
Abstract
The ATM (ataxia-telangiectasia, mutated) protein kinase is a major regulator of cellular responses to DNA double-strand breaks (DSBs), DNA lesions that can be caused by ionizing irradiation (IR), oxidative damage, or exposure to certain chemical agents. In response to DSBs, the ATM kinase is activated and subsequently phosphorylates numerous downstream substrates, including p53, Chk2, BRCA1, and KAP1, which affect processes such as cell cycle progression and DNA repair. Numerous studies have demonstrated that loss of ATM function results in enhanced sensitivity to ionizing irradiation in clinically relevant dose ranges, suggesting that ATM kinase is an attractive therapeutic target for enhancing tumor cell kill with radiotherapy. Previously identified small-molecule ATM kinase inhibitors, such as CP466722 and Ku55933, were identified using in vitro kinase assays carried out with recombinant ATM kinase isolated from mammalian cells. Since it has not been feasible to express full-length recombinant ATM in bacterial or baculovirus systems, a robust in vitro screening tool has been lacking. We have developed a cell-based assay that is robust, straightforward, and sensitive. Using this high-throughput assay, we screened more than 7000 compounds and discovered additional small molecules that inhibit the ATM kinase and further validated these hits by secondary assays.
Introduction
ATM (ataxia-telangiectasia, mutated) is a serine/threonine protein kinase and a key mediator of signal transduction cascades controlling responses of mammalian cells to DNA breakage.1,2 ATM is a member of the PI3K-like kinase (PIKK) family, which also includes ATR (ATM and Rad3 related), DNA-PKcs (DNA-dependent protein kinase, catalytic subunit), mTOR (mammalian target of rapamycin), and SMG1 (suppressor with morphological effect on genitalia family member), all of which have been implicated in DNA damage responses. 1 ATR is activated primarily by single-strand DNA breaks and stalled DNA replication forks, whereas ATM and DNA-PKcs are involved in the response and repair of double-strand breaks (DSBs). 3 DNA-PKcs promotes the repair of DSBs by nonhomologous end-joining, whereas ATM is thought to facilitate the repair of DSBs by homologous recombination. 3
Previous work from our laboratory had demonstrated that ATM is sequestered in an inactive state as a homodimer in unstressed cells. 4 DNA breakage induces the kinase domain of one ATM molecule to phosphorylate serine 1981 of the interacting ATM molecule, causing dimer dissociation and release of active monomers. 4 The activated monomers are then free to phosphorylate substrates involved in DNA damage response pathways, including p53, Chk2, SMC1, NBS1, BRCA1, and KAP1. 4 The target phosphorylation sites of the ATM kinase are serines or threonines located adjacent to a glutamine (S/TQ). 5
Cells derived from patients or mice lacking ATM are extremely sensitive to ionizing radiation. 6 In addition, cells treated with small-molecule ATM kinase inhibitors, such as CP466722 or Ku55933, are hypersensitive to ionizing radiation.7,8 CP466722 and Ku55933 were both identified in medium-throughput enzyme-linked immunosorbent assay (ELISA)–based in vitro kinase assays.7,8 Since it has not been possible to express full-length ATM protein in either bacteria or baculovirus and isolated ATM kinase domains have limited to no detectable kinase activity, the relatively small amounts of ATM protein used in the kinase assays have been immunoprecipitated or purified after multiple chromatographic steps from human cells. 9 This puts a strong logistical bottleneck on use of such assays for large screens. Other limitations of in vitro kinase assays include an inability to predict the permeability, toxicity, and in vivo activity of a compound.
In an attempt to overcome such problems, we developed a mechanism-based cellular screen that combines a targeted approach with a biologically integrated end point. 10 We used the In-Cell Western (ICW), a cell-based immunoassay that can analyze protein signaling pathways by measuring protein expression or phosphorylation both sensitively and quantitatively, for this purpose.11,12 This method is amenable to high-throughput screening (HTS) for ATM kinase inhibitors while ensuring that the compounds detected trigger an appropriate signaling response in situ to DNA damage through the ATM pathway.
Materials and Methods
Chemicals
Compound CP466722 was synthesized at the St. Jude Children’s Research Hospital by the condensation of 4-chloro-6,7-dimethoxyquinazoline with 3-(pyridin-2-yl)-1H-1,2,4-triazol-5-amine in the presence of Cs2SO3. Both reactants used for this final condensation, the quinazoline13,14 and triazole, 15 were prepared according to reported procedures.
1H NMR (400 MHz, Chloroform-d) δ 9.21 (s, 1H), 8.93 (s, 1H), 8.79 (s, 1H), 8.16 (d, J = 7.9 Hz, 1H), 7.82 (t, J = 7.7 Hz, 1H), 7.38 (s, 2H), 7.06 (s, 2H), 4.15 (s, 3H), 4.10 (s, 3H). LC/MS (ESI): m/z 350.3 [M+ + H]+.
CP466722 was dissolved in DMSO at a concentration of 4 mM (its solubility limit). Ku55933 and Ku60019 were purchased from Sigma (St. Louis, MO) and were dissolved in DMSO at a concentration of 10 mM.
Cell Culture
MCF7 cells were obtained from ATCC (American Type Culture Collection, Manassas, VA), and cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum at 37°C in a humidified atmosphere (5% CO2).
