugs, the majority of the kinase inhibitors were equally sensitive or marginally less active under hypoxia conditions in the tested T-ALL cell lines. This was consistent with the observation of reduced cell survival at the maximum tested dose under hypoxia ( Suppl. Fig. S2 ). The results of our study indicate that hypoxic T-ALL cells are highly responsive to kinase inhibitors and should be further investigated in the context of hypoxia-sensitive therapies in T-ALL. Further studies will be required to determine the effects of other tumor microenvironment factors, such as adhesion molecules or extracellular matrix components, on drug sensitivity in leukemia. Our results underscore the potential importance of hypoxia testing in drug profiling studies and provide compelling evidence for the application of HTFC to search for hypoxia-selective drug candidates.
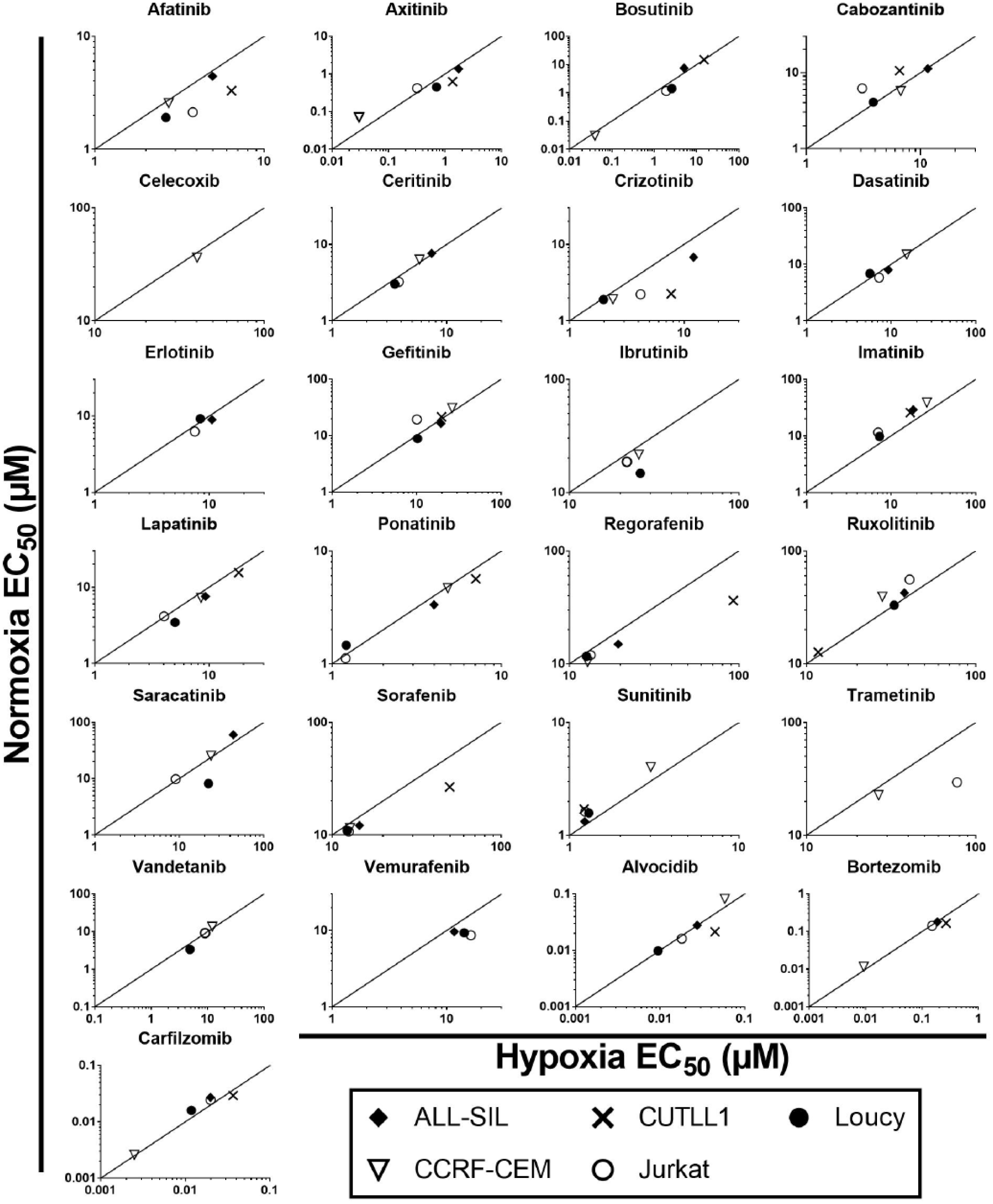
HCRs for five T-ALL cell lines treated with 31 small-molecule inhibitors. T-ALL cell lines were incubated with the tested inhibitors for 72 h under normoxia (20% O2) and hypoxia (1% O2), respectively. The HCR values were calculated as EC50normoxia/EC50hypoxia values obtained from three independent experiments. The detailed HCR values are reported in Supplemental Figure S1. The straight line represents HCR 1, indicating equitoxic responses of the cells under hypoxia and normoxia. The area above the line indicates sensitivity under hypoxia (HCR >1), while the area under the line (HCR <1) indicates resistance under hypoxia.
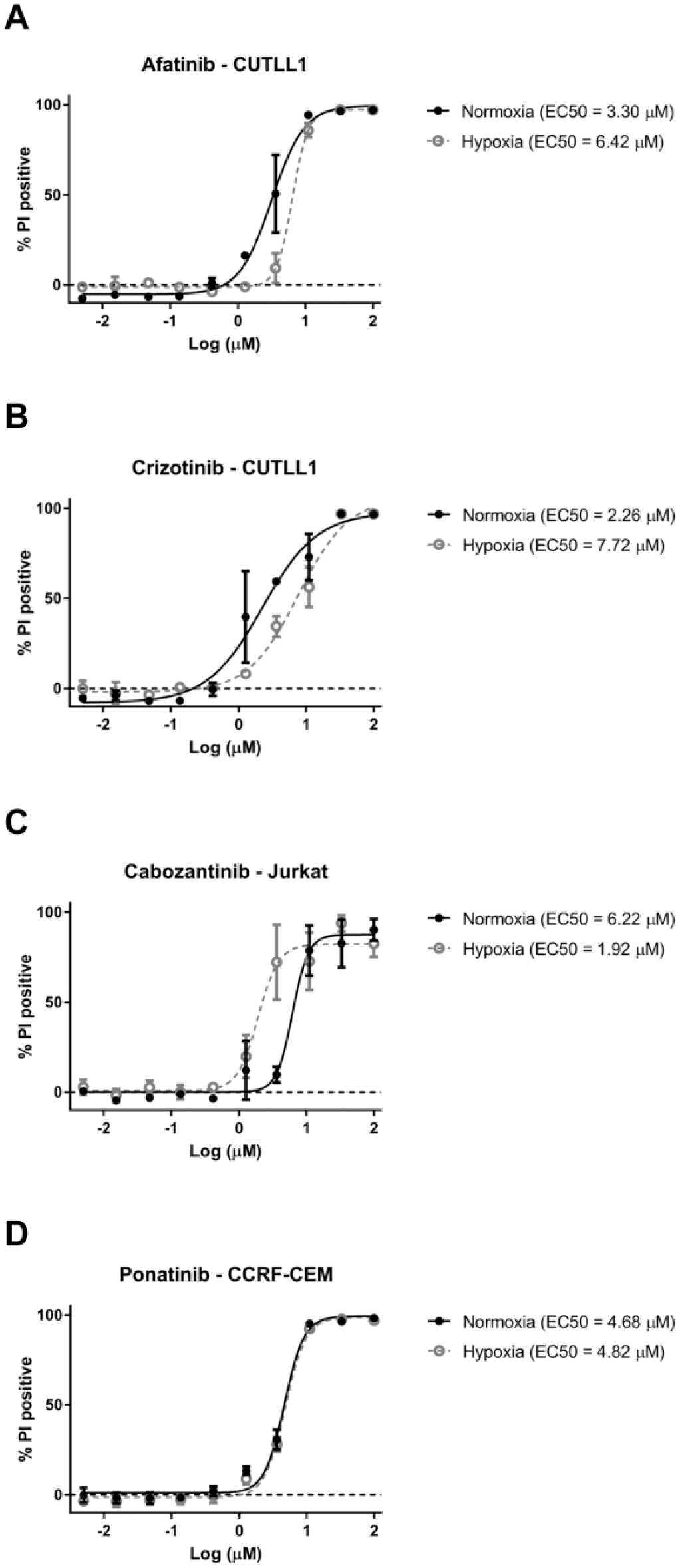
Representative patterns of T-ALL sensitivity to kinase inhibitors in an oxygen-deprived environment: (
Drug Sensitivity Screening in T-ALL Primary Samples and PDXs
