
Abstract
The LINE-1/L1 retrotransposon is a transposable element still active in the human genome. Most retrotransposons in the genome are inactive or repressed by several host mechanisms. In specific contexts, active L1 retrotransposons may evade repression and copy themselves into new genomic loci. Despite a general knowledge of the L1 life cycle, little was known about the dynamics of L1 proteins and function during the different stages of the host cell cycle. Our work highlighted a well-orchestrated localization of L1 proteins and mRNA that take advantage of mitotic nuclear membrane breakdown. Once in the nucleus, L1 ribonucleoproteins (RNPs) are able to retrotranspose during the S phase when L1 retrotransposition peaks. Our conclusions highlight previously unappreciated features of the L1 life cycle, such as the differences between cytoplasmic and nuclear RNPs and the cycling of L1 ORF1 protein and L1 activity during progression through the cell cycle. These new observations are discussed here in light of the evolutionary arms race between L1 retrotransposons and the host cell.
The human genome is full of sequences derived from transposable elements (TEs), which are DNA units that have (or had, once upon a time) the ability to move to new sites within a host genome. 1 Retrotransposons are a class of TEs, able to spread through a “copy-and-paste” process, that take advantage of an mRNA intermediate. In the modern human genome, the only TE able to move autonomously is the LINE-1/L1 retrotransposon. The life cycle of L1 starts with transcription by RNA polymerase II. In the cytoplasm, the bicistronic L1 mRNA is translated into two L1 proteins, ORF1p with RNA chaperone activity and ORF2p, which has endonuclease (EN) and reverse-transcriptase (RT) activities. Many ORF1p proteins and perhaps just one or two copies of ORF2p bind the L1 mRNA that encoded them, to form L1 ribonucleoprotein (RNP) particles. The L1 RNPs enter the nucleus and, via the ORF2p EN and RT activity, L1 mRNA is copied into cDNA and “retrotransposed” into a new genomic locus. Before our work,2,3 this series of events that generally describes the life cycle of L1 was largely “de-contextualized” from the cellular environment and cell cycle dynamics of the host cell ( Fig. 1 ).
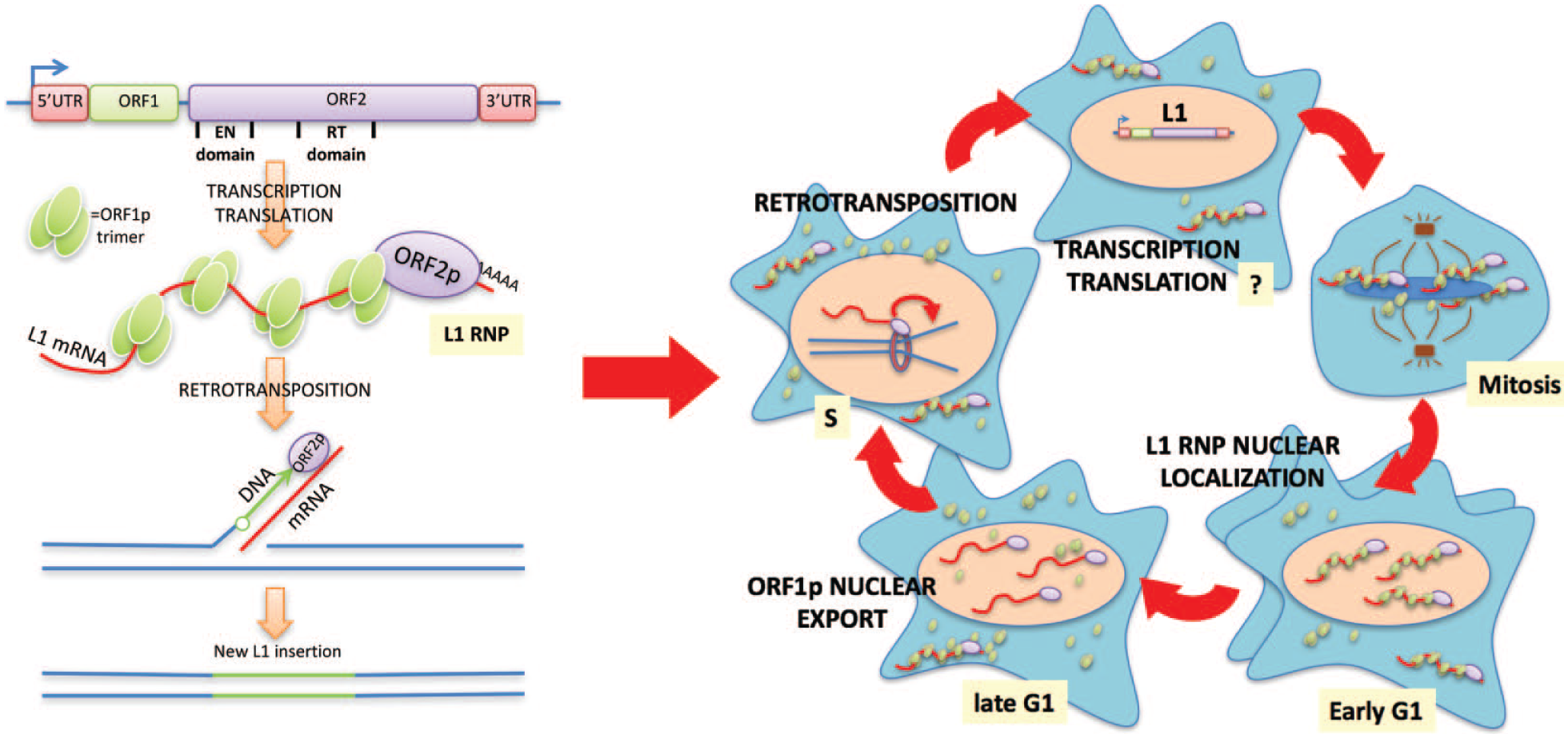
L1 life cycle in the context of the cell cycle. Schematic representation of the main steps in L1 life cycle in a “cell-free” scenario (left) and in the context of the cell cycle (right).