Irradiation
Cells were irradiated using a 137Cs-source (Gammacell 40-exactor, MDS Nordion, Ottawa, Ontario, Canada) at a dose of 10 Gy.
Western Blotting
Cells were harvested, lysed in RIPA buffer, quantitated, and prepared for Western blotting as previously described. 8 Primary antibodies were prepared in 1:1000 dilutions in 1× Tris-buffered saline with Tween (TBST) buffer containing 5% bovine serum albumin (BSA). Antibodies used in this experiment include anti–phospho-ATM(S1981) (cat. 2152-1; Epitomics, Burlingame, CA), anti–phospho-p53(S15) (cat. 9284L; Cell Signaling, Danvers, MA), anti–phospho-KAP1(S824) (cat. A300-767A; Bethyl Laboratories, Montgomery, TX), and anti–β-actin (cat. A5316; Sigma). One anti–phospho-ATM(S1981) 4 and one anti–phospho-SMC1(S957) 16 antibody were made by our laboratory.
ICW Assay for Screening of ATM Kinase Inhibitors
In total, 25 µL of a suspension of 4 × 105 MCF7 cells/mL in DMEM was dispensed to each well in 384-well plates using a Wellmate liquid dispenser (Matrix Technologies, Maumee, OH). The plates were incubated overnight in a 5% CO2, 37°C, 100% relative humidity atmosphere. After incubation, 25 nL of each test compound was transferred into the wells of the plates from stock plates (10 mM in DMSO) using a 384-pin array (V&P Scientific, San Diego, CA). DMSO was used as a negative control and CP466722, Ku55933, and Ku60019 as positive controls. A DNA DSB-inducing agent, etoposide, was then added using the 384-pin array to give a final concentration of 25 µM in each well to activate ATM kinase. The cells were incubated at 37°C, 5% CO2, 100% relative humidity for 1 h and then fixed by dispensing 25 µL of 8% formaldehyde using a Wellmate liquid dispenser (Matrix Technologies) to a final concentration of 4% (vol/vol) and incubated for 20 min at room temperature. Then, the cells were permeabilized by washing the plates (3 × 50 µL) with a microplate washer (ELx405; BioTek, Winooski, VT) using phosphate-buffered saline (PBS) containing 0.1% Triton X-100. After permeabilization, the plates were washed three times with PBS containing 0.1% Tween-20 and then incubated overnight at 4°C with 20 µL of pKAP1 antibody (1:3000 dilution; Bethyl Laboratories) in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE). The next day, plates were washed three times with PBS containing 0.1% Tween-20. Subsequently, IRDye 800CW goat anti–rabbit secondary antibody solution (1:5000 dilution; LI-COR Biosciences) containing DNA stain DRAQ5 (1:5000 dilution) was added into each well. After a 1-h incubation at room temperature, the plates were washed three times with PBS containing 0.1% Tween-20. After the final wash, the washing buffer was discarded. The plates were then scanned on an Aerius automated Infrared Imaging system (LI-COR Biosciences) for quantitative analysis at 700 and 800 nm at 200 microns of scan resolution and 3.0 mm of focus offset. Images for DRAQ5 and pKAP1 staining were obtained by using the 700- and 800-nm channels of the Aerius Infrared imaging system, respectively. Fluorescence intensities were measured using the software provided with the imaging system.
For assay optimization, we optimized the washing parameters (height for aspiration and dispensing, washing cycles, and dispensing volume and rate) of the microplate washer to prevent cell loss. The timing after IR or etoposide treatment was also optimized to obtain maximum activation of pKAP1. To save cost on pKAP1 antibody, the dilutions of pKAP1 antibody were optimized to use minimal antibody without compromising the signal window.
After optimization, HTS was performed on a customized system developed by High Resolution Engineering (Woburn, MA) with integrated subsystems that exactly match those used for optimization. Plates were transferred from instruments to instruments by a Staübli T60 robot arm (Staübli, Duncan, SC).
Dose-Response Experiments
For dose-response experiments, screening compounds were diluted 3-fold over 10 steps using a Tecan Evo liquid handling system (Tecan, Männedorf, Switzerland), transferred from a compound plate to a 384-well assay plate using a Biomek FX (Beckman Coulter, Brea, CA) liquid handler equipped with a pin tool, and then assayed as described above.
Data Processing and Hit Selection
Primary screening data were analyzed using our in-house Robust Interpretation of Screening Experiments (RISE) application written in Pipeline Pilot (version 8.5; Accelrys, San Diego, CA) and the R program (R Development Core Team, Vienna, Austria). Percentage inhibition was calculated by first log10-transforming the fluorescence intensity observed at 800 nm and then normalizing per plate to the 25% trimmed mean value of the positive control, CP466722, and the 25% trimmed mean value of the negative control, DMSO, according to the following formula: %inhibition = 100 * (screening compound – negative control)/(positive control – negative control). The top 1% of compounds from the primary screen were defined as “hits” and were selected for further study. Dose-response curves were fit with Prism (version 6; GraphPad Software, La Jolla, CA) using the standard four-parameter Hill equation.
