
Abstract
Lung diseases impose a significant socioeconomic burden and are a leading cause of morbidity and mortality worldwide. Moreover, respiratory medicine, unlike several other therapeutic areas, faces a disappointingly low number of new approved therapies. This is partly due to lack of reliable in vitro or in vivo models that can reproduce organ-level complexity and pathophysiological responses of human lung. Here, we examine new opportunities in application of recently emerged organ-on-chip technology to model human lung alveolus and small airway in preclinical drug development and biomarker discovery. We also discuss challenges that need to be addressed in coming years to further enhance the physiological and clinical relevance of these microsystems, enable their increased accessibility, and support their leap into personalized medicine.
Keywords
The discovery of new therapeutics is a lengthy and costly process. Moreover, the pharmaceutical industry faces numerous challenges in their research and development (R&D) that contribute to a high project attrition rate. 1 The drug development process can be broken down into four distinct phases: (1) discovery and development, (2) preclinical research, (3) clinical research, and (4) regulatory review and monitoring2,3 ( Fig. 1 ). In its entirety, this complex and resource-intensive process is entrenched with a high degree of uncertainty regarding whether a drug will eventually reach regulatory approval. From inception to production, creating a new therapeutic agent can take up to 15 years and cost well more than US$1 billion.4,5 Therefore, streamlining the process, while relying on robust and significant model systems, will contribute to increasing the pace and efficacy of identifying new drugs, and minimizing the associated costs required to bring novel therapies to patients.
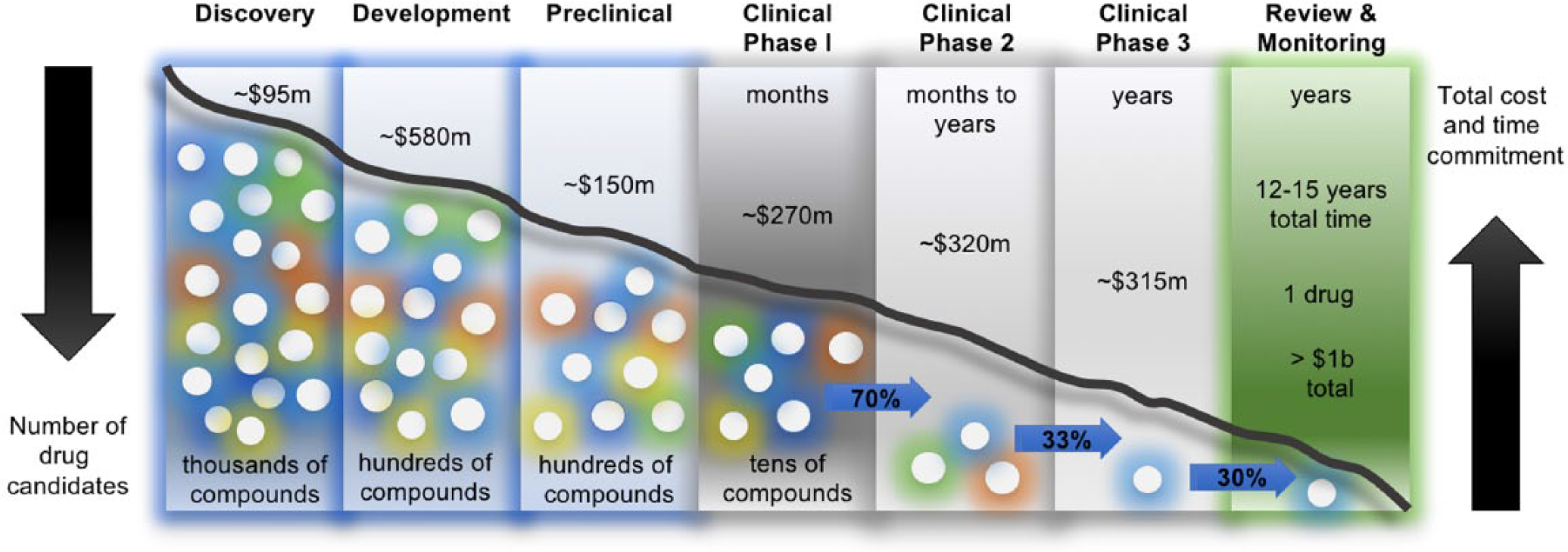
Drug discovery process. The drug discovery process is composed of multiple distinct and sequential phases. Potential drug candidates, therapeutic targets, and biomarkers are initially identified and validated during the discovery phase, often through the use of high-throughput (cellular and/or acellular) screening platforms. The target-specific (e.g., inhibitory) compounds will then be subjected to efficacy and safety testing using cell culture systems and animal models during the preclinical stages of development. Next, lead compounds with desired efficacy and safety profiles will move to the clinical trial, which consists of three separate phases. Once a drug successfully completes a phase 3 clinical trial, its manufacturer applies for its regulatory approval to enter the market, and the drug will be continuously monitored for postmarketing effects. The whole process can take up to 15 years from start to finish, and monitoring after the drug launch can last an additional 15 years. Furthermore, the combined cost associated with developing a single drug is estimated to be more than US$1 billion. Amounts shown represent average capitalized costs per new compound per development phase. Input data were obtained from Refs. 2,3,5.
Drug candidates have an approximately 10% likelihood of approval among all indications, and safety issues and insufficient efficacy have been reported as the principal causes of drug candidate attrition during clinical development.6–8 A major contributor to this extraordinarily high level of failure is the challenge in translating findings from conventional two-dimensional (2D) cell culture systems and animal models to humans. Widely used animals such as rodents often fail to reliably and accurately predict human responses, in part due to considerable interspecies differences.9,10 In addition, there is a legitimate and growing ethical concern regarding the use of animals in biomedical research, particularly in areas in which they almost completely fail to recapitulate human-relevant pathophysiology. 11 Furthermore, 2D in vitro cell cultures are unable to support differentiated, complex, and tissue-specific functions of many cell types, and as such they cannot accurately predict critical drug activities.12,13 Thus, the use of poorly predictive preclinical models often results in costly drug failure during clinical trials.