In our work, 2 we followed the cellular localization of L1 components (ORF1p, ORF2p, and L1 mRNA) during the cell cycle. ORF1p protein is mainly localized in the cytoplasm.4,5 However, the observation that some cells, usually in closely spaced pairs, displayed clear nuclear ORF1p staining inspired us to interrogate the cell cycle to explain the heterogeneous nuclear localization within an asynchronous cell population. Markers of the cell cycle clearly revealed that cells, with nuclear ORF1p and nuclear L1 mRNA, were in the early G1 phase and therefore had recently exited mitosis, strongly suggesting that the L1 RNPs formed in the cytoplasm were able to enter the nucleus thanks to mitotic nuclear membrane breakdown. The molecular mechanisms driving this localization are unclear. L1 RNPs may be randomly trapped in the nucleus after the reformation of the nuclear membrane, or some specific biochemical process might facilitate this process. The positively charged ORF1p may weakly interact with chromatin, therefore increasing the chances for L1 RNPs to accumulate at chromatinized DNA during mitosis and then remain in the nucleus after cell division. Unexpectedly, we showed that ORF1p cellular localization cycled during progression through the cell cycle. ORF1p exits the nucleus during the late G1 phase through a CRM1-mediated process that maintains the low level of nuclear ORF1p even after multiple cell divisions. These observations imply that the nuclear L1 RNPs mainly consist of ORF2p and L1mRNA and lack ORF1p during cell cycle phases following G1 (S and G2/M phase). These conclusions were also demonstrated by an independent study 3 that, through mass spectrometry analysis of different L1 RNPs, demonstrated the existence of a class of nuclear L1 RNPs depleted of ORF1p that was highly enriched for a set of co-immunoprecipitating nuclear proteins. In retrospect, this result is not surprising, considering that in vitro L1 retrotransposition reactions and Alu elements, which hijack L1 ORF2p for their retrotransposition, do not require ORF1p protein. These results are consistent with a “chaperone” activity for ORF1 in delivering the RNA-ORF2 complex to chromatin.
We also tracked L1 retrotransposition during cell cycle progression. Through the development and implementation of a novel L1 retrotransposition reporter based on the expression of a fluorescent timer, we show that L1 retrotransposition peaks during the S phase. These observations are in line with cell cycle block and release biochemical experiments showing that ORF2p binds chromatin preferentially during the S phase, and that ORF2p and L1 mRNA, mostly depleted of ORF1p, bind the nuclear DNA clamp proliferating cell nuclear antigen (PCNA) 6 and components of the minichromosome maintenance (MCM) complex, and localize with a subset of DNA replication forks during the S phase. These exciting and unexpected observations suggest that L1 retrotransposition may be functionally connected to the DNA replication fork.