Chemical Library and HTS
The screening library consisted of 7152 unique compounds with a minimum of 90% purity from commercial sources. The library includes Food and Drug Administration (FDA)–approved drugs, kinase inhibitors, CDK inhibitors, and other bioactive compounds. These compounds are stored at 1 to 10 mM in DMSO in 384-well plates.
Results
ICW Assay Development for the ATM Signaling Pathway
Before setting up the ICW assay, we tested the cellular activities of some ATM kinase inhibitors obtained from earlier screens (CP466722, Ku55933, and Ku60019) in the MCF7 breast cancer cell line to determine if this cell line would be an adequate model for screening for ATM inhibitors. MCF7 cells were pretreated with these inhibitors or DMSO for 1 h before exposure to 10 Gy of IR. Then cells were incubated for 1 h after IR exposure and lysed for Western blotting analysis. As expected, IR induced ATM autophosphorylation at serine 1981 and ATM-mediated trans-phosphorylation of p53 and KAP1 ( Fig. 1A ). CP466722 and Ku55933 caused dose-dependent inhibition of phosphorylation of both ATM and its downstream targets p53 and KAP1 with potencies similar to those previously reported. Ku60019 is a more potent ATM kinase inhibitor than CP466722 and Ku55933, completely inhibiting ATM-dependent phosphorylation at 1 µM.
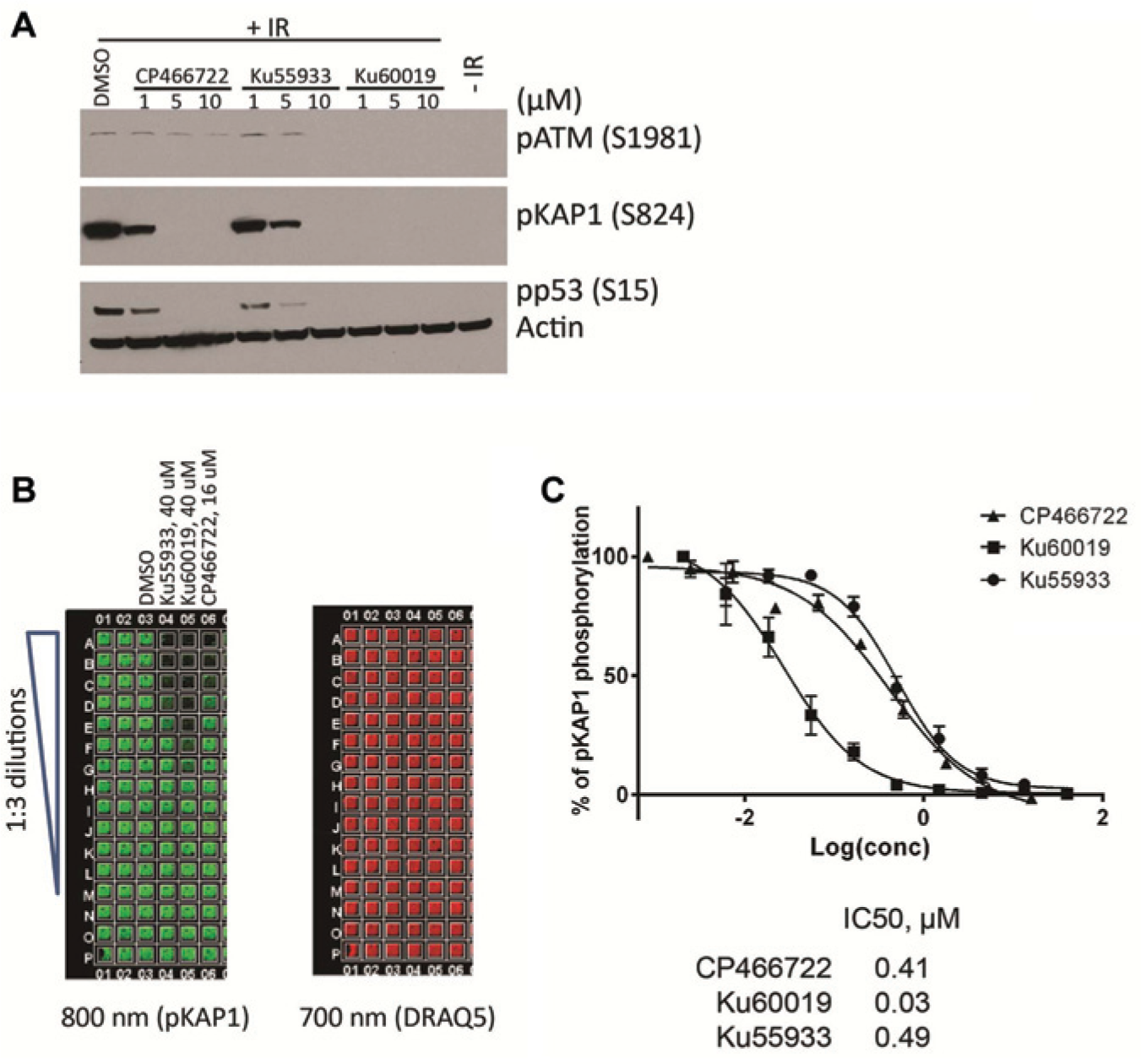
In-Cell Western (ICW) assay development for ATM (ataxia-telangiectasia, mutated) kinase inhibition. (
For the screening assay, cells were grown in 96- or 384-well plates, treated with test compounds, and then stimulated with DNA damaging agents. After treatment, cells were fixed with formaldehyde, permeabilized, and stained with phospho-specific primary antibodies that recognize target proteins. The cells were then incubated with an infrared fluorescent dye–labeled secondary antibody and a DNA binding dye, DRAQ5, for normalizing cell numbers in each well. The plates were scanned simultaneously at 700 nm (for signal of DRAQ5) and 800 nm (for signal of fluorescence dye labeled secondary antibody) with an Aerius instrument (LI-COR Biosciences), which has separate lasers and florescence detectors for each dye and is able to simultaneously detect two targets at 700 and 800 nm for two spectrally distinct dyes.