With evidence that kinase inhibitors are effective against T-ALL cell lines, we next utilized our platform to evaluate drug sensitivity in T-ALL primary samples (n = 2) and PDXs (n = 5). Primary tissue is often considered a limiting factor in high-throughput studies, which require a high number of cells. In this study, we utilized PDX models of T-ALL, which were shown to better preserve both the genomic integrity and the tumor heterogeneity observed in patients. 14 Therefore, PDX models are proposed as an attractive alternative strategy for drug profiling studies when sufficient primary tissue is not available. We tested a small set of T-ALL samples for which phenotypic and genomic data were available ( Suppl. Table S1 ). Patient and PDX samples were subjected to HTFC screening for sensitivity to 31 small-molecule inhibitors under conditions described in Material and Methods. Untreated cells demonstrated excellent cell viability in serum-rich monoculture, which varied between 70% and 95% up to 72 h incubation ( Suppl. Table S4 ), which was comparable to the viability of the T-ALL cell lines.
We observed that the sensitivity of T-ALL cells to kinase inhibitors was heterogeneous but also sample dependent ( Fig. 4 ). Primary cells and PDX models were less sensitive to kinase signaling inhibition than cell lines ( Figs. 1 and 4 ). This is consistent with previous studies showing that primary samples or relevant PDXs are more resistant, and often respond differently, to investigational agents in comparison with cell lines.15,16 T-ALLs with ETP phenotype and/or harboring MLLT10 and/or KMT2A gene rearrangements are slowly proliferating leukemias, which often present stemlike features. PDXs obtained from such patients were included in our sample set ( Suppl. Table S1 ). Cell stemness and a low proliferation rate may affect cellular responses to drugs whose cytotoxic activity relies on the proliferative activity of the leukemic cells. Such differences in cell proliferative activity may also affect engraftment kinetics. 