
Abstract
The modification of a diverse array of substrates by Fe(II)/2-oxoglutarate-dependent dioxygenases is central to the modulation of distinct biological processes such as epigenetics, hypoxic signaling, and DNA/RNA repair. Of these, JumonjiC domain–containing histone lysine demethylases (JMJCs) and prolyl hydroxylases are potential drug targets due to their relevance to human diseases. Thus, assays to interrogate this enzyme superfamily are needed to identify selective and potent inhibitors as leads for drug development and that could also be useful research tools. Since succinate is a common product to all Fe(II)/2-oxoglutarate-dependent dioxygenase reactions, a method that detects succinate would be suitable to all members of this enzyme superfamily. We therefore developed a bioluminescent and homogenous succinate detection assay and validated its use with diverse sets of enzyme classes. We evaluated the substrate specificities of these enzymes, their apparent kinetic constants, and inhibition profiles and mode of action of reported and novel inhibitors. Our results indicate that succinate detection is a useful readout for the monitoring of enzymatic activities with distinct substrate entities, as well as for the discovery of novel inhibitors. By investigating a large number of Fe(II)/2-oxoglutarate-dependent enzymes, this method could have a significant impact on the field of dioxygenase research.
Introduction
The structurally conserved iron (Fe2+ or Fe(II)) and 2-oxoglutarate (2-OG, or α-ketoglutarate [α-KG])-dependent dioxygenase superfamily is composed of approximately 60 enzymes that catalyze the hydroxylation of a diverse array of substrates, resulting in protein side-chain modifications, 1 epigenetic regulation,2,3 alkylated DNA/RNA repair, 4 oxygen sensing, 5 lipid metabolism, 6 and the synthesis or degradation of several metabolites. 7 Details regarding the active sites and reaction mechanisms of these enzymes have been well described.7,8
The JumonjiC domain–containing histone lysine demethylase (JMJC) family are members of the Fe(II)/2-oxoglutarate-dependent dioxygenase superfamily. To date, two families of N-methyl-lysine demethylases (KDMs) have been identified: the lysine-specific demethylase 1 family (LSDs or KDM1s) that are Flavin-dependent demethylases that couple substrate oxidation to the reduction of flavin adenine dinucleotide (FAD) and the larger family of JMJCs. 9 Only the latter are ferrous iron-, 2-oxoglutarate-, and oxygen-dependent demethylases. These enzymes have a catalytic JumonjiC domain and are grouped in seven lysine demethylase subfamilies (i.e., KDM2–8), comprising a total of more than 30 members. These enzymes demethylate lysines containing different methylation states (i.e., mono-, di-, and trimethylated lysines) in the flexible tails of histones H3 and H4. Hydroxylation of the methyl group leads to the formation of an unstable lysyl hemiaminal intermediate, which is nonenzymatically released as formaldehyde.7,10 In general, the degree of lysine methylation and its identity affect gene transcription levels. For instance, methylation of H3K4, H3K36, and H3K79 correlates with active gene expression, whereas methylation of H3K9, H3K27, and H4K20 is associated with gene silencing. 11 In addition to protein demethylases, this superfamily of dioxygenases includes mRNA demethylases, such as the fat mass and obesity-associated protein FTO, which is a target for obesity and type 2 diabetes drug development;12,13 ALKBH5, which impacts mRNA translation and transport; and the proyl- and asparaginyl-hydroxylases (i.e., EGLN1–3 and the hypoxia-inducing factor 1 alpha inhibitor FIH), which are targets for the treatment of inflammatory and ischemic processes. 14
Due to their relevance to human cell health and disease, members of the Fe(II)/2-oxoglutarate-dependent dioxygenases are attractive drug target candidates for the treatment of a number of pathological conditions. In particular, the JMJCs and mRNA demethylating enzymes have generated a considerable interest for drug development. Since these enzymes function as general regulators of the transcriptional machinery and are overexpressed or mutated in several cancers, JMJCs are currently being pursued as targets for inflammatory diseases and cancer therapies.15,16
Different approaches have been developed to elucidate the enzymatic activities of the JumonjiC histone demethylases. These include radiometric or fluorescent- or colorimetric-based enzyme-coupled methods for the detection of formaldehyde release, 2,17 mass spectrometry–based methods for the direct detection of histone peptide methylation,18,19 and anti-methyl lysine-specific antibodies for the detection of methylation states.2,16 AlphaScreen and time-resolved fluorescence resonance energy transfer (TR-FRET) assays were also developed for KDMs and rely on antibodies specific for particular histone methylation states.20,21 This restricts the universality of these assays and can lead to assay interference by methyl-lysine mimetic inhibitors or other chemical compounds. 22 Other limitations of the methods mentioned above include sensitivity, risks of false hits resulting from compound interference, elevated costs associated with instrumentation or reagents, and low assay throughput. Furthermore, there are limited assay options that would enable the characterization of the other Fe(II)/2-oxoglutarate-dependent dioxygenases that modify DNA, RNA, or metabolites in a high-throughput manner.23–26 Therefore, there is a need to develop assays that can detect the activity of these enzymes and address the shortcomings of the currently available technologies. An ideal assay would be homogeneous to be applicable to high-throughput screening, be sensitive enough to detect a broad range of enzyme activities, be resistant to chemical compound interference, and show reproducible performance. Moreover, to avoid a need for multiple assay formats, it would be ideal if the assay were universal for all Fe(II)/2-oxoglutarate-dependent dioxygenase enzyme/substrate pairs, and this would be a particular advantage for profiling inhibitors against a panel of enzymes with diverse enzymatic activities.
