
Abstract
Major depressive disorder (MDD) is a multifactorial psychiatric disorder with obscure pathophysiology. A biomarker-based approach in combination with standardized interview-based instruments is needed to identify MDD subtypes and novel therapeutic targets. Recent findings support the impairment of the mammalian target of rapamycin complex 1 (mTORC1) in MDD. No well-established biomarkers of mTORC1 disease- and treatment-modulated activity are currently available for use in early phase antidepressant drug (AD) development. This review aims to summarize biomarkers of mTORC1 activity in MDD and to suggest how these could be implemented in future early clinical trials on mTORC1 modulating ADs. Therefore, a PubMed-based narrative literature review of the mTORC1 involvement in MDD was performed. We have summarized recent pre-clinical and clinical findings linking the MDD to the impaired activity of several key biomarkers related to mTORC1. Also, cases of restoration of these impairments by classical ADs and novel fast-acting investigational ADs are summarized. The presented biomarkers may be used to monitor pharmacological effects by novel rapid-acting mTORC1-targeting ADs. Based on findings in the peripheral blood mononuclear cells, we argue that those may serve as an ex vivo model for evaluation of mTORC1 activity and propose the use of the summarized biomarkers for this purpose. This could both facilitate the selection of a pharmacodynamically active dose and guide future early clinical efficacy studies in MDD. In conclusion, this review provides a blueprint for the rational development of rapid-acting mTORC1-targeting ADs.
Keywords
Introduction
Major depressive disorder (MDD) is a multifactorial psychiatric disorder that is primarily characterized by a sustained and pathologically depressed mood and anhedonia and accompanied by disturbances in cognitive function, psychomotor activity, and vegetative symptoms. MDD is one of the top four causes of years lived with disability worldwide, and patients suffering from MDD are at risk of suicidal behavior or death by suicide.1,2 Besides, up to 30% of MDD patients fail to respond to currently registered antidepressant drugs (ADs), a majority of patients experience unacceptable side effects or residual symptoms despite antidepressant treatment, and therapeutic effects with conventional monoaminergic ADs only occur after several weeks. 3 The future development of novel ADs for the treatment of MDD should therefore pursue rapid mood improvement that is associated with a more favorable efficacy and safety profiles compared with currently available drugs.
Currently, the classification of MDD is primarily based on diagnostic criteria of the Diagnostic and Statistical Manual of Mental Disorders (5th edition; DSM 5). 4 Importantly, the most widely used method to assess the severity of depressive symptoms and the response to pharmacological interventions in virtually all clinical MDD studies is one or a combination of depression severity rating instruments. These include the clinician-rated Montgomery–Åsberg Depression Rating Scale (MADRS) and the Hamilton Rating Scale for Depression (HAMD), and the self-reported Inventory of Depressive Symptomatology (IDS-SR).5–7 These instruments are composed of scoring items that are predominantly derived from the DSM diagnostic criteria, with the severity of each item being rated using a 4- to 6-point Likert-type scale, and ultimately resulting in a total score.5–7 Despite being useful in clinical practice to track treatment response over time, the validity of such phenomenology-based assessments depends on various factors such as rater experience, MDD-mediated recall bias, and unintended non-specific therapeutic effects as a result of interactions with raters or the research team during the study. Moreover, these instruments are not sufficient to distinguish between different biological subtypes or endophenotypes that may underlie the heterogeneous MDD syndrome. Also, as these have been validated using predominantly monoaminergic ADs, they might not be sufficiently sensitive to grasp rapid dynamic central nervous system (CNS) molecular changes induced by drugs with novel mechanisms of action such as N-methyl-D-aspartate receptor (NMDAR) antagonists or α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR) modulators. Clearly, there is a need for biomarkers that complement the current interview-based evaluations, as biomarkers are expected to objectify the effects of novel compounds in early phase psychiatric drug development and ultimately personalized MDD treatments. 8
Although neuroscience has contributed greatly to a better understanding of the neurobiology of psychiatric diseases, the pathophysiology of MDD remains largely obscure. Drug development for MDD is currently still dominated by a phenomenological approach, which ultimately results in the selection of heterogeneous patient populations for inclusion in drug trials and may contribute to the dilution of treatment effects with novel compounds. A biomarker-based approach for MDD presents obvious advantages, such as the identification of patient subgroups that may benefit from drugs with a specific mechanism of action (MOA) and the drug development guided by pharmacological biomarkers.3,9 Several different biomarkers have been proposed, ranging from CNS-based cerebrospinal fluid (CSF) and neuroimaging profiles to peripheral changes involving the neuroendocrine system and inflammatory responses. 9 However, so far, none of these biomarkers have been adequately validated for application in the complementary diagnosis and treatment of MDD, or in drug development programs of novel ADs. 9
An increasing number of molecular findings suggests a relationship between stress-related synaptic dysfunction, one of the putative pathophysiological factors in MDD, and inhibition of the mammalian target of rapamycin complex 1 (mTORC1). 10 Therefore, mTORC1 has recently emerged as a novel surrogate target for emerging rapid-acting ADs, as well as a potential pharmacological and diagnostic biomarker in drug development.11–13 Although mTORC1 activity can readily be quantified in dissected brains of laboratory animals, and molecular and morphological changes can be linked to behavioral response to AD treatment, this approach is not feasible in living human subjects.14,15
This literature review, therefore, aims to summarize the evidence for molecular mTORC1 biomarkers in MDD, and to suggest how these could be implemented as pharmacological biomarkers in future early clinical trials with rapid-acting ADs that target the reversal of stress-related synaptic dysfunction in MDD. Ultimately, development of new ex vivo models may demonstrate the mTORC1-related pharmacological activity of drugs by monitoring relevant biomarkers within the signaling pathway.
mTORC1 involvement in MDD
mTOR signaling governs cell growth and metabolism. mTOR complex is divided into two distinct multiprotein complexes named mTORC1 and mTORC2. The signaling of both complexes is intertwined as they share multiple proteins, that is, mLST8, the Tti1/Tel2 complex, and Deptor.16,17 mTORC1 additionally associates with the nonenzymatic scaffolding protein Raptor, the inhibitory PRAS40, which itself is inhibited by Akt, having in total six protein components. 18 mTORC1 plays a central role in controlling cell growth, protein synthesis, lipogenesis, energy metabolism, and autophagy, whereas mTORC2 facilitates cell survival and cytoskeletal organization. 19 Activity of both complexes is modulated by growth factors, and mTORC1 is additionally affected by cellular energy status, oxygen, and nutrients, such as glucose and amino acids.14,19–24 Figure 1 shows a simplified representation of major mTORC1 signaling pathways. Under the scope of this review, only the mTORC1 signaling is discussed in more detail.