An Evolutionary Prospective
Non-long-terminal-repeat (LTR) retrotransposons date back to the Precambrian era and the origin of eukaryotes 7 and have even much older ancestors in prokaryotes, namely, group II introns, which also mobilize via a target-primed reverse transcription (TPRT) reaction. The L1 clade of non-LTR retrotransposons has been evolving with primate genomes for the past 80 million years, and as a consequence, more than 30% of the modern human genome is composed of these TEs. As “selfish DNA,” L1 retrotransposons are engaged in a continuous arms race with the host cell that tries to silence and “eliminate” their dangerous activity. 8 Considering this long-lasting fight and coevolution, it is not surprising that the L1 life cycle is now intimately intertwined with fundamental mechanisms of mammalian cells. 9 Leveraging essential cellular mechanisms, such as the cell cycle, for its spread, L1 makes it very difficult for the host cell to evolve new defense mechanisms.
The evolution of the nuclear membrane must have created a strong evolutionary pressure for retrotransposons to evolve mechanisms to adapt to the new cytoplasmic environment and to overcome the nuclear membrane barrier. ORF1p may have evolved its chaperone activity to protect and increase the stability of the L1 mRNA in the cytoplasm. Our finding that ORF1p is seemingly not directly involved in L1 TPRT in the nucleus is in line with this view. Non-LTR retrotransposon progenitors (group II introns) and several non-LTR retrotransposons perform retrotransposition without an ORF1-like protein. 7 It is possible that ORF1p trimers, coating L1 mRNA approximately every 50 nt, may actually exert a negative effect during reverse transcription by ORF2p; for efficient reverse transcription, there might be a need to strip all the ORF1p from the L1 mRNA before or during cDNA polymerization.
We also showed that L1 retrotransposition peaks in the S phase. This phase, at least in concept, represents a highly favorable time for the RT (or any other DNA polymerase) because of the higher availability of nucleotides necessary for the polymerization of L1 cDNA before reinsertion into the genome. This idea is supported by the finding that enzymes involved in the regulation of dNTP concentration, such as ribonucleotide reductase (RNR) and SAMHD1 (sterile alpha motif and HD-domain-containing protein 1), also affect L1 retrotransposition in mammalian cells.2,10
Interestingly, the apurinic/apyrimidinic-type endonuclease (APE) of L1 ORF2p, characterized by low targeting compared with the restriction enzyme-like (REL) EN of other non-LTR retroelements, has been suggested to more strongly rely on DNA repair/replication machineries of the host cell. 7 This hypothesis is supported by our finding that L1 prefers the S phase to retrotranspose and that ORF2p interacts with components of the replication fork (namely, MCM proteins and PCNA).
If L1 evolved to prefer the S phase and use DNA replication for its retrotransposition, do DNA damage response mechanisms such as the homologous recombination (HR) or the Fanconi anemia (FA) mechanism affect L1 retrotransposition? Very recently published data and unpublished data from our laboratory suggest that this is the case. 11 It is compelling to suggest that some DNA damage response pathways implemented during DNA replication may have evolved to suppress L1 retrotransposition.
Open Questions and Future Investigations
The conclusions highlighted by our work open the way to the study of new questions, some of which are discussed below:
We showed a well-orchestrated regulation of ORF1p nuclear localization and the cell cycle. The molecular mechanisms that regulate these processes are still unknown and are waiting to be uncovered. What are the differences between nuclear and cytoplasmic L1 RNPs? Is ORF1p the target of nuclear posttranslational modifications that trigger ORF1p to leave the L1 RNP complexes? Does nuclear ORF1p exert an inhibitory role toward L1 retrotransposition?
We showed that ORF2p interacts with components of the replication fork.2,6 However, the molecular nature of this interaction is unclear. L1 proteins may be recruited on the replication fork during the S phase and use the MCM proteins to scan the DNA and find A/T-rich sites amenable for L1 retrotransposition ( Fig. 2 , right). If this is the case, L1 may retrotranspose before the replication fork, propagating its genetic material into two daughter cells with a single retrotransposition event. Alternatively, retrotransposition may happen downstream of the replication fork, possibly favoring one of the two strands, leading or lagging. Alternatively, the interaction between L1 and components of the replication fork could be explained by collision of the replication fork with the DNA nick created by the retrotransposing L1 ( Fig. 2 , left). In the latter case, L1 may trigger replication fork stalling and recruitment of DNA damage repair proteins.
Similarly to ORF1p localization and L1 retrotransposition, other steps of the L1 life cycle could be regulated by the cell cycle. The transcription of L1 mRNA or the still puzzling mechanism of ORF2p translation could also oscillate within the cell cycle ( Fig. 1 , question mark).