An area with significant unmet medical needs, for which only a few new classes of safe and effective therapeutics have emerged during the past four decades, is respiratory medicine. 14 Lung diseases impose a significant socioeconomic burden and are a leading cause of morbidity and mortality. 15 Lower respiratory tract infections, chronic obstructive pulmonary disease (COPD), and lung cancers collectively formed three of the top five killers in humans globally in 2015 and accounted for more than 8 million deaths. 16 In the United States alone, more people die from COPD than from any other condition except cancer and cardiovascular illnesses. 17 In fact, statistics released by the COPD Foundation illustrate that COPD affects around 30 million Americans, 18 and the total annual cost of the disease in the United States in 2010 was projected at $49.9 billion. 19 Because COPD is a progressive lung disorder that is linked to cumulative exposure to inhaled noxious gases and particles, especially tobacco smoke, it is anticipated that its prevalence and hence its burden on healthcare systems will grow in coming years, particularly in developing countries. In addition, lung tumors are the most common type of diagnosed cancer and the leading cause of cancer-related death worldwide. 15 Furthermore, during the past 20 years, new respiratory pathogens have emerged, including severe acute respiratory syndrome coronavirus (SARS-CoV) and influenza virus strains H5N1, H7N9, and the 2009 pandemic swine H1N1, with epidemic or pandemic potential that could or did threaten global health security. 20
Despite its enormous burden, respiratory medicine (unlike other therapeutic areas, such as hematology, cardiovascular, dermatology, and HIV/AIDS) faces a disappointingly low number of new approved therapies. 14 This is partly due to lack of reliable in vitro or in vivo models that can reproduce the organ-level complexity and pathophysiological responses of human lung. Availability of such model systems can remarkably advance the drug development process from target identification and validation to late-stage preclinical toxicity profiling. Several animal models for human pulmonary conditions have been developed;21–23 however, a key limitation is that lung tissue microstructures, airway branching, and cellular distribution (e.g., percentage frequency of mucosa-lining epithelial subtypes [ciliated, mucin-producing, basal, and club cells]), as well as immune system functions, differ significantly between humans and widely used rodents.23–26 For example, neutrophils—innate immune cells with critical activities during the acute phase of infection and inflammation—comprise 50–70% of circulating blood leukocytes in humans, whereas they represent only 10–25% in mice.25,26 In addition, the number of nonciliated small airway epithelial cells in the bronchioles of rats and mice is approximately tenfold greater than in humans. 23 Moreover, rodents are obligate nose breathers; in fact, their respiratory system has evolved to create highly intricate and developed nasal turbinates that, when compared with the human system, can lead to a different particulate deposition pattern during smoke or environmental particle exposure studies. 27 Rodent models of COPD are limited by their variable and inconsistent response to cigarette smoke (including among littermates), the mild phenotype, and the lack of robust airway disease to reflect the processes involved in chronic bronchitis. Thus, it is crucial to acknowledge and take into account limitations of animal models for respiratory research. Notably, several state-of-the-art three-dimensional (3D) in vitro models exist, which relative to 2D cultures better represent the spatial and chemical complexity of the living tissues, for translational studies in respiratory medicine; however, their use is hampered by a number of drawbacks. For instance, they cannot recapitulate dynamic interaction between the lung structural cells (e.g., epithelium and endothelium) and the innate or adaptive immune defense (e.g., recruitment and activation of circulating immune cells) at homeostasis or in response to pathogenic and nonpathogenic airborne challenges. In addition, for tobacco smoke or electronic cigarette vapor exposure studies, these platforms cannot recreate exposure of the airway epithelium to inhaled particles and volatile components under physiologically relevant dynamic breathing airflow. Importantly, due to lack of blood-like fluidic flow, even if endothelial cells are cultured in these model systems, they do not experience physiological vascular shear.
In this review, we will highlight the latest advances of in vitro modeling of human lung physiology and pathobiology. We will briefly outline current methodologies used to recreate the lung microarchitecture with an emphasis on “organ-on-chip” technology. We will describe how the development of the microengineered lung alveolus/small airway-on-a-chip has enabled a new strategy to accurately model several lung pathologies, including COPD, asthma, and drug-induced edema.26,28,29 In addition, we provide proof-of-concept evidence on how these microsystems can be used for disease-specific biomarker identification and drug efficacy testing. The aim of this review is to highlight unique and clinically relevant capabilities of these emerging technologies for accelerating the drug discovery process.