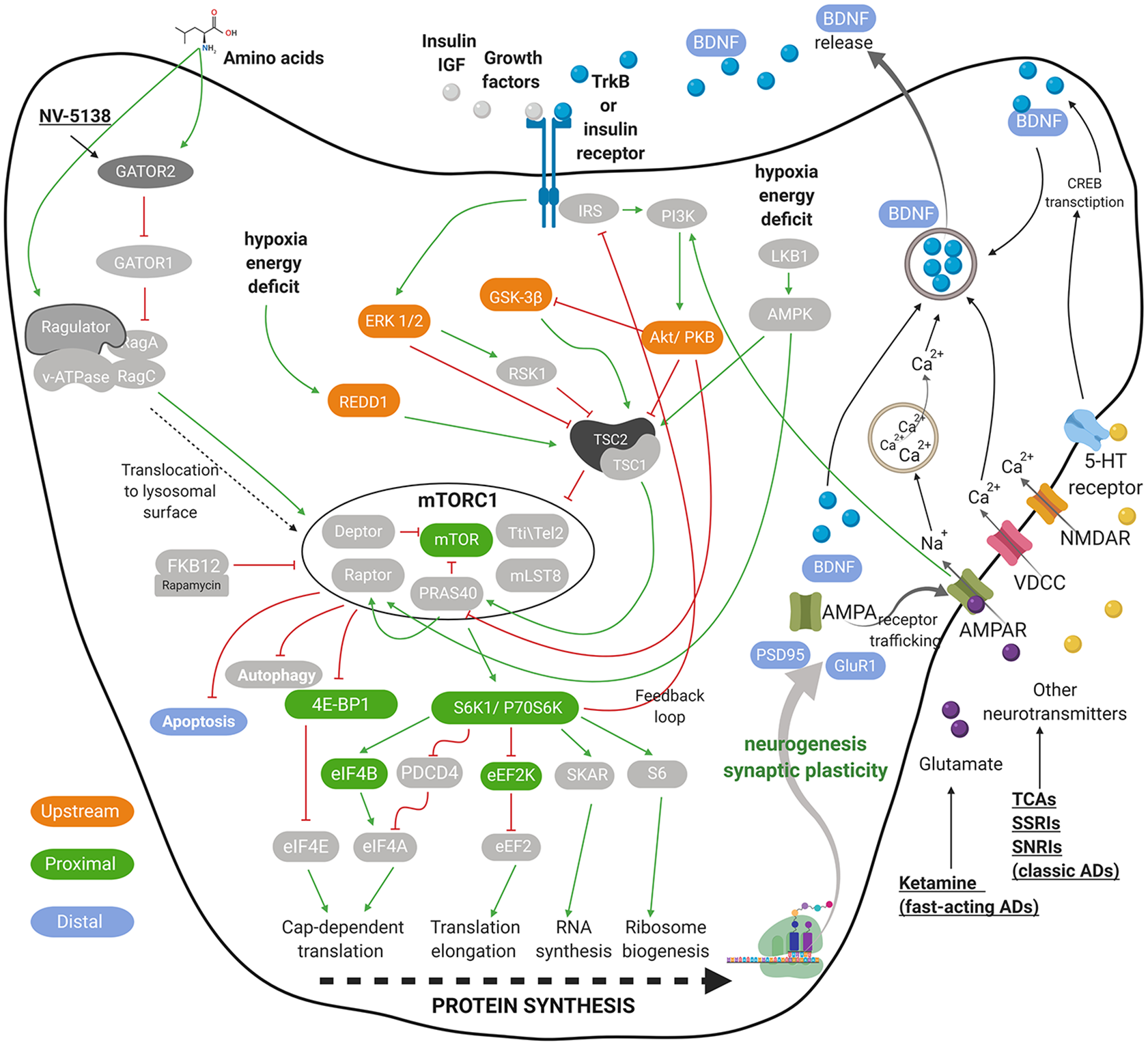
Graphical representation of neuronal mTORC1 signaling. Presented are the key elements that regulate mTORC1 signaling and play a role in depression pathophysiology mediated by impaired synaptogenesis. The crucial inputs regulating mTORC1 activity include amino acids, growth factors, stress, energy and oxygen status, as well as several neurotransmitters and their receptors. A few targets of AD are presented in a simplified form. The green pointed arrowheads indicate activation and the red flat indicate inhibition. mTORC1-related biomarkers, upstream of, close relative to (mTORC1-proximal), farther downstream of the mTORC1 (mTORC1-distal) are indicated in orange, green, and blue, respectively. For all abbreviations, see list of abbreviations.