Primary antibodies used in ICW assays need to be very specific to the studied target protein. To select the ideal antibodies for the ICW assay, we screened the specificity and efficiency of several antibodies that detect ATM-dependent phosphorylation events, including pATM(S1981), pp53(S15), pKAP1(S824), and pSMC1(S957) in immunoblot analyses (
To evaluate whether the pKAP1 antibody worked in ICW assays, MCF7 cells were plated in a 384-well plate and incubated overnight at 37°C. The next day, cells were pretreated with CP466722, Ku55933, or Ku60019 at a final concentration of 10 µM or with DMSO as a control for 1 h before IR. Cells were then exposed to 10 Gy IR, incubated for 1 h after IR, and fixed and processed for the ICW assay. The phosphorylation signal (800 nm, green signal) of KAP1 showed a dose-dependent reduction in the presence of the inhibitors ( Fig. 1B , C ), indicating a dose-dependent inhibition of ATM signaling. These ATM inhibitors did not affect cell survival by themselves during the 2-h treatment since the DRAQ5 signals (700 nm, red signal) was consistent across all wells. The IC50 values for CP466722, Ku55933, and Ku60019 in this cell-based assay were 0.41, 0.49, and 0.03 µM, respectively.
Use of Etoposide as the DNA Damaging Agent in the Screening Assay
Since irradiators are normally located in separate secured locations for safety and security reasons and are typically not part of HTS facilities, cell plates would have to be transferred between rooms or buildings to allow irradiation of this type of screening assay. Even if co-localized, at the very least, plates would have to be transferred between an irradiator and the plate reader, which would be suboptimal. Irradiation would thus become a bottleneck for HTS. For ease of screening, we explored the option of replacing the irradiation step with a DSB-inducing chemotherapeutic agent, such as etoposide, a topoisomerase II inhibitor.17,18 The activation of ATM and pKAP1 in MCF7 cells treated with 25 µM etoposide peaked at 1 h after treatment, and pretreatment with 10 µM CP466722 inhibited both pATM and pKAP1 signals ( Fig. 2A ). Thus, the ATM specificity of the signaling pathway with this chemical agent is functionally equivalent to irradiation for this assay and makes etoposide a viable replacement for irradiation as the DSB-inducing agent. As confirmation of this conclusion, IC50 values for the various inhibitors in the ICW assay with etoposide were similar to those seen following IR ( Fig. 2B , C ). Therefore, IR was replaced by etoposide as the DNA damaging agent in the HTS assay.