Finally, the connection of L1 retrotransposition and cell division highlights some discrepancies with previous literature that demonstrated L1 retrotransposition in terminally differentiated and not cycling cells.12–14 Despite the fact that L1 “hopping” has been described mainly in organs or compartments with high proliferation potential, such as the germ line, transformed cells, or human neuronal and other progenitor cells, it is possible that L1, if highly expressed, may still gain access to the nucleus and be able to retrotranspose in noncycling cells using alternative mechanisms to the one that we described. 2 As for several exogenous retroviruses, a transient rupture of the nuclear membrane could be one of the mechanisms that would allow the entrance of L1 into the nucleus and possible retrotransposition in nondividing cells. Intracellular vesicles, recently connected with L1 activity, 15 may also represent a new route to the nucleus used by L1.
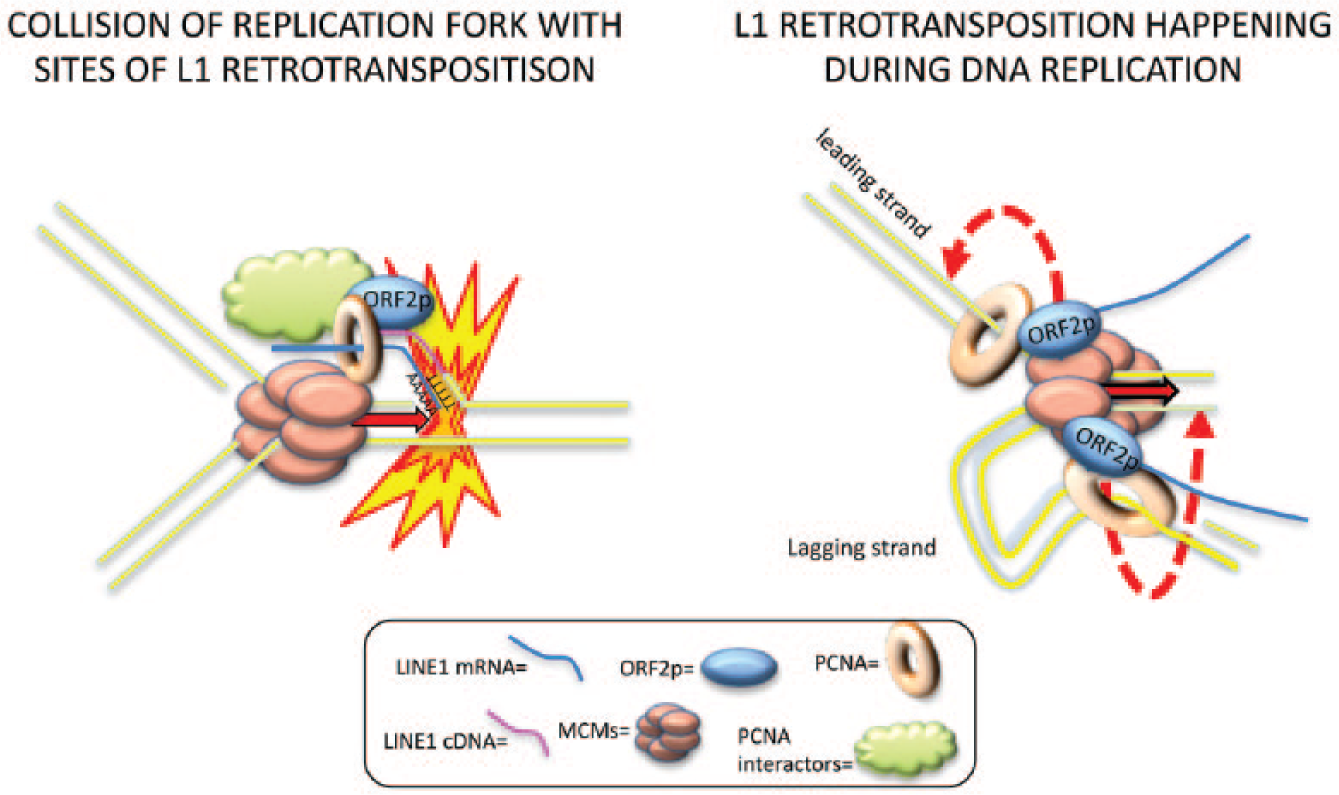
Models of L1 interaction with DNA replication fork.
Altogether, our new data demonstrating the synchronous cycling of the LINE-1 life cycle with the host cell cycle may shed new light on previously not appreciated evolutionary selected dynamics of LINE-1 and the host cell.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by NIH grant P50GM107632 to JDB.