List of abbreviations

mTORC1 is of crucial importance in various CNS-related physiological processes, where it is primarily responsible for the regulation of neuronal development, neurogenesis, and synaptic plasticity.10,25–27 Synaptic plasticity is an adaptive process that regulates synaptic strength, numbers, and density by which neurons and circuits change their excitability and connectivity. Such processes help to adjust to developmental changes, such as aging and environmental stimuli, for example, learning and stress, which influence thoughts, mood, and behavior. 28 It is hypothesized that chronic stress-related mood disorders such as (certain subtypes of) MDD may be associated with loss of synaptic plasticity, which compromises neuronal function.26,29,30 The number of synapses and the cell body size have been found to be significantly decreased postmortem in the prefrontal cortex (PFC) of MDD patients. 31 Such structural neuronal changes are believed to negatively influence modulatory effects and functional connectivity within and between neuro-circuits involved in the physiological regulation of emotion, such as the PFC and subcortical structures, hypothalamus, amygdala, and hippocampus (HIP).32,33 Alongside morphological changes, PFC of postmortem MDD patients also exhibits a significant reduction in expression of mTORC1-dependent translation initiation factors; mTOR, p70S6K, and eIF4B, which may be involved in the regulation of brain function and pathophysiology of MDD.27,34
Animal models of chronic stress that exhibit depressive-like behavior also show neuronal atrophy in the PFC and HIP as a result of reduced expression levels of synaptic proteins.14,22,35 Studies suggest that these changes are associated with stress-induced prolonged excessive extracellular glutamate, which causes excitotoxicity, reduction of spine density, and synaptic strength and overall dendritic atrophy within the PFC.36,37 Moreover, direct inhibition of mTORC1 through overexpression of REDD1 in rodents can induce depressive-like symptoms without exposure to stress. 22 Similar observations are found in MDD patients. Changes in postmortem brain tissue morphology co-occur with decreased levels of phosphorylated (phospho) mTORC1-associated molecules, such as mTOR, p70S6K, and ribosomal protein S6.13,14,22,38,39
These findings indicate a strong connection between stress-related morphological changes in the brain, depressive-like behavior, and reduction of mTORC1 activity. Therefore, mTORC1 signaling may be a putative (indirect) target for monitoring ADs evaluated in a variety of mTORC1-related biomarkers.
The role of mTORC1 in the MOA of novel ADs
Recent pre-clinical discoveries, in the search for novel therapeutic targets, indicate that mTORC1 plays a crucial role in the rapid onset of antidepressant action. One of the emerging rapid-acting ADs, with a MOA relying on mTORC1, is the non-competitive NMDAR antagonist ketamine. 40 Through inhibition of both presynaptic and postsynaptic NMDARs, ketamine blocks the spontaneous activity of GABAergic interneurons, which leads to an acute glutamate release and its binding to pro-synaptogenic AMPARs. Especially, the GluN2B subunit of NMDARs on GABAergic interneurons is proposed to be essential for the rapid antidepressant actions of ketamine. 41 As depicted in Figure 1, this mechanism leads to a subsequent downstream mTORC1 activation, triggering synaptogenesis which contributes to synaptic plasticity resulting in an antidepressant effect.10,26 mTORC1-dependent effect of several investigational ADs with a fast onset of action, including ketamine, has been reported in multiple pre-clinical studies. Table 1 summarizes mTORC1-related molecules altered by chronic stress exposure in pre-clinical studies and the reversed effects after pharmacological treatment. In all cases, the investigational ADs reversed the stress-induced depressive-like behavior.
Potential mTORC1-related biomarkers in animal model of MDD after pharmacological treatment. Summarized are mTORC1-related biomarkers, which are close relative to mTORC1 (mTORC1-proximal), farther downstream to mTORC1 (mTORC1-distal), and upstream to mTORC1. Presented data are based on pre-clinical studies in animals subjected to chronic stress paradigms in which the pharmacological treatment deemed successful, unless stated otherwise.
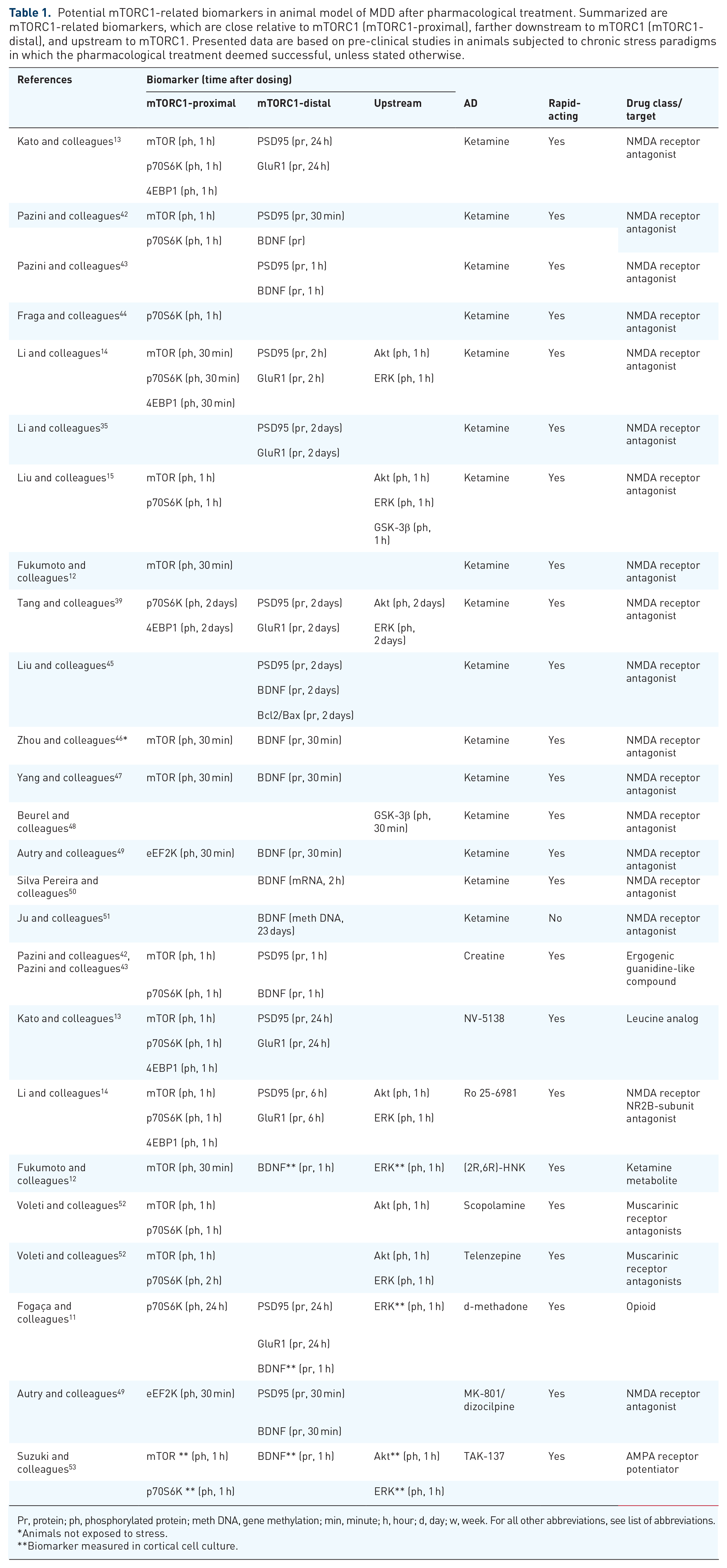
Pr, protein; ph, phosphorylated protein; meth DNA, gene methylation; min, minute; h, hour; d, day; w, week. For all other abbreviations, see list of abbreviations.