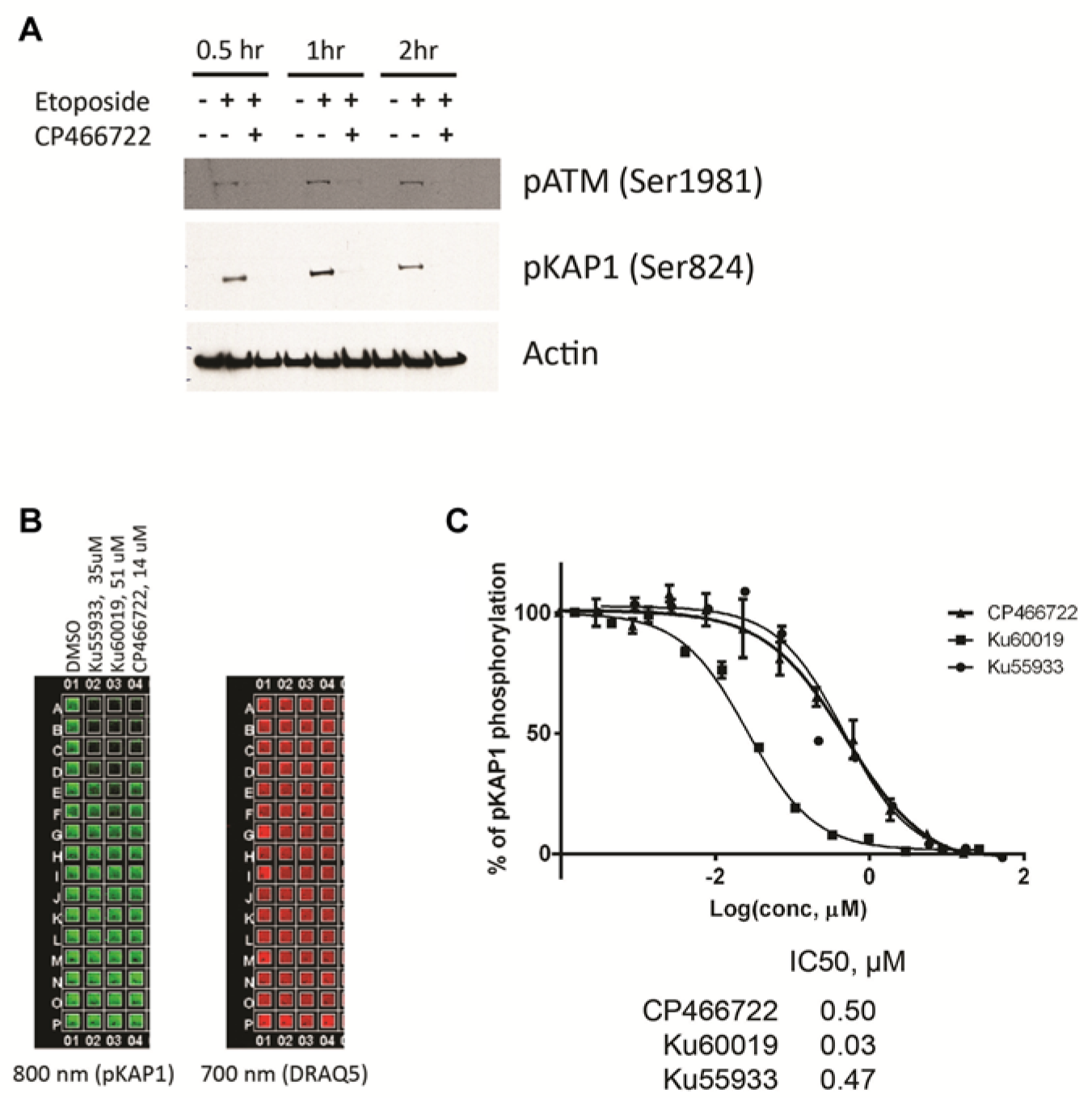
Evaluation of the activation of ATM (ataxia-telangiectasia, mutated) kinase by etoposide in MCF7 cells. (
Assay Validation and Optimization of the High-Throughput Assay
The variability of the assay in 384-well plates was assessed by running triplicate plates with alternating max/min/mid signals on 3 days. After etoposide treatment, the maximum signal (cells treated with DMSO only), minimum signal (cells treated with 10 µM CP466722), and the mid-signal (cells treated with 0.8 µM CP466722) were plotted for these plates ( Fig. 3A ). We observed that the raw signal scattered the most for the DMSO-treated negative control cells and only minimally for cells treated with the highest concentration of the positive control. This is not unusual in a situation where the negative control represents pathway activation (maximal triggered signal, amplified by pathway) and can be corrected by applying a log10-transform to the raw signal. This processing step equalizes variance across the dynamic range of the assay ( Fig. 3B ) and enables the use of parametric statistical methods for hit selection. Following data transformation, inter- and intraplate variability was consistent and reasonable: median Z′ (comparing the max and min signals) across the nine plates in the assay validation experiment was 0.61, with a minimum of 0.32 and maximum of 0.66. Assay drift was negligible: median column drift was 11% and median row drift was 7%. We did observe one instance each of row and column drift exceeding >20%, but in these cases, the values abruptly shifted for only a single row or column at the outer edge as opposed to gradually shifting across the plate ( Fig. 3C ). This variation did not correlate with cytotoxicity as measured by DRAQ5. We recommend that plate heatmaps for this assay be closely examined for row and column bias.
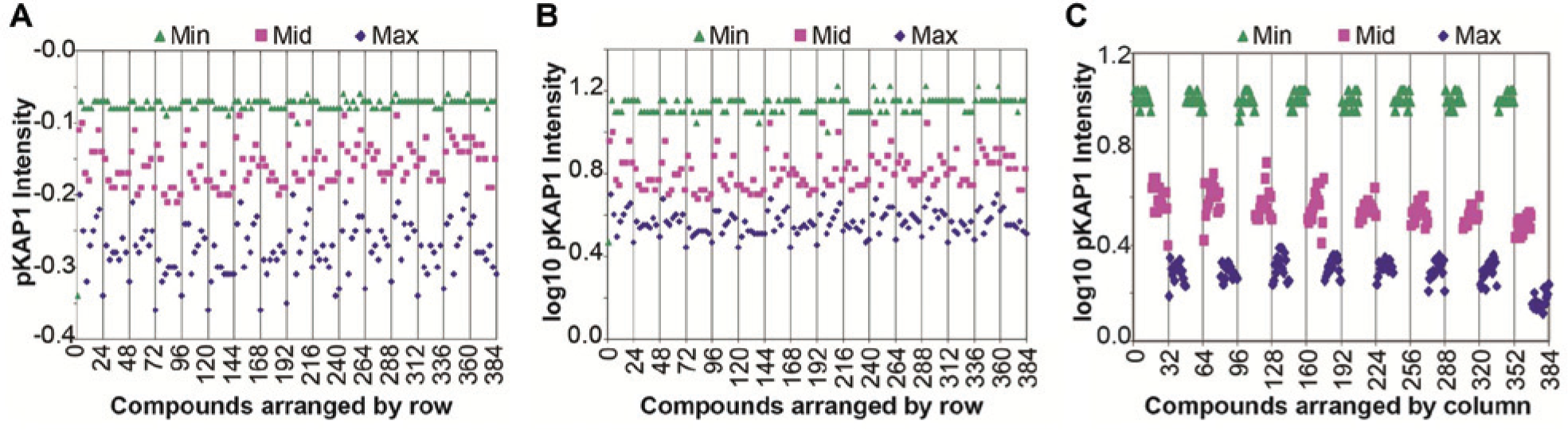
Validation of In-Cell Western (ICW) assay for ATM (ataxia-telangiectasia, mutated) kinase inhibitor in the high-throughput screening format. (
We had intended to normalize all data using the 700-nm cytotoxicity control channel, but it is clear that this data set is noisy relative to the antibody binding data and does not give reliable normalization. Instead, it can be used to filter the data, removing compounds with the 700-nm signal below the lower bound of the 95% confidence limit as potentially cytotoxic.