Modeling Lung Pathophysiology in Three Dimensions
3D Cell Culture Technologies for Lung Biology
Although there is an urgent and growing need for better models of the human lung, the complexity and intricate nature of this organ presents many challenges for the development of reliable and meaningful models in vitro. Current 2D cell-culturing techniques are not sufficient to mimic a significant part of tissue- and organ-level microarchitecture and dynamic intercellular interactions in vitro. In response to the need for improved in vitro models, several 3D cell-culturing techniques have been developed that better simulate the intricacies found in the whole lung organ in vivo. A number of these techniques have been recently reviewed by Fang and Eglen. 30 One of the initial 3D systems was spheroids. Spheroids are aggregates of cells that are grown on top of each other and create a spherical geometry. These were first used for modeling malignancy development in vitro and are often created using patient-derived tumor cells or immortalized cancerous cell lines. In the lungs, spheroids have been used to model stem cell niches of lung progenitor cells and to study non-small-cell lung cancer.31,32 Spheroids have a marked advantage over conventional 2D systems due to their physiologically relevant cell–cell and cell–extracellular matrix (ECM) interactions. Despite the obvious benefits of spheroid cultures, several obstacles can be found in these models, including challenges in growing spheroids of a uniform size and controlling cell ratios in co-cultures. 30
The limitations found in spheroid models necessitated the development of other 3D in vitro systems, such as organoids. Organoids are culturing systems in which organ-specific progenitor cells rapidly expand and generate a 3D structure composed of cell types that are specifically found within that organ. 30 These systems provide a unique solution to modeling some of the complexities found in the lung, because they can be produced using an array of cell types such as airway epithelial basal progenitor cells, airway secretory cells, and type 2 alveolar epithelial cells. 33 Recently, Lee et al. developed organoids using 3D co-culture methods by culturing endothelial cells with lung bronchioalveolar stem cells. 34 This system was then applied to dissect contributions of the lung environment niche to cell lineage commitment. In another study, Dye et al., via manipulating developmental signaling pathways, generated human lung organoids from embryonic and induced human pluripotent stem cells, which consisted of upper airway–like epithelial (immature ciliated cells and basal cells) and mesenchymal (smooth muscle cells and myofibroblasts) compartments along with an alveolus-mimicking compartment. 35 The authors showed that their organoid resembled human fetal lung. Similarly, Nikolić et al. used the tip epithelium of branching lung to generate lung organoids for the comparison of human and mouse developmental features, work that culminated in the generation of a genetically tractable organoid system. 36 Unlike spheroids, organoids are able to mimic several basic functions of the lung in vitro, such as functional signaling pathways and generation of cells capable of ciliary beating.33,37 As such, organoids are highly useful for modeling organ development, disease modeling, and cell–cell interaction. 38 Unfortunately, organoids are still restricted in their ability to reproduce key dynamic processes (e.g., blood-like flow, immune cell–endothelium interaction under vascular shear, inhalation–exhalation-associated airflow, and mechanical expansion–retraction that occurs during breathing) of the whole lung, and they often fail to generate mature and fully functioning cells and tissues.28,30 Lastly, organoids generally lack specific cell types found within their respective organs in vivo. 30
Although spheroids and organoids can be useful in modeling lung physiology and pathobiology, their 3D architecture is relatively limited; therefore, generating cell–cell or tissue–tissue co-culture in a defined manner can be challenging. To address these, researchers have explored the use of hydrogel-based scaffolding systems, which can provide well-defined matrices for culturing cells and creating co-cultures.39,40 Hydrogels are produced from various water-soluble polymers (synthetic, natural, or combined41,42) and typically include proteins from the ECM.30,43 Hydrogel technology has been harnessed to interrogate lung wound healing and fibroblast activation, and to model pulmonary diseases such as emphysema.40,44 Moreover, it has been applied as a therapeutic delivery system in a model of emphysema. 40 Hydrogels have the added benefit of being degradable;30,40 however, they have relatively weak mechanical strength properties, which to some extent limits their applications. 45
The latest technological advances have culminated in the advent of 3D bioprinting, which enables layering of living cells and biomaterials on top of one another, in very defined orientations, and in a uniform manner.30,46,47 This technology allows investigators to create reproducible cell culture models in well-controlled 3D microarchitectures.46,48 Bioprinting has enormous potential for mass production of culture systems in a high-throughput format with a wide range of applications, such as drug screening, disease modeling, and particularly tissue repair and regeneration. 49 The current trajectory of the field is toward fabricating functional tissues with high levels of complexity, such as vascularized tissue containing a complex but precisely defined arrangement of cells.47,50 In the context of lung, bioprinting has been used to model the air–blood barrier of lung alveoli in vitro. 46 Despite these advances, the broad application of bioprinting has been significantly hindered due to the relative dearth of available materials, and the pressure and biomechanical forces associated with cell extrusion.51–53
Most recently, two new technologies have emerged that can enable better modeling of human lung pathophysiology and overcome several limitations of spheroid, organoid, and bioprinting systems: (1) precision-cut 3D lung slides (PCLS)54,55 and (2) organs-on-chips.26,29,56 PCLS are thinly sliced lung tissues (usually ~250 µm) that retain viable cell, tissue, and organ structure throughout a short-term period ex vivo.55,57 PCLS have been used to analyze the lung cell response to various stimuli, including cytotoxicity, immune-modulating molecules, and cells in disease states such as asthma.57,58 One benefit of PCLS is that the organ’s structural integrity and function are maintained, but the fleeting viability of the cells poses an obstacle in their application. Similar to organoids, PCLS are limited in their ability to reproduce key dynamic organ-level processes, and it is very challenging, if not impossible, to recreate physiological air–liquid interface of the human lung.