Animals not exposed to stress.
Biomarker measured in cortical cell culture.
In the PFC of animal models, acute ketamine treatment was able to reverse the chronic stress-induced deficits in phospho-mTOR, p70S6K, and 4E-BP1, as well as the expression of PSD95 and glutamate/AMPA receptor subunit, GluR1, responsible for synaptic plasticity.12–15,22,35,39,42 A similar effect on phospho-p70S6K was observed in the HIP alongside an increased dendritic spine density in the dentate gyrus. 44 Ketamine could also lower the level of phospho-eEF2K, which inhibits translation elongation. 49 When administered 1 week prior a corticosterone treatment, ketamine prevented corticosterone-induced deficits of PSD95 and GluR1 in the HIP. 54 The mTORC1-dependent action of ketamine was also demonstrated after pretreatment with a selective mTORC1 inhibitor, rapamycin, which blocked any effect of ketamine on the levels of phospho-mTOR, 4E-BP1, PSD95, and GluR1 as well as on animal behavior, compared with a non-pretreated group.35,55
Besides the rapid increase of phospho-mTOR and PSD95, a single dose of ketamine led to an increase of brain-derived neurotrophic factor (BDNF) in the PFC and the HIP.43,45–47,49,50 Interestingly, this effect was abolished by rapamycin. 43 A long-term ketamine treatment led to less methylation of the BDNF gene in the PFC, which increased its translation and BDNF protein levels. 51
In contrast to ketamine, as a rapidly acting AD, conventional ADs such as sertraline require chronic administration to decrease depressive-like behavior that corresponds to increased BDNF in rodent PFC.56,57 Chronic antidepressant treatment activates a transcription factor CREB which regulates transcription of BDNF. 58
BDNF is essential for neuroplasticity and brain development. It is initially transcribed into two forms, proBDNF and mature BDNF, which activate different processes. proBDNF, expressed constitutively in low levels, binds to neurotrophin receptor P75 (p75NTR) and leads to disrupted plasticity, whereas mature BDNF undergoes neuronal activity dependent release and binds to TrkB. Therewith, mature BDNF positively regulates translation of i.a. synaptic proteins through activation of the PI3K–Akt pathway, followed by activation of mTORC1, as shown in Figure 1. 10 Moreover, proBDNF contains a common single-nucleotide polymorphism (SNP), Val66Met, which hinders its processing to the mature form. A summary of the role of BDNF in the pathophysiology and treatment of MDD is provided in a recent review. 27 As demonstrated by several studies, inhibition of BDNF by infusion of BDNF neutralizing antibodies or a knock-in of Val66Met blocks any antidepressant and molecular effect of investigational ADs.12,13,59 Taken together, a rapid elevation of mature BDNF upon fast-acting AD administration can be considered a potential biomarker signifying mTORC1 activity, given its dependence on mTORC1 and its co-occurrence with other mTORC1-related biomarkers.
In addition, an increase of apoptosis, indicated by i.a. a lower ratio between the pro-apoptotic B-cell lymphoma 2 (Bcl2) and anti-apoptotic Bcl-2-associated X protein (Bax) in the HIP of rats subjected to chronic unpredictable stress (CUS), was alleviated 24 h after ketamine treatment. 45 Considering that energy stress regulates apoptosis through mTORC1 (Figure 1), and direct inhibition of mTORC1 leads to altered neuronal cell death in developing mice, Bcl2: Bax ratio may also be related to mTORC1-mediated action of ketamine.60,61
Stress-induced deficits of several mTORC1-related molecules were also reversed by different investigational drugs that exhibit rapid antidepressant-like effects in pre-clinical animal behavior studies. Similar to ketamine, a leucine analog (NV-5138) and an NMDAR NR2B-subunit antagonist (Ro 25-6981), both increased phospho-mTOR, p70S6K, and 4E-BP1 and expression of PSD95 and GluR1.13,14 Interestingly, the increase in PSD95 and GluR1 was observed with a 5–23 h delay compared with mTOR, p70S6K, and 4E-BP1, confirming that PSD95 and GluR1 are modulated by mTORC1, as depicted in Figure 1.13,14 A similar result was observed after creatine, an ergogenic guanidine-like compound, which increased phospho-mTOR and p70S6K and PSD95 and BDNF protein levels in the HIP.42,43 In a different pre-clinical study, treatment with muscarinic receptor antagonists, either scopolamine or telenzepine also rapidly increased phospho-mTOR and p70S6K. 52 Treatment with d-methadone, an opioid, increased p70S6K, PSD95, and GluR1 in PFC and BDNF levels in in vitro cortical cells. 11 A non-competitive NMDAR antagonist dizocilpine, also known as MK-801, decreased phospho-eEF2K and increased BDNF levels in the same fashion as ketamine in an animal model. 49 A ketamine metabolite, (2R,6R)-HNK, also increased phospho-mTOR in rat PFC and BDNF levels in vitro. 12 Moreover, an AMPAR potentiator (TAK-137) rapidly increased phospho-mTOR and p70S6K and BDNF levels in vitro. 53
Interestingly, the aforementioned investigational ADs targeting mTORC1 also lead to a rapid increase in phosphorylated molecules upstream of the mTORC1, Akt, and ERK in the PFC of animal models and in in vitro experiments.11,12,14,15,39,52,53 The activity of the mTORC1-inhibiting GSK-3β was also found to be decreased after ketamine treatment.15,48 These observations suggest that the signaling pathways linked to mTORC1 are also worth investigating alongside the mTORC1 to better understand the MOA of the novel ADs.