Since succinate is a common reaction product to all members of the Fe(II)/2-oxoglutarate-dependent dioxygenase superfamily, we developed a universal bioluminescent assay to monitor their activities based on succinate detection. We characterized the enzymatic activity of several JumonjiC histone demethylases and Fe(II)/2-oxoglutarate-dependent hydroxylases, their substrate specificities, and their apparent kinetic parameters. Finally, we validated our method as a screening tool for inhibitors using the library of pharmacologically active compounds (LOPAC) 1280 compound library and studied the mode of action of selected compounds against members of this enzyme family. Our results demonstrate that succinate detection with bioluminescence is effective for characterizing multiple Fe(II)/2-oxoglutarate-dependent dioxygenases with diverse substrate structures, and that it enables the investigation of a large number of enzymes in a single format.
Materials and Methods
Enzymes and Substrates
Recombinant enzymes representing the JumonjiC histone demethylase subfamilies KDM3 (JMJD1A and JMJD1B), KDM4 (JMJD2A, JMJD2B, JMJD2C, JMJD2D, and JMJD2E), KDM5 (JARID1A, JARID1B, and JARID1C), KDM6 (JMJD3), and KDM7 (JHDM1D and PHF8) and the DNA demethylase TET1 were purchased from BPS Bioscience (San Diego, CA). UTX (KDM6A) was purchased from Cayman Chemical (Ann Arbor, MI). The prolyl hydroxylase EGLN1, DNA/RNA dioxygenase/demethylase ALKBH3, and RNA demethylase FTO were purchased from Origene Technologies (Rockville, MD). The peptide substrates histone H3 (1–21) N-terminal, [Lys(Me2)9]-histone H3 (1–21), [Lys(Me3)9]-histone H3 (1–21), [Lys(Me2)4]-histone H3 (1–21), [Lys(Me3)4]-histone H3 (1–21), [Lys(Me2)27]-histone H3 (23–34), and [Lys(Me3)27]-histone H3 (23–34) and the EGLN1 substrate HIF-1α (556–574) were purchased from Anaspec (Fremont, CA). The recombinant histone H3K4me3, histone H3K9me2, and histone H3K27me2 were purchased from Active Motif (Carlsbad, CA). The ALKBH3, FTO, and TET1 custom substrate sequences3,12,27 2(3mC) ssDNA (5′-CATGATAA(3-meC)CGCGA(3-meC)TACACT GAC-3′), 6mA RNA (5′-CUUGUCA(m6A)CAGCAGA-3′), and 5-methylcytosine ssDNA (5′-TGCTACCTCCTCAACGT (5mC)GACCA CCGTCTCCTGCA-3′) were purchased from IDT Technologies (Coralville, IA).
Chemicals and Assay Components
Ascorbate ((+)-sodium
The Succinate-Glo JmjC Demethylase/Hydroxylase Assay kit from Promega Corporation (Madison, WI) is composed of a succinate standard solution and components for two succinate detection reagents. Reagent I uses succinate as a substrate in a coupled enzyme reaction to produce ATP, and Reagent II detects ATP as light output from an ATP-dependent luciferase enzyme ( Fig. 1 ).