14 In our study, PASGJG, which was insensitive to the tested inhibitors, engrafted more slowly than PATZZM and PASNXS, which were more sensitive to the tested agents (data not shown). Second, we observed that the primary specimens (12-089 and 15-093) had an overall better response to kinase inhibitors than PDXs ( Fig. 4B ). This interesting observation needs to be further explored, considering the relatively small sample size in our study. Recent data indicate that PDXs maintain oncogenic translocations and preserve ~75% of the genomic lesions initially identified in patients. 14 However, PDXs undergo mouse-specific tumor evolution, which is comparable to that of cell lines, but still may affect drug response. 17
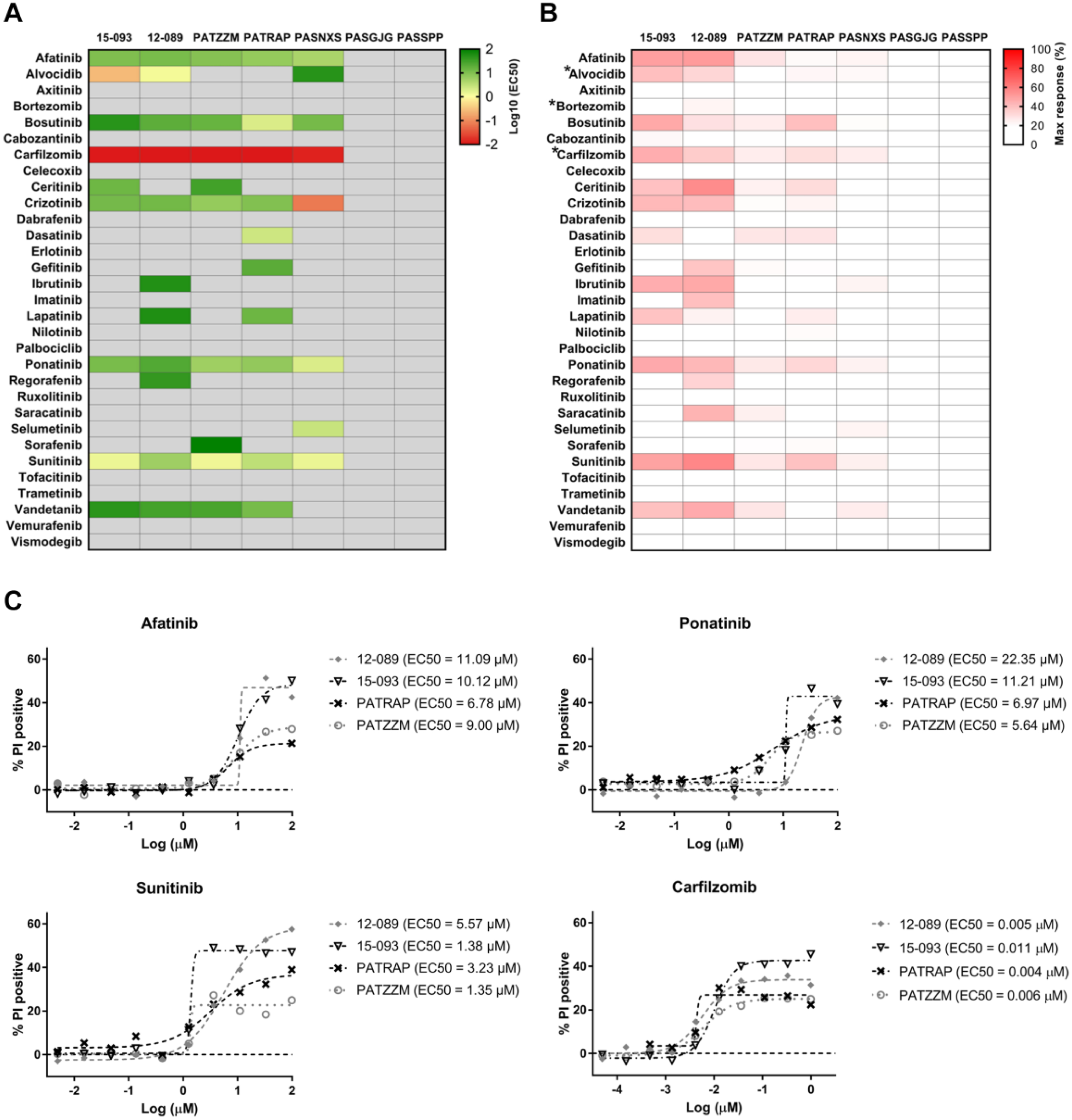
Response of T-ALL primary samples (n = 2) and PDXs (n = 5) to small-molecule inhibitors (n = 31). (
Among the PDX samples, PATRAP and PATZZM were more sensitive to the tested drugs than PASNXS ( Fig. 4 ). PASGJG and PASSPP were resistant to all the tested inhibitors, suggesting that some T-ALL samples demonstrate high resistance to kinase signaling inhibition, which can be associated with kinome reprogramming and/or adaptive bypass response, leading to drug resistance. 18 This may also be related to the genetic and molecular features of the tested cells. Some of our PDX models were developed from samples that harbor genomic lesions, for example, KMT2A gene rearrangements, which are associated with high-risk and refractory disease ( Suppl. Table S1 ).1,19
Interestingly, several samples demonstrated decreased sensitivity at 72 h compared with 48 h, which might be related to the heterogeneous and clonal architecture of the tested samples, and/or induction of spontaneous cell death, and/or induction of quiescence in some cell subpopulations of T-ALL ( Fig. 4 , Suppl. Fig. S3 ). Sensitivity