Primary Assay: HTS of a Compound Library
A relatively small collection of compounds (7152), including bioactive compounds, FDA-approved compounds, and kinase inhibitor collections, was used in a pilot screen with the assay. MCF7 cells were seeded in 384-well plates, cultured overnight, treated with compounds at a final concentration of 10 µM, and subsequently exposed to 25 µM etoposide. Then, 10 µM CP466722 was included as a positive control and DMSO was used as a negative control. The scatterplots for percentage inhibition of the library are shown in Figure 4A . The boxplot of log10-transformed anti-pKAP1 fluorescence intensity values shows consistent and stable separation between positive and negative controls ( Fig. 4B ). Median Z′ was 0.52, with a minimum of 0.27 and a maximum of 0.82 ( Fig. 4C ). The top 1% of compounds tested, or 72 compounds, had percentage inhibition ≥43.4% and were defined as “hits” in the primary screen.
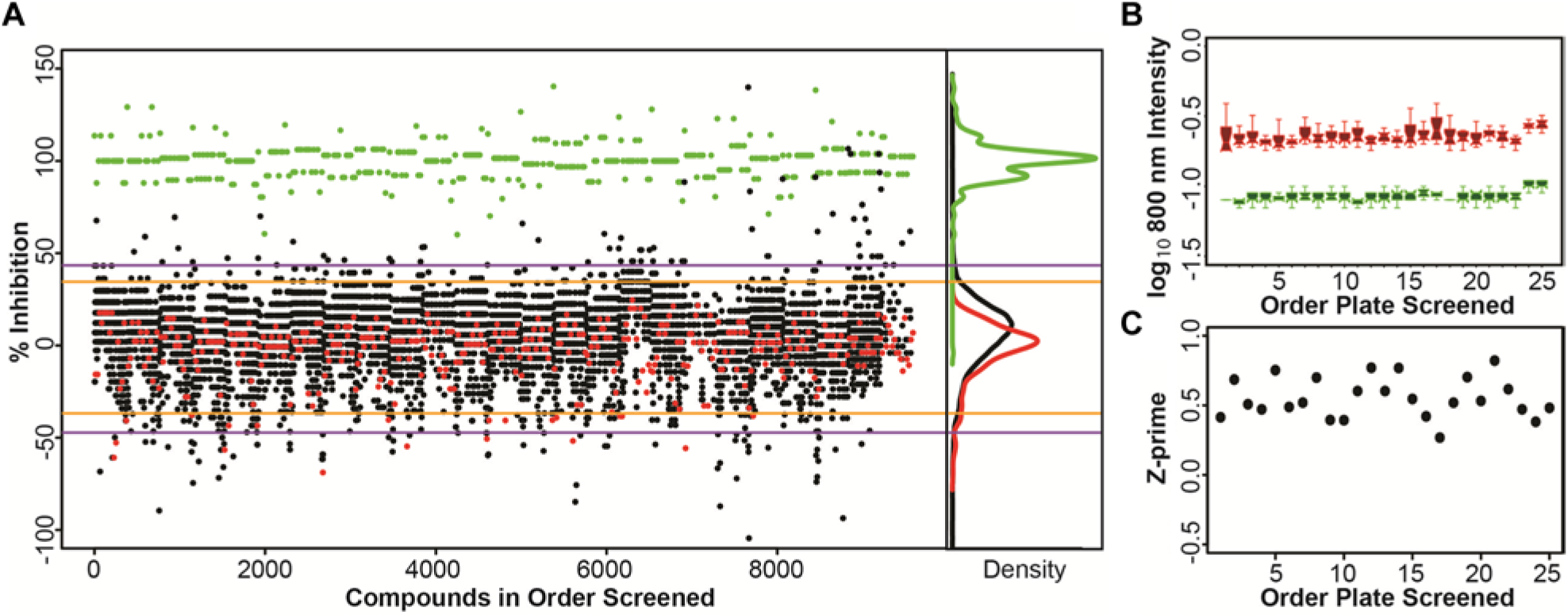
Results of the primary screen. (
Hit Validation
To validate the hits, two commercially available known kinase inhibitors ( Fig. 5A ) were repurchased as pure powders, reformulated, and tested in a dose-response assay to confirm the results of the primary screening. Both hits showed a dose-dependent inhibition of the pKAP1 signal with an IC50 value of 0.48 µM and 4.72 µM, respectively ( Fig. 5B ).