Conclusively, a significant body of experimental evidence provides support for the involvement of mTORC1 in the MOA of investigational rapid-acting ADs. Monitoring of mTORC1 activity through the evaluation of molecular biomarkers could, therefore, provide a helpful insight into novel ADs of which the MOA is not fully elucidated.
CNS-based mTOR-related MDD biomarkers
Postmortem human brain
Animal models have enabled us to thoroughly study the molecular effects of investigational CNS drugs at the cellular level directly in dissected brain tissue combined with behavioral experiments. Whereas evidence for the role of mTORC1 in the activity of ADs can be easily obtained from animals, the same kind of mechanistic data are much more difficult to obtain from humans, where we are limited to postmortem brains and the CSF. Therefore, to date, there are merely a handful of clinical studies focused specifically on mTORC1-related molecules in relation to MDD. Table 2 contains a summary of postmortem studies that indicate differences in levels of mTORC1-related molecules between MDD patients and controls.
mTORC1-related biomarkers detected centrally in postmortem human brain. Summarized are the measured levels of mTORC1 components in postmortem brain tissue of depressive patients.

C, healthy control; P, patient; m, male; f, female; DF, drug free; NR, not reported; NS, not significant; amit, amitriptyline; ser, sertraline; par, paroxetine; flu, fluoxetine. For all other abbreviations, see list of abbreviations.
Protein levels normalized to actin.
Kinase activity expressed in pmol/min/mg of brain tissue.
Fold change relative to control.
Amount of protein expressed in ng/g brain tissue.
Protein immunoreactivity.
mRNA level relative to neuron-specific enolase (NSE) mRNA.
All presented p values are statistically significant unless indicated otherwise.
One prominent postmortem study has reported a significant reduction of mTOR, p70S6K, eIF4B, and phospho-eIF4B in the PFC of MDD patients. 34 In the same group of deceased patients, also the level of synaptic protein PSD95 was lower compared with healthy controls. 62 Moreover, BDNF was lowered in MDD as reported by three independent studies.65–67 BDNF protein level was lowered in the PFC and HIP of drug-free MDD patients relative to healthy control. However, there was no significant difference from patients who received (classic) antidepressant treatment. 66 A similar result was observed in a different study, where a lower BDNF was measured in the HIP of unmedicated MDD patients, compared with medicated ones. 67 BDNF protein, as well as mRNA expression, was also reduced in PFC and HIP of patients in a study, where, unfortunately, the drug type was not reported. 65 The activity of Akt was found to be lowered and the inhibitory GSK-3β and REDD1 increased in depressive patients.22,63,64
As can be seen in Table 2, the common problems with the presented studies are (1) the very narrow window between the biomarker levels measured in patients and controls, and (2) the relatively small sample sizes and not equal male: female ratios due to unavailability of donor tissue. Also, medication use within a study varies between patients, and some studies do not report such information. Therefore, it is not possible to differentiate between true disease-related and drug-related effects.
CSF
Despite that the CSF provides the closest approximation of the CNS and can be obtained from living subjects, to date, no attempts have been made for a systematic evaluation of mTORC1-related molecules in this matrix. The only biomarker related to mTORC1-signaling that has been researched is BDNF, however with inconsistent outcomes. Levels of CSF BDNF in MDD patients were found to be notably lower than in healthy controls. Citalopram treatment led to a reduction in MADRS and HAMD scores, coinciding with trends of increasing CSF BDNF. However, the presented control group was not matched to the patients and the observed changes were not statistically significant. 68 Stronger evidence was presented in a different study, which reported lower BDNF levels in MDD patients and a significant correlation between BDNF levels and severity of depression. 69 In two other studies, no significant differences were found in CSF nor plasma BDNF levels between depressed and control subjects, and no effect of ADs was observed.70,71 The ratio of the BDNF pro-peptide to the total protein level in CSF of male MDD patients was found to be significantly lower than in male controls; however, the mature BDNF levels alone were below the limit of detection. This difference was not found in female subjects, which may be explained by the difference in gene methylation between sexes.72–74
Overall, notwithstanding the limitations, these findings suggest that the activity of mTORC1 in the CNS is impaired in MDD patients. Therefore, it is safe to assume that restoration of mTORC1 activity, upon an AD treatment, signified by relevant biomarkers, could serve as an indication of its molecular MOA.
Peripheral mTOR-related MDD biomarkers
Peripheral blood
Reminiscent to the CNS, a limited number of publications report peripheral alterations in the expression or activity of mTORC1-related molecules in MDD. Table 3 presents studies that report alterations in peripheral levels of mTORC1-related molecules after pharmacological treatment.
mTORC1-related biomarkers in clinical studies. Summarized are mTORC1-related biomarkers, which are close relative to mTORC1 (mTORC1-proximal), farther downstream the mTORC1 (mTORC1-distal), and upstream of mTORC1. Presented data are based on clinical studies in MDD in which the pharmacological treatment deemed successful and resulted in fast mood improvement, unless stated otherwise..