to afatinib and sunitinib (EC50 ≤ 10 μM) was detected in five out of seven tested T-ALL samples (~70% samples) after 48 h incubation ( Fig. 4 , Suppl. Table S5 ). Bortezomib, which is currently being tested in the COG AALL1231 clinical trial (NCT02112916), had no activity in the tested cells (the highest concentration tested was 1 μM), compared with carfilzomib, the second-generation proteasome inhibitor that was highly efficacious (EC50 ≤ 1 μM) in the T-ALL patient samples and PDXs (five out of seven samples) ( Fig. 4A , C , Suppl. Table S4 ). In addition, ponatinib and crizotinib were efficacious against at least three out of seven samples (~42%), with the lowest EC50 values observed for PASNXS (ponatinib, EC50 = 1.92 µM; crizotinib, EC50 = 0.69 µM) ( Fig. 4A , Suppl. Table S5 ). Strikingly, selumetinib was uniquely selective against PASNXS (EC50 = 2.70 µM), while dasatinib was preferentially effective in PATRAP (EC50 = 2.43 µM) ( Fig. 4 , Suppl. Table S5A ). The analyses of the maximum responses of the tested inhibitors at the highest concentration tested further revealed the high heterogeneity of the drug responses ( Fig. 4B ). Although the patient and PDX samples were overall less sensitive to the tested drugs compared with the cell lines, similar trends of drug response were observed ( Figs. 1B and 4B ). With a few exceptions (e.g., axitinib and erlotinib), the inhibitors that were found to be the most potent against T-ALL cell lines induced a decrease in T-ALL cell survival at the highest tested concentration across the majority of the samples (e.g., afatinib, ponatinib, and sunitinib). In addition, we investigated the sensitivity of normal peripheral blood mononuclear cells (PBMCs) to the tested inhibitors. We found that the most potent inhibitors were also cytotoxic toward normal cells, indicating that they were not specific to leukemic cells ( Suppl. Fig. S5 ).