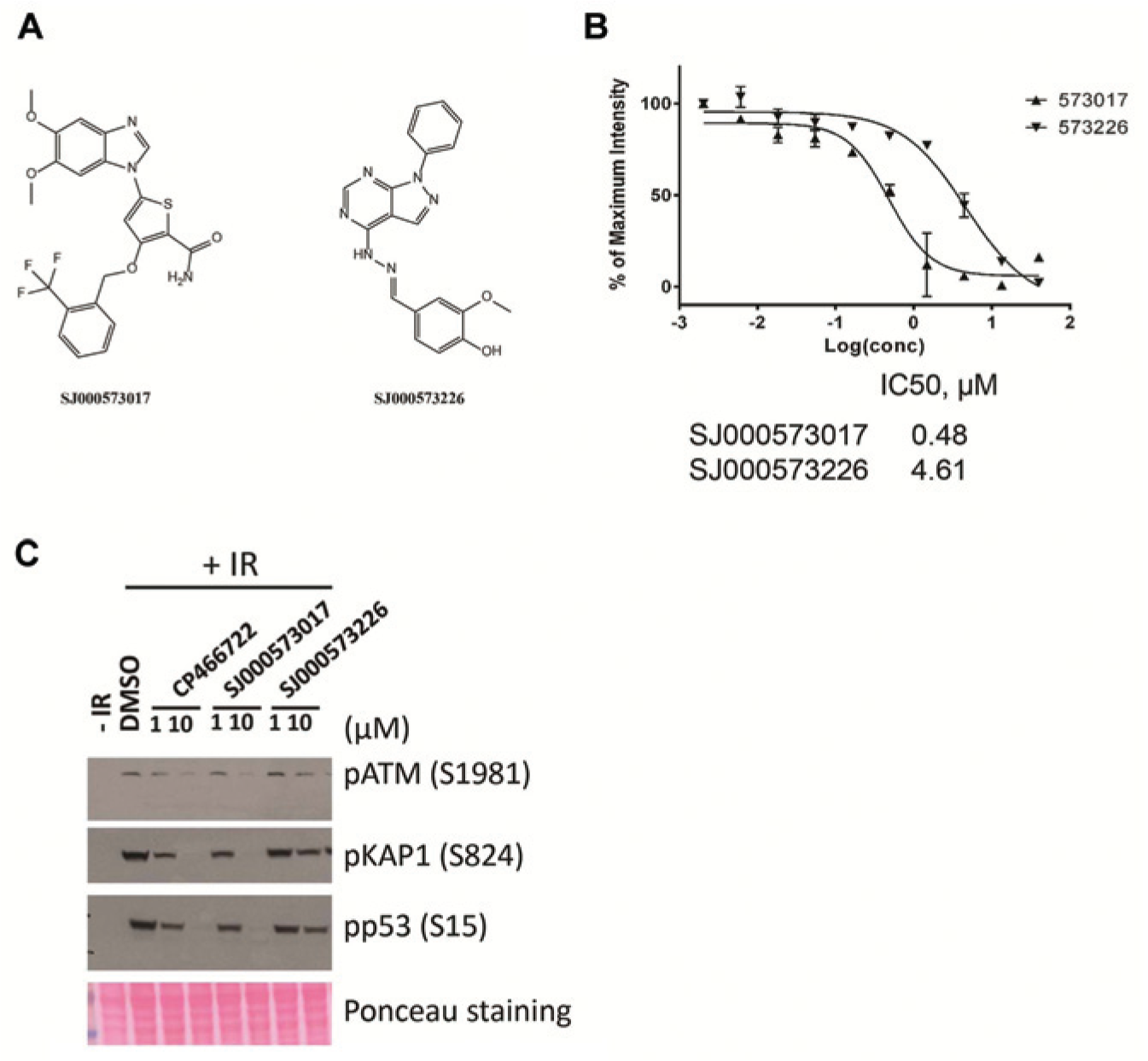
Further identification of two hits from screening. (
To further characterize the two initial positive hits, we tested them in confirmatory immunoblot assays. SJ000573017 (10 µM) inhibited IR-induced formation of pATM, pKAP1, and pp53, consistent with inhibition of the ATM signaling pathway. However, SJ000573226, which was ~10× less active than CP466722 based on IC50 values in the ICW assay, only showed a partial inhibition on pATM and its downstream targets pKAP1 and pp53. Thus, the potencies observed in the immunoblot assays correlated well with those from the ICW results.