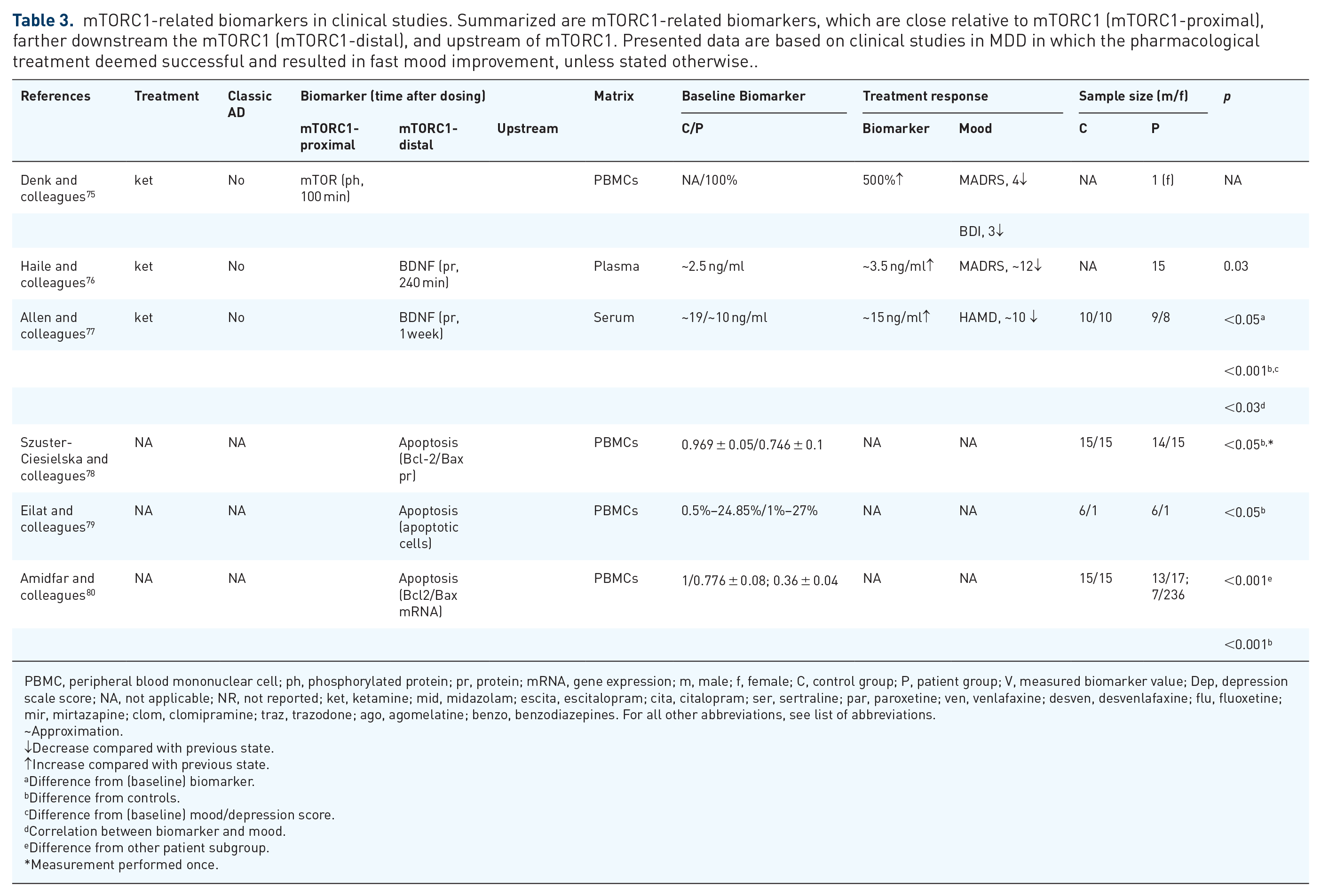
PBMC, peripheral blood mononuclear cell; ph, phosphorylated protein; pr, protein; mRNA, gene expression; m, male; f, female; C, control group; P, patient group; V, measured biomarker value; Dep, depression scale score; NA, not applicable; NR, not reported; ket, ketamine; mid, midazolam; escita, escitalopram; cita, citalopram; ser, sertraline; par, paroxetine; ven, venlafaxine; desven, desvenlafaxine; flu, fluoxetine; mir, mirtazapine; clom, clomipramine; traz, trazodone; ago, agomelatine; benzo, benzodiazepines. For all other abbreviations, see list of abbreviations.
~Approximation.
↓Decrease compared with previous state.
↑Increase compared with previous state.
Difference from (baseline) biomarker.
Difference from controls.
Difference from (baseline) mood/depression score.
Correlation between biomarker and mood.
Difference from other patient subgroup.
Measurement performed once.
In a study involving a treatment-resistant depression patient, treatment with ketamine was found to cause an acute increase in phospho-mTOR in peripheral blood mononuclear cell (PBMCs). This change coincided with a decrease in MADRS and HAMD scores. 75 However, this observation should be interpreted with caution as this study was conducted on only one patient, besides lacking a healthy control. 75 Another study, performed in an adequate number of subjects, supports part of previous outcomes by showing increased phospho-GSK-3β in platelet-rich plasma of MDD patients compared with controls. In the same study, the phospho-GSK-3β decreased after monoaminergic AD treatment and correlated with lowered HAMD scores. 81
Several authors showed evidence of accelerated apoptosis in PBMCs of patients with mood disorders, which may be triggered by an impaired activity of mTORC1. Indicators of apoptosis such as elevated expression levels of several cell death–regulating proteins such as Bax in T-lymphocytes and a higher release of reactive oxygen species have been reported in depressed patients relative to healthy controls.78,79 Therewith, the Bcl2: Bax ratio was significantly decreased in PBMCs and specifically CD14+ leukocytes of depressed patients.78,80 Combined with the findings in animal models of chronic stress, where the (mTORC1-activating) ketamine increased the Bcl2: Bax ratio, these observations imply that Bcl2: Bax ratio could serve as an indicator of mTORC1 activity. 45
Despite the lack of well-grounded evidence, the available limited clinical findings suggest that alterations in mTORC1-related signaling can be detected in the periphery and especially in the PBMCs.
BDNF
Being closely related to the regulation of brain function and morphology, BDNF expression has been extensively explored in MDD animal studies. Likewise, numerous attempts have been made to use peripheral BDNF either as a complementary diagnostic biomarker of MDD or to monitor the pharmacological effects of ADs in humans. Due to the positive association to mTORC1 signaling, as described in the context of pre-clinical studies, BDNF could be considered as a marker of mTORC1 activity. Supplementary Table S1 summarizes the evidence of peripheral detection of BDNF found in the literature.