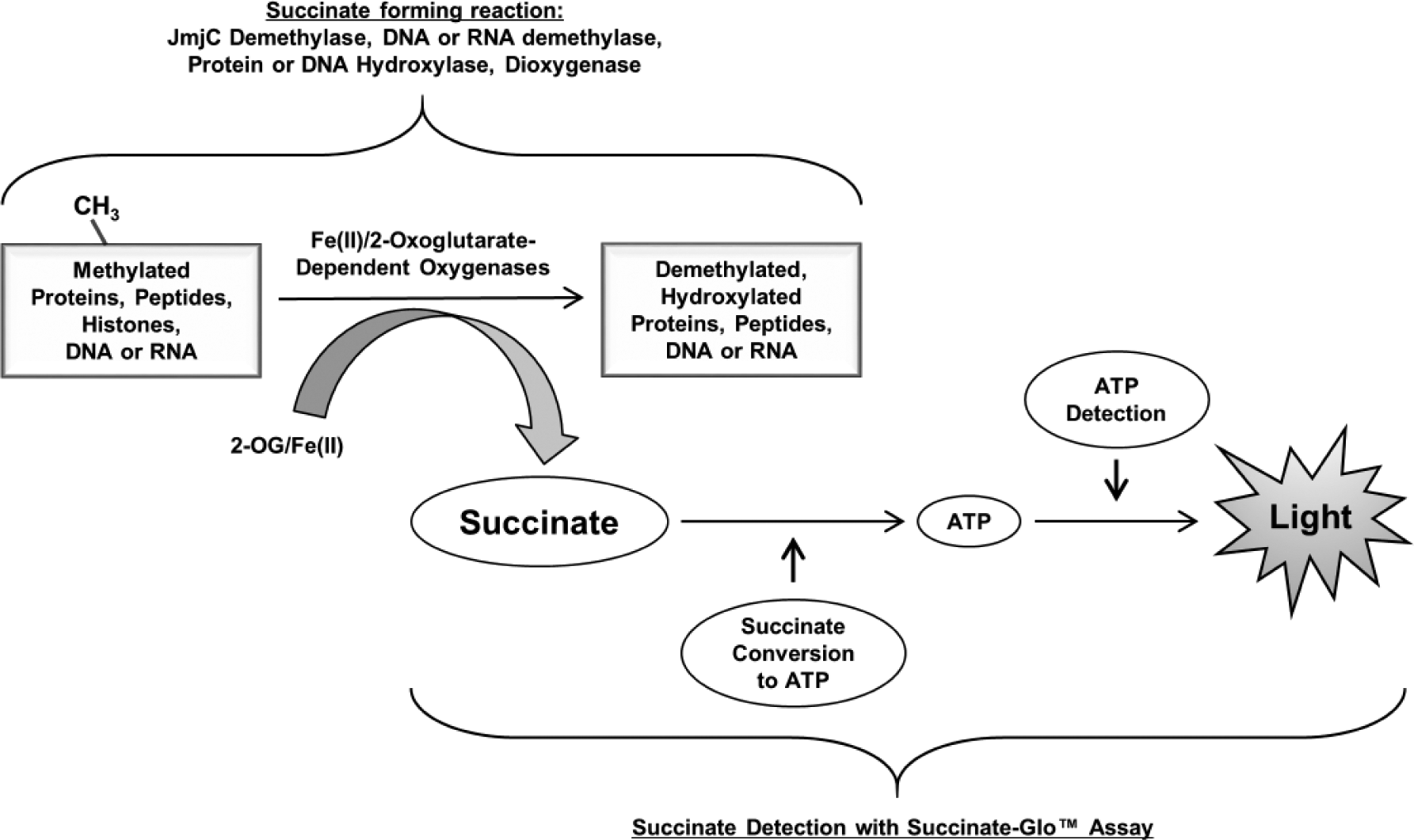
Succinate-Glo JmjC Demethylase/Hydroxylase Assay principle. Succinate-Glo assay detects succinate generated as a result of demethylase/hydroxylase activity. In the first step, Reagent I uses succinate as a substrate in a coupled enzyme reaction to produce ATP. In the second step, ATP is detected by Succinate Detection Reagent II containing the luciferase/luciferin system. The light generated correlates to succinate present and demethylase or hydroxylase activity.