Organs-on-Chips
Recent advances in microsystems engineering have made it possible to create biomimetic microfluidic cell culture devices, known as organs-on-chips. These devices are fabricated using computer microchip manufacturing techniques, are the size of a USB memory drive, and contain continuously perfused microchannels that are lined by living cells. These systems can recapitulate the multicellular architecture, physiochemical microenvironment, and tissue–tissue interface of the human lung in vitro. Moreover, their micrometer-sized features and their unique ability to have microflow enable them to recreate in vivo–like vascular perfusion and leukocyte circulation.12,26,59 Importantly, organs-on-chips offer new opportunities for modeling physiology, pathobiology, therapeutic target and biomarker discovery, and drug efficacy assessment in vitro. 60
The first major milestone in the development and application of organs-on-chips for in vitro modeling of human organ-level functions and disease was reported in 2010, when a lung alveolus-on-a-chip was created. 29 Since then, the technology has been rapidly adapted to engineer microfluidic mimetics of several other tissues or organs, including liver;61–67 kidney;68–70 intestine;56,71,72 bone;73–75 blood vessels;29,76–79 fat;62,63 smooth, striated, and cardiac muscle; 80 skin; 81 the blood–brain barrier;76,82–85 bone marrow;73–75,86 and neuronal tissue and cornea.87–91 The initial organ-on-chip systems predominately relied on cell lines to populate the chips (e.g., cancerous distal lung cell lines A549 and NCI H441 in a lung alveolus chip, 29 an intestinal epithelial cell line of neoplastic origin Caco-2 in a gut-on-a-chip, 71 etc.). Most recently, however, there has been a growing interest to culture induced pluripotent stem cells (iPSCs) and primary patient-derived cells in microfluidic chips.26,28,92 Using cell lines is a cost-effective strategy and is generally associated with more robust cell culture than primary cells or iPSCs. Biological pathways are often heavily altered in cell lines, however, resulting in cell types that poorly represent normal physiological functions and/or microarchitecture seen in vivo. Importantly, when studying healthy normal conditions, it is critical to avoid using cell lines of neoplastic origin. In contrast, primary human cells obtained from healthy donors or diseased individuals are more relevant because their physiology and pathobiology more closely mimic that found in our bodies and better distinguish responses in normal versus diseased states; however, the availability of these cells is often a bottleneck for in vitro experimentation. iPSCs offer an alternative to primary patient-derived cells because they can be adapted to fit the needs of organ-on-chip technologies. 92 iPSCs have a remarkable capacity to self-renew indefinitely and differentiate into a range of different cell types under appropriate conditions. 92 They also can provide a potentially unlimited source of cells, eliminate the need for invasive procedures to obtain human lung tissue (e.g., biopsy, bronchoscopy brushing, etc.), and create cells when their abundance is very low (e.g., extremely low levels of mesenchymal progenitor cells in COPD lungs).
Regardless of the cell source, two key features of organs-on-chips are their exceptional and valuable ability to (1) reproduce nongenetically imprinted molecular defects that lead to tissue and organ pathologies in vivo, and (2) reverse-engineer biological components that are critical in recreating organ-level function(s). These enable investigators to mimic phenotypes in the absence of a disease inducer (e.g., the inherent hyperreactivity of COPD airway epithelia on-chip to secrete pro-inflammatory cytokines in response to pathogen-like stimulation 26 ). In addition, by leveraging already known processes, pathways, and mechanisms that constitute the fundamental principles of normal functioning of human cells, tissues, and organs, they allow scientists to reverse-engineer organ functions in vitro. 28
Microphysiological Models of Human Lung-on-a-Chip