Our observation that several T-ALL samples were generally more susceptible to multiple kinase inhibitors ( Fig. 4 ) strongly supports the rationale for targeting the deregulated kinome in T-ALL. However, it is worth noting that most of the tested samples were highly selective toward one or a few specific inhibitors, suggesting that high-throughput drug screening may rapidly detect unique sensitivities of individual patient cells to the tested drugs. 5 Our data underscore the roles of personalized medicine in the development of modern therapies in T-ALL.
Recently, two independent reports implicated dasatinib as a novel targeted therapy in a subset of T-ALL.14,15 While dasatinib was effective against our tested T-ALL cell lines, only one T-ALL sample was preferentially sensitive to this inhibitor in our study ( Figs. 1 and 4 ). Interestingly, there were other promising inhibitors that demonstrated greater activity against T-ALL cells than dasatinib and are worthy of further investigation ( Figs. 1 and 4 ). One interesting observation in this study was sensitivity to cyclin-dependent kinase inhibition. Alvocidib (CDK7/CDK9 inhibitor), which is currently under development as a combination therapy for AML (NCT01349972 and NCT02520011), 20 was efficacious against T-ALL cell lines and some patient samples ( Figs. 1 and 4 ).
The results of our study also indicate that T-ALL cells show good response to several TKIs, which target multiple kinases, including PDGFR, VEGFR (ponatinib, sunitinib, and axitinib), EGFR (afatinib), and ALK (ceritinib, crizotinib) ( Figs. 1 and 4 ). Although preliminary, these findings provide a foundation for future studies on targeting the T-ALL kinome by FDA-approved multitarget inhibitors. It will be critical to test active agents in murine models of T-ALL, either genetically engineered or injected with PDXs.