Serum BDNF
The levels of serum BDNF in subjects suffering from MDD were repeatedly reported to be lower compared with healthy controls.29,73,77,82–94 However, several studies did not demonstrate any differences.85,95–98
As shown in Table 4, in the studies in which the effect of ADs was tested, the serum BDNF increased alongside a decrease in HAMD or MADRS scores after treatment with several different classic ADs.29,85,87,89,90,99–101 Also, ketamine (see Table 3) caused an increase of serum BDNF and an amelioration of symptoms in MDD patients. 77 Following the pre-clinical observations, the elevation of serum BDNF required chronic dosing of several weeks with classic ADs, in contrast to ketamine after which BDNF was elevated in serum within 1 week after dosing. 77 As mentioned before, this is accounted by the difference between the relatively fast mTORC1-related BDNF elevation and the slower CREB-mediated elevation. 58
BDNF as biomarker in clinical studies. Presented data are based on clinical studies in MDD in which chronic pharmacological treatment with classical ADs deemed successful and resulted in slow mood improvement..

pr, protein; mRNA, gene expression; m, male; f, female; C, control group; P, patient group; V, measured biomarker value; Dep, depression scale score; NA, not applicable; NR, not reported; ket, ketamine; mid, midazolam; escita, escitalopram; cita, citalopram; ser, sertraline; par, paroxetine; ven, venlafaxine; desven, desvenlafaxine; flu, fluoxetine; mir, mirtazapine; clom, clomipramine; traz, trazodone; ago, agomelatine; benzo, benzodiazepines. For all other abbreviations, see list of abbreviations.
Correlation between biomarker and mood.
Difference from controls.
Studied on Alzheimer’s disease patients with and without depression.
Difference from (baseline) biomarker.
Difference from (baseline) mood/depression score.
Difference in biomarker between treated and untreated patients.
~Approximation.
↓Decrease compared with previous state.
↑Increase compared with previous state.
Measurement performed once.
Measurement performed two times.
The inconsistencies in the BDNF-based discrimination between MDD patients and healthy controls are of concern. These can be attributed to the fact that BDNF is ubiquitously expressed and the circulating protein originates from several sources including neurons, as it can pass the blood-brain barrier, as well as platelets.106,107 BDNF in the blood is mainly stored at high levels in platelets that store and release continuously in plasma and after activation due to a traumatic injury or in serum during clotting. 107
Plasma BDNF
Overall, the literature on BDNF levels in plasma shows the same picture as serum BDNF, with a decreased BDNF level in MDD patients compared with healthy controls.94,108–110 Plasma BDNF was also found increased after chronic AD treatment (Table 4), and as fast as 2 h after a single ketamine administration (Table 3).76,102,103 Interestingly, responders to the treatment had a higher baseline plasma BDNF than treatment-resistant patients.
Contradicting results were reported by other studies, in which no differences in plasma BDNF levels were found between MDD patients and controls.71,111 Moreover, in a different study, ketamine reduced the MADRS scores but did not affect plasma BDNF. 112
BDNF in peripheral cells
In line with the observations based on plasma and serum, BDNF (mRNA) expression was found to be significantly decreased in PBMCs of MDD patients, when compared with healthy controls.90,105,113,114 The same was reported for adult as well as pediatric depressed patients. 115 An 8- and 12-week escitalopram treatment normalized the decreased BDNF expression in leukocytes from MDD patients who responded to the treatment.90,105 In a different study, however, despite reduced neurotrophins, such as glial cell line–derived neurotrophic factor, no significant difference in the expression levels of BDNF mRNA was found between patients with MDD and healthy subjects. 116
It is also worth noting that the epigenetic regulation of the BDNF gene may play an important role in depressive disorders. Two independent studies in MDD patients found a significantly enriched methylation of BDNF promoter, which represses gene transcription, along with impaired BDNF expression in PBMCs.114,117 The same correlations between BDNF methylation and prevalence of MDD, bipolar disorder, or schizophrenia were found in multiple other studies.72,74,117–121 Methylation of the BDNF gene was found to be sex-dependent with a higher degree in male subjects in schizophrenia and bipolar disorders.72,74 Moreover, an elevated BDNF methylation was observed in parallel in the PFC and peripheral muscle tissue of deceased BD patients, which correlated with the values measured in PBMCs of living patients. 74
However, it has to be noted that just like the evaluation of depression severity in most studies, the levels of BDNF were measured at only one timepoint after weeks-long antidepressant therapy. (Continued) Because of this, it cannot be determined whether the used therapy has a slow or rapid effect on BDNF. Moreover, levels of BDNF protein in the periphery differ among sexes and are sensitive to circadian rhythm, which is not accounted in most of the studies, considering that the samples are never obtained longitudinally.71,111,122,123 Also, expression of BDNF is regulated by i.a. glucocorticoid receptors, and depends therewith on activity of the hypothalamic-pituitary-adrenal axis (HPA-axis). 124 The discrepancies between different studies may be also explained by an earlier treatment as most of the enrolled patients used ADs in the past. This makes it even more difficult to distinguish between the drug- and disease-related effects.
Importantly, BDNF blood alterations are not MDD-specific as those are also found in several other disorders such as bipolar disorders or schizophrenia. 90 Still, peripheral BDNF can be used to recognize a common BDNF and mTORC1-mediated pathophysiology shared by disorders in which synaptic plasticity and mTORC1-mediated pharmacological effect play an important role.
Summary and discussion
We aimed to identify and summarize potential molecular biomarkers of mTORC1 activity in MDD and to suggest how these could be implemented in future early clinical trials with rapid-acting ADs that target the reversal of stress-related synaptic dysfunction in MDD.
The current interview-based MDD severity rating instruments are prone to bias and are not sufficiently sensitive to the downstream molecular effects of emerging rapid-acting ADs. Therefore, a complementary biomarker-based approach is needed to unravel neurobiological mechanisms underlying different MDD endophenotypes and to link these to the molecular actions of ADs. Pre-clinical and clinical data support the involvement of mTORC1 signaling in the pathophysiology of MDD, as well as being a molecular target of clinically effective monoaminergic ADs. mTORC1-related molecules could, therefore, serve as potential biomarkers of an mTORC1-related endophenotype in MDD on one hand, and pharmacological biomarkers for antidepressant action of rapid-acting ADs specifically on the other hand. Here, we present and clarify the relationship between mTORC1 and MDD and the effect of AD treatment based on the available evidence.