The lung alveolus-on-a-chip model that was developed by Huh et al. was used to specifically recreate the human alveolar–capillary interface. 29 This chip was composed of two closely apposed microchannels separated by a thin porous polydimethylsiloxane (PDMS) membrane—a biocompatible, gas-permeable, and transparent polymer, coated with ECM proteins fibronectin and collagen. Human alveolar epithelial cells and human pulmonary microvascular endothelial cells were cultured on the opposite sides of the membrane. On applying vacuum via two adjacent microchannels on the sides of the main alveolar–capillary channel, the device could successfully mimic breathing motions of the living human lung alveoli in vitro. The authors applied this platform to study nanoparticle-induced cytotoxicity, and revealed that cyclic mechanical strain accentuates toxic and inflammatory responses of the lung to silica nanoparticles. This mechanically active microdevice expanded the capabilities of cell culture models for modeling alveoli.
In an effort to apply the lung alveolus-on-a-chip system for disease modeling, Huh et al. later used this platform to mimic drug-induced pulmonary edema. 77 Following reconstitution of the alveolar–capillary interface and exposure of the microdevice to physiological mechanical strains, the authors observed that interleukin-2 (IL2), a cytokine that is therapeutically administered for the treatment of malignant melanoma and metastatic renal cell carcinoma, leads to vascular leakage, fibrin deposition, and excessive fluid accumulation in the alveolar air space. 77 Moreover, they found that the mechanical forces associated with physiological breathing motions played a crucial role in IL2-induced edema.
In spite of alveoli involvement in a number of respiratory conditions, such as pulmonary edema and emphysema, a significant portion of debilitating lung disorders, like COPD, 93 asthma, 94 and cystic fibrosis, 95 primarily affect the small airways. Small airways are defined as noncartilaginous conducting airways with an internal diameter of ≤2 mm, and they are normally located at generations 8–15 of the airway branching. 93 Recently, we leveraged the same tissue-microengineering principles used to develop the alveolus chip, and we created a human lung small airway-on-a-chip to study lung inflammatory diseases ( Fig. 2 ).26,96 This microdevice was made of PDMS and contained two parallel microfluidic channels separated by a porous polyester membrane that was lined on one side by human primary lung bronchiolar epithelial cells that formed a pseudostratified, polarized, mucociliary epithelium at the air–liquid interface (ALI), and on the opposite side by human pulmonary microvascular endothelial cells that experienced continuous blood-like fluid flow and formed a cobblestone morphology. The device was designed in a way that the top (airway lumen mimic) and the bottom (postcapillary venule mimic) microchannels reproduced similar dimensions to what is observed in vivo. For instance, the height of the airway lumen channel matches the radius of a human small airway (1000 µm). Biologically validating the model, the authors demonstrated highly similar distribution of well-differentiated epithelial subtypes (ciliated, mucin-producing, basal, and club cells) and physiologically important functionalities, such as ciliary beat frequency (CBF) and mucociliary transport (MCT), on-chip compared with those observed in human airways. 26
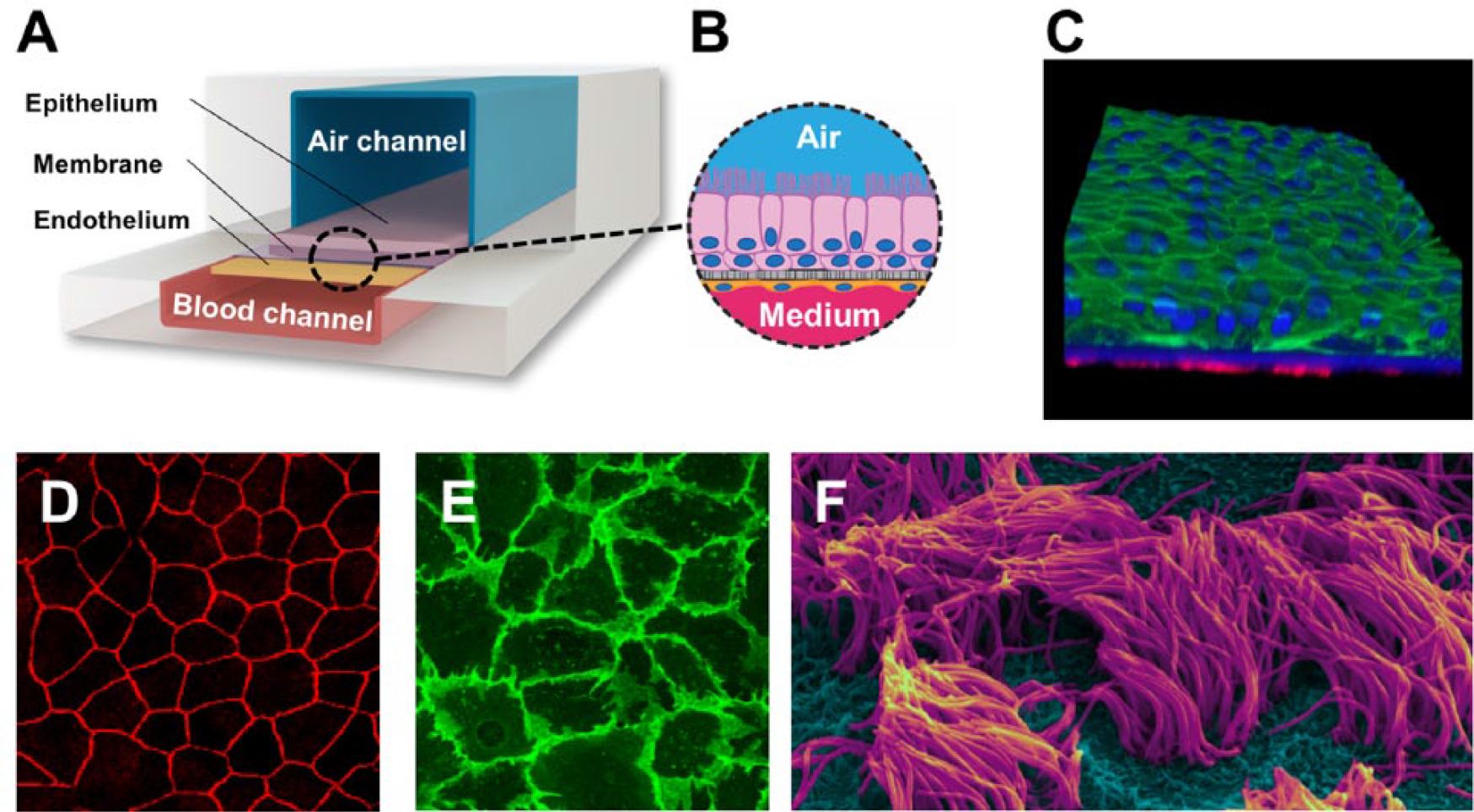
Human lung small airway-on-a-chip. (
This biochip tackled two major limitations of the state-of-the-art, human-derived in vitro models (Transwell insert culture systems; Corning) by (1) offering a unique ability to flow media and immune cells through the vascular microchannel, and (2) enabling inhalation exposure via airflow through the airway channel to air, gases, and particles. This allowed the investigators to recreate dynamic intercellular, tissue–tissue, and tissue–environment interactions at an organ level.