Discussion
The ATM kinase is a central mediator of cellular responses to DNA breakage. Inhibition of ATM kinase activity significantly enhances tumor cell kill after irradiation and certain types of chemotherapy. To provide better illumination of cellular responses directly and circumvent limitations of in vitro kinase assays using immunoprecipitated or purified cellular ATM protein, we developed a cell-based ICW assay as an alternative way to screen small-molecule libraries for inhibitors of the ATM kinase in a high-throughput manner. Rather than using the logistically cumbersome ionizing radiation to induce ATM activity, we switched to use of etoposide as the DSB-inducing agent. Both IR and etoposide result in DNA DSBs and activate ATM and the entire ATM DNA damage signaling pathway. The critical issue is whether the etoposide-induced phosphorylation of KAP1 is dependent on ATM, and we demonstrated that it is. We also showed that ATM kinase inhibitors (CP466722, Ku55933, and Ku60019) have the same IC50 values after IR and etoposide. There are many advantages to using etoposide over IR for the screening, including the following: (1) it can cut down the HTS running time since the transferring of etoposide from a compound plate to an assay plate is much faster than irradiating an assay plate, and (2) with etoposide, you do not need an irradiator with attendant security control. Therefore, using etoposide is equivalent functionally to and operationally advantageous over IR. This opens the assay up for a much wider range of facilities. The most critical factor in optimizing this type of assay is to have an antibody that is highly specific for ATM activation in its binding to a modified target protein. After testing antibodies that recognize the phosphorylated ATM(S1981) or ATM-dependent phosphorylation of p53, KAP1, SMC1, and others, we found that anti–phospho-KAP1 and anti–phopho-p53 antibodies were the only ones that bound to no other proteins in immunoblots of whole-cell lysates. Since the anti–phospho-KAP1 antibody displayed a higher signal window than the anti–phopho-p53 antibody, this antibody was adapted to ICW assays and used for subsequent screening.
The ICW assay can identify ATM inhibitors of multiple possible mechanisms by monitoring a direct downstream substrate of ATM kinase. Theoretically, this approach is able to discover ATM inhibitors with diverse mechanisms of action, including active site inhibitors, allosteric inhibitors, and even substrate binding site inhibitors. Compared with the monitoring of the phospho-ATM signal, monitoring ATM substrate KAP1 phosphorylation is able to detect inhibitors that target the active ATM monomers or prevent activation of the ATM homodimer. On the other hand, the direct monitoring of the phospho-ATM signal itself would miss inhibitors that only target the active ATM monomer. Interestingly, testing of many ATM inhibitors has demonstrated that most of the inhibitory compounds (including Ku55933 and CP466722) are much more potent at inhibiting transphosphorylation by the active ATM monomer than autophosphorylation of ATM dimers (data not shown). This could be due either to a conformational difference of the ATM kinase domain in the inactive dimer versus the active monomer or it being much harder for small molecules to insert themselves into the tight ATM dimer and inhibit autophosphorylation than it is for the molecules to inhibit the unencumbered kinase domain of the ATM monomers that are involved in transphosphorylation of substrates. Therefore, in addition to its improved ease and scale for ATM inhibitor screening, this strategy has certain advantages over a recently published high-content screening (HCS) assay for ATM kinase inhibitors that targeted ATM autophosphorylation itself. 19 Such HCS methods definitely provide in-depth information on the effects of compounds on cellular targets and cellular processes. However, our ICW assay has three advantages. First, high-content imagers generally generate far larger file sizes than ICW assays (for a two-channel HCS assay, 12 MB/well, giving 4.6 GB/384-well plate), whereas the same data set for a 384-well plate for ICW is about 1 MB. In the long run, the ICW files are likely much smaller, faster to analyze, and easier to maintain in archives. Second, the ICW generally provides faster data acquisition. The HCS takes about 4× the time to scan a 384-well plate in comparison to an ICW assay (15–20 min for HCS vs. 3–5 min for ICW). This can be a significant issue with longer screening campaigns. Third, the scanner used in ICW is relatively cheap compared with an HCS scanner. It is worth noting that non–LI-COR scanners having two-channel detection should also be able to run this assay.
This mechanism-based cellular HTS assay offers sensitive and quantitative detection of ATM kinase inhibition. To validate the assay for automated screening, we have screened more than 7000 compounds and identified two validated hits as novel ATM kinase inhibitors. We identified and further characterized hits as novel ATM kinase inhibitors: SJ000573017, a potent inhibitor of Polo-like kinase 1 (PLK1) and PLK3 kinases, and SJ000573226, a GSK-3 kinase inhibitor.20,21 It is interesting to note that a Kinome scan of ATM kinase inhibitor CP466722 at 10 µM against 442 human kinases (unpublished data) showed that CP466722 does not inhibit GSK-3 kinase but inhibits PLK1 kinase by 86%, PLK2 by 80%, PLK3 kinase by 38%, and PLK4 kinase by 98%. These results suggest that the kinase domains of ATM and PLK may share some similarities. Thus, we appear to have identified a dual inhibitor of ATM kinase and PLK1 kinase in this screen. It is conceivable that a dual ATM and PLK1 kinase inhibitor could have certain antitumor therapeutic advantages by simultaneously inhibiting tumor cell growth through PLK1 and radiosensitizing with ATM inhibition during radiotherapy. Previously published studies of the two validated hits indicate neither is a pan-active kinase inhibitor (
Footnotes
Acknowledgements
We thank Dr. Taosheng Chen and Jimmy Cui for assistance with the St. Jude high-throughput screening center. We thank Dr. Jonathan Low and Dr. Jaeki Min for comments and suggestions on the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the American Lebanese Syrian Associated charities (ALSAC), St. Jude Children’s Research Hospital (SJCRH), and the Abraham J. & Phyllis Katz Foundation.