Overall, our analyses show that T-ALL samples were less sensitive than the cell lines. Importantly, our approach includes culturing T-ALL primary samples and PDXs in 10% FBS and 10% human AB serum concentration. Serum may sequester drugs via their binding to serum proteins and therefore limit their cytotoxic response. Furthermore, the growth factors present in the culture medium may accelerate pro-survival signaling as opposed to serum-free culturing systems, which may affect cell metabolism and eventually sensitize the cells to the tested agents. Taking this into account, it would be critical to establish the minimum levels of serum-facilitating drug response profiling without compromising cell viability in our monoculture systems. We successfully utilized a conditioned medium from stromal cells to maintain the viability of B-ALL cells in our previous reports. 16 Frismantas et al. 14 have recently reported serum-free co-cultures of PDXs and/or primary cells on hTERT-immortalized mesenchymal stroma cells as a novel strategy to facilitate the survival of ALL cells, including T-ALL. The authors postulated that stromal co-cultures are better predictors of drug response in the context of in vivo validation studies. Still, they were able to demonstrate a significant correlation between both serum-free co-culture and serum-supplemented monoculture systems.
The results of this study demonstrate that a flow cytometry-based PI screen can be effectively used in HTFC to identify small-molecule inhibitors as a targetable therapy in T-ALL. Common plate reader-based viability assays, such as MTT and CellTiter Glo, require cell lysis and thus provide only one metric from a treated sample. In contrast, a flow cytometry approach with PI allows for rich information to be collected from a treated sample in addition to simple viability assessments. Flow cytometry enables particle count and analysis of a sample on a cell-by-cell basis, and hence drug effects within a heterogeneous population may be determined ( Suppl. Fig. S4 ). The PI assay used here provided phenotyptic analysis of the TKI-treated T-ALL cells. Our data suggest that primary patient samples can be screened within 72 h and yield results that may inform personalized therapies with the potential to compare drug activity on PBMC ( Suppl. Fig. S5 ) and patient samples ( Fig. 4 ) with circulating drug levels ( Suppl. Table S2 ). (See, for example, the potential of sunitinib to provide a therapeutic window between PBMC and patient samples with an achievable dose.) Thus, our approach provides a reproducible tool for functional screening and novel therapeutics.
Supplementary Material
Supplementary Material, DS_DISC774248 – High-Throughput Flow Cytometry Identifies Small-Molecule Inhibitors for Drug Repurposing in T-ALL
Supplementary Material, DS_DISC774248 for High-Throughput Flow Cytometry Identifies Small-Molecule Inhibitors for Drug Repurposing in T-ALL by Dominique R. Perez, Christian K. Nick, Anna Waller, Cristina Delgado-Martin, Travis Woods, Nitesh D. Sharma, Michelle L. Hermiston, Mignon L. Loh, Stephen P. Hunger, Stuart S. Winter, Alexandre Chigaev, Bruce Edwards, Larry A. Sklar and Ksenia Matlawska-Wasowska in SLAS Discovery
Footnotes
Acknowledgements
We would like to acknowledge the Shared Flow Cytometry Resource, Animal Resource Facility, and Animal Models Shared Resource at the UNM Comprehensive Cancer Center. Material was provided in part by the Children’s Oncology Group (AALL15B1-Q to K.M.W.). We apologize to all authors whose work could not be cited due to space constraints.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported in part by the National Center for Research Resources and the National Center for Advancing Translational Sciences of the NIH through grant number 8UL1TR000041, and by the National Cancer Institutes and the Cancer Center Support Grant through grant number P30CA118100. Other grant support includes Dedicated Health Research Funds from the University of New Mexico School of Medicine (K.M.W.). D.R.P. was supported by funding from NIH Minority Institutional Research Training Program Award T32 HL007736. S.P.H. has received honoraria from Jazz PHARMACEUTICALS, Spectrum Pharmaceuticals, and Erytech, and consulting fees from Novartis.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.