Cigarette smoking is a major risk factor for the development and exacerbation of COPD. 97 Benam et al. in a follow-up study developed a multicompartment, individually designed, and developed yet microfluidically integrated system to “breathe” freshly produced whole cigarette smoke in and out of the small airway-on-a-chip under conditions that mimic natural inhalation–exhalation cycles ( Fig. 3 ).28,98,99 To functionally validate the model, the authors demonstrated that acute exposure to cigarette smoke leads to significant increase in oxidative stress, as determined by upregulation of the antioxidant heme oxygenase 1 (HMOX1) gene expression and enhanced phosphorylation of the transcription factor nuclear factor (erythroid-derived 2)-like-2 (Nrf2), which is a hallmark of cigarette-induced lung damage. 100 Moreover, the authors performed whole-genome transcriptomic analysis and compared their in vitro results against those obtained by similar analysis of clinically isolated (by bronchoscopy brushing) small airway epithelial cells from a published dataset. They identified functional enrichment of the oxidation reduction pathway in both studies, with striking similarities in expression changes of genes involved in this pathway (e.g., induced expression of aldo-keto reductase family 1 member-B10 [AKR1B10] and cytochrome P450 family 1 subfamily B polypeptide-1 [CYP1B1] after smoke exposure on-chip and in human smokers vs. healthy nonsmokers). 28
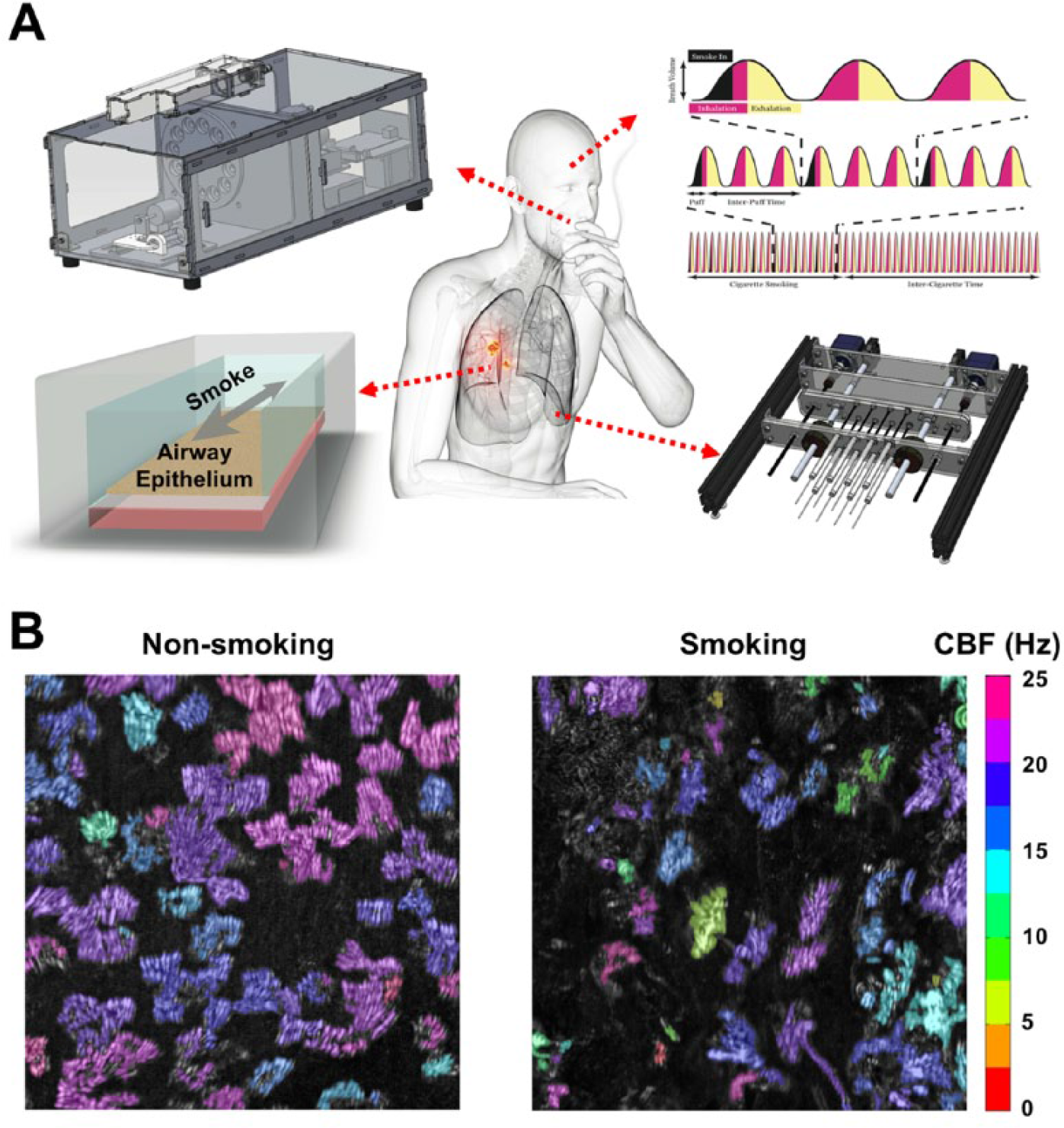
Breathing-smoking human lung-on-a-chip technology. (
Most recently, Barkal et al. engineered a microscale fluidic organotypic model of the human bronchiole to examine inflammatory responses to respiratory fungal pathogens. 101 The authors developed a complex multicellular platform that contained primary human bronchial epithelium, tightly packed lung microvascular endothelium, fibroblasts, ECM, and whole-blood extracted neutrophils. The microsystem enabled real-time analysis of leukocyte extravasation and migration through the endothelial layer and collagen matrix toward fungal hyphae during infection, and it allowed study of airway response to microbial volatiles. 101 A key advantage of this platform was inclusion of ECM and stromal cells; however, the airway epithelial cells on-chip were not differentiated into pseudostratified mucociliated epithelium, the co-culture was not maintained beyond 7 days in culture, and there was no demonstration of applying the model for therapeutic target and biomarker discovery or drug efficacy testing.
Organ-on-Chip-Enabled Biomarker Discovery and Target Validation
Biomarker Identification: Validation to Discovery
The ability to reproducibly detect biomarkers in a disease- and condition-specific manner in preclinical model systems has great potential to enhance the speed and efficacy of the drug development process. Simulating COPD acute exacerbation in vitro using the lung small airway-on-a-chip technology and human-derived cells, it was found that a bacterial-like stimulus (lipopolysaccharide [LPS], which activates Toll-like receptor-4 [TLR4]) and viral mimic (poly I:C, which activates TLR3) induce secretion of, respectively, interleukin-8 (IL8) and macrophage colony-stimulating factor (M-CSF) in the COPD, but not healthy, small airway chips. 26 The LPS-triggered IL8 secretion was consistent with past findings that showed that bronchial epithelial spheroids from COPD-diagnosed individuals exhibit higher IL8 release in response to LPS challenge compared with healthy controls. 102 The enhanced levels of M-CSF in response to poly I:C (not LPS) challenge in COPD (not healthy) airways on-chip had not been reported previously, and as such M-CSF may act as a novel biomarker of COPD viral exacerbation. Stimulation with poly I:C, however, led to considerable increase in secretion of the cytokine interferon-γ (IFNγ)-inducible protein-10 (IP10) in both healthy and COPD small airway chips. This observation was consistent with prior findings that serum IP10 is an excellent clinical biomarker for acute viral exacerbations in COPD,103,104 discriminating the pathogenic cause but not the underlying diseased status. Altogether, these results indicate the selective upregulation of cytokines IL8 and IP10 and clinically validate the model, and the induction of M-CSF demonstrates the ability of small airway-on-a-chip technology for biomarker discovery.