There is abundant evidence of mTORC1-mediated regulation of neuronal development and especially synaptic plasticity.10,25,26 Deficits in synaptic plasticity followed by a decreased number of synapses and cell body size in neuro-circuits involved in the physiological regulation of emotion have been linked to the pathophysiology of MDD. 31 On top of that, postmortem studies in MDD patients show deficits in the expression of several mTORC1-related molecules (see Table 2).22,34,62–65,67,94 Pre-clinical studies on animals exposed to challenges such as CUS confirm those morphological and cell-based findings in humans.13,14,22,35,38,39 Moreover, a multitude of pre-clinical studies demonstrate a clear correlation between antidepressant effects and restoration of the impaired mTORC1 activity in animals after administration of investigational and conventional ADs (see Table 1).12–15,35,39,45–47,49 Together, these findings demonstrate mTORC1 involvement in both the pathophysiology of MDD and the therapeutic effects of not only investigational rapid-acting but also conventional ADs. In vivo quantification of the activity of mTORC1-related molecules could therefore serve as pharmacological biomarkers in clinical AD development.
Based on postmortem studies, mTOR, p70S6K, eIF4B, phospho-eIF4B, PSD95, and BDNF, all lowered in the PFC of MDD patients, could be considered as potential mTORC1-related MDD biomarkers (see Table 2).34,62,65–67 Yet, to be applicable in a clinical trial setting, the biomarkers should be also available in matrixes proximal to the CNS that can be sampled in living subjects, such as CSF.
There is some human evidence indicating that decreased phospho-mTOR in PBMCs has a potential to recognize mTOR-related MDD endophenotypes. 75 Moreover, the occurrence of apoptosis in the PBMCs signified by a lowered Bcl2: Bax ratio could serve the same purpose.78,80 These biomarkers could be also used to monitor the effect of pharmacological treatment in general and rapid-acting antidepressants in particular, provided that the mTORC1 signaling is involved.75,90,105 Obviously, this stresses the need to identify patient subtypes within DSM categories that might benefit from mTORC1 modulation. However, the existing literature does not report any studies focused specifically on mTORC1-related molecules in the CSF, emphasizing the need for such clinical studies.
To date, BDNF is the most prominently researched mTORC1-related molecule with a potential to be used as a peripheral biomarker. However, BDNF levels in peripheral plasma, serum, or PBMCs suffer from high intraindividual variability attributable to many factors such as circadian rhythm, HPA-axis activity, an unidentifiable source of the free circulating protein as well as the possible occurrence of the Val66Met SNP.27,71,90,105–107,111,113–115,117,122–124 Measuring BDNF directly in the periphery without taking into account the factors that contribute to its variability, such as the presence of platelets in serum, is of a low informative value. Therefore, an ex vivo approach is needed.
The findings in the PBMCs suggest that patient- or healthy volunteer (HV)-derived material could be considered as a valuable ex vivo model to study the molecular effect of novel mTORC1-targeting ADs.78,80,90,105,113–115,117 Application of an ex vivo model provides a simplified representation of a complex system, in which any external stimuli, such as HPA-axis activity or circadian rhythm, can be controlled or eliminated. Therefore, in theory, all of the presented biomarkers could be used to study mTORC1 activity ex vivo (see Tables 2 and 3).
We argue that PBMCs may serve as a peripheral model for evaluation of physiological pathways involved in mental pathologies, given the strong physiological resemblance in relevant pathways between circulating immune cells and brain cells.125,126 Also, in a recent drug development program, PBMCs served as a matrix for the evaluation of the pharmacological activity of mTORC1-targeting ADs. 127 Conceptually, the presented mTORC1-related biomarkers (see Tables 2 and 3) can be used to establish an ex vivo challenge model based on HV-derived PBMCs. In such a model, the mechanistic profile of mTORC1 signaling responses to targeted mTORC1 impairment by known inhibitors, such as rapamycin, should be defined. Once established, it can be implemented in early clinical investigation of mTORC1-targeting ADs. Here, the mechanistic profile of mTORC1 signaling responses to such ADs could be defined, following exposure to (CSF-based) pharmacokinetic concentration relevant to in vivo safety studies on HV. Ultimately, ex vivo methodology could contribute to a more precise selection of pharmacologically active dose for early first-in-patient studies.
In conclusion, based on an extensive literature review, we identified several biomarker candidates for monitoring mTORC1 activity in humans. These can be already implemented to study the mechanistic profile of mTORC1 responses to investigational ADs in ex vivo settings. Once validated, these biomarkers may also facilitate both identification of altered mTORC1 signaling in MDD and dynamic pharmacological effects by rapid-acting, novel mTORC1-targeting ADs. From a drug development perspective, such biomarkers can aid selection of ‘mTORC1 impaired’ MDD (sub)populations, to characterize the pharmacodynamics of novel ADs that target mTORC1 on a molecular level, and to ultimately relate pharmacology to behavioral effects in the context of a pathophysiological cascade of events. 128 This approach could be of particular value for early clinical proof-of-pharmacology (safety) and proof-of-mechanism (pharmacodynamic) trials. Here the availability of such biomarkers would facilitate not only an efficient selection of a pharmacodynamically active dose or dosing regimen for proof-of-concept studies but also provide guidance to future efficacy studies in psychiatric patient populations. Herewith, this review is a first step toward the rational development of rapid-acting mTORC1-targeting ADs.
Supplemental Material
sj-docx-1-tpp-10.1177_20451253211036814 – Supplemental material for MTORC1 signaling as a biomarker in major depressive disorder and its pharmacological modulation by novel rapid-acting antidepressants
Supplemental material, sj-docx-1-tpp-10.1177_20451253211036814 for MTORC1 signaling as a biomarker in major depressive disorder and its pharmacological modulation by novel rapid-acting antidepressants by Tomasz Cholewinski, Diana Pereira, Matthijs Moerland and Gabriel E. Jacobs in Therapeutic Advances in Psychopharmacology
Footnotes
Acknowledgements
We would like to thank Karen Broekhuizen, PhD, who provided medical writing assistance on behalf of the Centre for Human Drug Research, Leiden, The Netherlands.
Conflict of interest statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.