Studies using the breathing-smoking human lung-on-a-chip technology identified a unique set of disease-specific molecular signatures by comparing the responses of on-chip differentiated mucociliated bronchiolar epithelia using lung cells obtained from healthy versus COPD individuals. 28 Several genes were differentially upregulated in response to whole cigarette smoke in COPD airway epithelia in this platform, such as metallothionein-1H (MT1H), transmembrane protease serine-11E (TMPRSS11E), matrix metallopeptidase-1 (MMP1), small proline-rich protein-3 (SPRR3), and repetin (RPTN). Moreover, smoke exposure led to upregulation of IL8 secretion from COPD, not healthy, airway epithelia on-chip. Disease-specific responses in expression of MMP1 and IL8 are in line with published clinical reports on the involvement of these proteins in COPD pathogenesis 105 and thus functionally validate the model. The selective induction of MT1H, TMPRSS11E, SPRR3, and RPTN in COPD chips, however, may reveal new disease biomarkers that have potential therapeutic or diagnostic applications and need to be tested and validated.
Drug Efficacy Testing
Organs-on-chips offer unique potential in biomarker identification; however, their application goes beyond the very initial stages of the drug development process to cover critical preclinical drug efficacy testing.
Lung-related organ-on-chip microsystems have been used in several instances to test the efficacy of candidate experimental therapeutics for various respiratory disorders. Huh et al. hypothesized that pharmaco-inhibition of transient receptor potential cation channel subfamily V member-4 (TRPV4), an ion channel that can be mechanically activated (and its stimulation has been associated with increased alveolar-capillary permeability), 106 might prevent IL2-induced vascular leakage in the lung alveolus-on-a-chip. 77 The authors observed that intravascular administration of a TRPV4 channel blocker, GSK2193874 (developed by GlaxoSmithKline 107 ), completely inhibited the IL2-mediated increase in alveolar-capillary permeability in chips that experienced cyclic strain. 77
Similarly, the lung small airway-on-a-chip was applied to evaluate therapeutic responses of experimental compounds for asthma and COPD. 26 Using this microsystem, Benam et al. tested the efficacy of a potent pan-inhibitor of Janus kinase-1 (JAK1), -2, and -3, tofacitnib (Selleckchem), in abrogating the effects of IL13-induced asthma in vitro, because IL13 has been shown to mediate its biological effects through the JAK–signal transducers and activators of transcription (STAT) pathway. 26 The authors observed that apical administration of tofacitnib to the airway epithelium on-chip, unlike the control drug corticosteroid dexamethasone (Sigma-Aldrich), significantly suppressed goblet cell hyperplasia, decreased secretion of the cytokine granulocyte colony-stimulating factor (G-CSF), and restored CBF to normal levels, 26 three key phenotypes associated with the disease. This finding was consistent with clinical data that asthmatic patients receiving dexamethasone often fail to respond to the treatment. 108
Microbial infections are the leading cause of acute exacerbations in COPD and a major contributor to the disease-associated morbidity, mortality, and hospitalization. 109 To test the breadth and robustness of the small airway-on-a-chip and truly apply the unique microfluidic feature of this device for drug discovery, Benam et al. studied whether a newly developed lead preclinical compound—an anti-inflammatory drug candidate [2-methoxy-N-(3-methyl-2-oxo-1,2-dihydroquinolin-6-yl) benzenesulfonamide] (Pfizer), which inhibits nuclear factor-κB (NFκB) signaling via specific blockade of bromodomain-containing protein-4 (BRD4)—would prevent neutrophilic recruitment from flow to inflamed endothelium in a viral-like exacerbated COPD small airway chip model. 26 Increased neutrophil accumulation in COPD lungs has been associated with enhanced severity of airflow limitation. 110
The authors observed that treatment with the BRD4 inhibitor, in contrast to the clinically prescribed glucocorticoid drug budesonide (Sigma-Aldrich), suppressed neutrophil adhesion to the endothelium from blood-like circulation by almost 75%. 26 Using a static cell culture system (Transwell inserts) that lacks physiologically relevant dynamic flow conditions, instead of the microfluidic small airway chip, it was found that the inhibitory effect of the BRD4 blocker was reduced by almost one-third, suggesting that the compound’s mechanism of action might involve preferential inhibition of early stages of the leukocyte adhesion and rolling, which cannot be mimicked in static models.
Future Directions for Human Lung-on-Chip Microsystems
Although this review highlights several key advantages of applying lung-related organ-on-chip technologies for translational pulmonary research, certain limitations of these microdevices need to be tackled as the field progresses to further improve their usability in preclinical studies. For instance, due to technical challenges in microassembly/fabrication and culture medium compatibility among multiple cell types and tissues, the alveolus- and small airway-on-a-chip in their current format lack natural (not synthetic) subepithelial basement membrane, ECM, and stromal cells such as fibroblasts, smooth muscle cells, and lung mesenchymal progenitor cells. We anticipate, however, that in the coming years, cumulative learned expertise throughout the field and the emergence of more advanced micro- and nanofabrication techniques will overcome these drawbacks. Other aspects that currently affect the use of alveolus and small airway chips in academic and industry settings are the complexity of operation, the low- to medium-throughput nature of experimentation, and the need for combined engineering and cell biology expertise when using these microsystems, hence restricting general accessibility of this technology. Thus, it is important to accelerate efforts to create off-the-shelf organ-on-chip products along with easy-to-operate device-associated accessories for end users (i.e., laboratory investigators). In addition, it is worth emphasizing that these microdevices are high-content, high-value platforms that enable high-resolution imaging and controlled manipulation of the organ microenvironment. Because of this, we advise potential users to consider them for addressing specific and refined scientific questions post initial analyses with simpler, higher throughput (e.g., cell line–based) systems.
Recently, Jain et al. used a primary human lung alveolus-on-a-chip model to perform quantitative analysis of organ-level contributions to inflammation-induced pulmonary thrombosis and test efficacy of a candidate antithrombotic therapeutic in vitro. 111 There is, however, a growing need to apply lung-related organs-on-chips in future to other respiratory disorders, such as cystic fibrosis (CF), pulmonary hypertension (PH), and idiopathic pulmonary fibrosis (IPF), which are unmet medical needs and are often associated with poor prognosis and high morbidity. Similarly, we anticipate that with increased accessibility of organs-on-chips and availability of biological samples, the alveolus and small airway chip microsystems will gradually be adapted for “personalized” disease modeling, drug testing, and biomarker discovery. Such strategy holds strong promise in revealing new discoveries based on “defined” patient populations (e.g., based on clinical endotypes, rather than the “average” patient) 112 and thereby can considerably enhance the pace and efficiency of translation.
Technically, in the next 5–10 years, we foresee integration of 3D bioprinting with organs-on-chips to facilitate construction of anatomically guided microstructures by precise patterning and layering of various biomaterials and cell–matrix compositions in vitro ( Fig. 4 ). Importantly, physiologically linking different organs-on-chips (e.g., lung and heart, lung and liver, lung and a hematopoietic organ, etc.) via microfluidic networks can provide new toolkits for researchers to dissect and interrogate dynamic organ–organ communications during health and disease. Moreover, detailed understanding of pharmacokinetics and pharmacodynamics while exploring disease- and organ-specific drug efficacy can create a more meaningful dataset on expected biological impact of a given therapeutic.
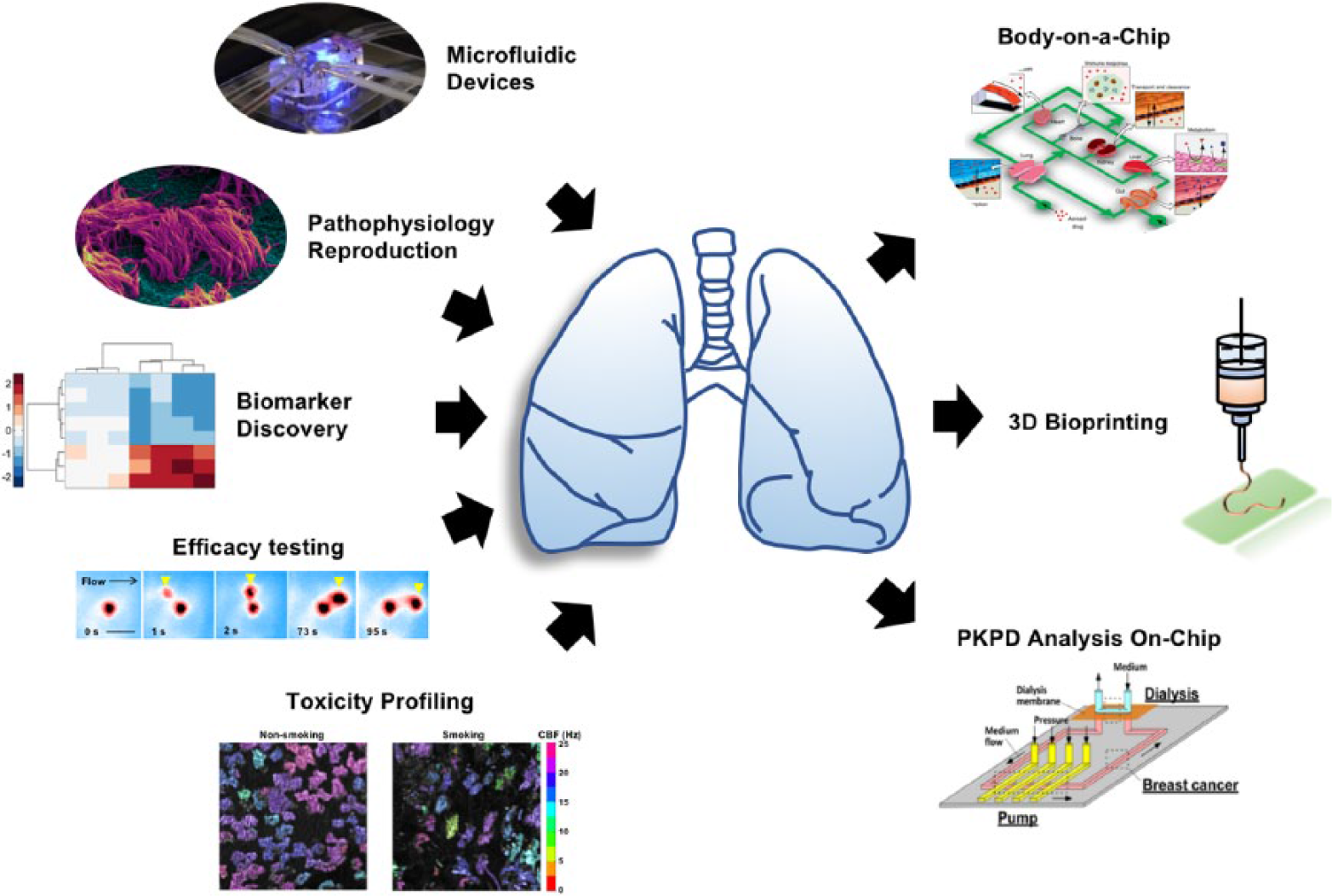
Lung-related organs-on-chips: current status and future directions for technological advancement. Although impactful advances have been made in recent years to microengineered lung organomimetics ranging from biomarker discovery and drug efficacy testing to toxicity profiling, it is anticipated that in the coming years, generation of physiologically more relevant linked multi-organ chips, integration with 3D bioprinting, and performing complex pharmacokinetics–pharmacodynamics (PKPD) analyses on-chip will further enable these technologies for clinical translation. Reprinted after modification with permission from Elsevier, Inc. (Cell Press), Macmillan Publishers Limited (Springer Nature), and American Chemical Society.8,13,26,28
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.