Succinate Standard Curve
Succinate standard curves were used to determine the sensitivity and linear range of the bioluminescent succinate detection. Succinate standards were prepared in a demethylase/hydroxylase enzyme assay buffer consisting of 50 mM HEPES (pH 7.5), 100 µM ascorbate, 10 µM Fe(II), and 10 µM α-KG. A 15 µM succinate solution was serially diluted twofold in 12 wells of a 96-well plate to produce a dilution series from 15 to 0.014 µM plus a 0 succinate blank. Ten microliters of each dilution was transferred to an assay plate, and succinate was detected using the Succinate-Glo assay following the manufacturer’s procedure. Briefly, 10 µL of Succinate Detection Reagent I was added to the standard curve samples and incubated for 60 min at room temperature (~23 °C). Then 20 µL of Succinate Detection Reagent II was added to the reactions and incubated for 10 min at room temperature before the luminescence was recorded on a plate-reading luminometer.
JumonjiC Histone Demethylase and Fe(II)/2-Oxoglutarate-Dependent Hydroxylase Assays
Demethylation and hydroxylation reactions were carried out in 384-well white plates in 10 µL volumes using the demethylase/hydroxylase buffer, plus 10 µM substrate and 1× acetoacetyl-CoA (supplied as 100× in the kit). JMJC histone demethylases and Fe(II)/2-oxoglutarate-dependent hydroxylases were serially diluted in the demethylase/hydroxylase buffer without α-KG, and 5 µL was transferred to an assay plate. Reactions were started by the addition of 5 µL of substrate mix containing 20 µM α-KG, 2× concentration of the corresponding enzyme substrate, and 2× acetoacetyl-CoA diluted in the demethylase/hydroxylase buffer. The substrates used, as well as the reaction incubation time, temperature, and starting concentration for each enzyme, are as described in each figure. Succinate formation was detected using the Succinate-Glo assay following the manufacturer’s procedure.
Substrate Specificity Studies
For determining substrate preferences of JMJD2C (KDM4C), 10 µL reactions were carried out in demethylase/hydroxylase buffer containing 0.01% Tween-20, 10 µg/mL bovine serum albumin (BSA), and 1× acetoacetyl-CoA in the presence of 0.08 µM JMJD2C and 10 µM of the specified substrates. Briefly, 5 µL of JMJD2C demethylase diluted in a buffer composed of 50 mM HEPES (pH 7.5), 0.01% Tween-20, and 10 µg/mL BSA was transferred to 384-well volume plates. Reactions were initiated by the addition of 5 µL of 2× substrate mix containing 200 µM ascorbate, 20 µM Fe(II), 20 µM α-KG, 2× acetoacetyl-CoA, and 20 µM substrates with diverse methylation status. These components were also in the JMJD2C dilution buffer consisting of 50 mM HEPES (pH 7.5), 0.01% Tween-20, and 10 µg/mL BSA. Reactions were incubated for 60 min at 23 °C.
Substrate Km Determinations
For determining the JMJD2C Km for histone H3 Me3K9 peptide and α-KG, 10 µL reactions were performed with 0.435 µM of enzyme and a titration of the studied substrate. Reactions were incubated at 23 °C for 20 min. For the α-KG titration, 60 µM histone H3 Me3K9 peptide, 60 µM Fe(II), and a serial dilution of α-KG were used. For the histone H3 Me3K9 peptide Km determination, a titration of the substrate was performed in the presence of 60 µM Fe(II) and 60 µM α-KG. After the indicated incubation times, 10 µL of Succinate Detection Reagent I was added to the reactions and incubated for 60 min at 23 °C. Then 20 µL of Succinate Detection Reagent II was added, and after an additional 10 min of incubation, luminescence was recorded. Km values were extracted from the data after fitting to the Michaelis–Menten equation using the nonlinear regression fit in GraphPad Prism, version 7.
Enzyme Inhibition Studies
Inhibition experiments were performed in low-volume 384-well plates at 23 °C. Compounds were dissolved in DMSO at stock concentrations ranging from 5 to 100 mM. The inhibitor IOX-1 was serially diluted twofold from 50 to 0.006 µM in a 5 µL reaction containing 0.318 µM JMJD2C in 50 mM HEPES (pH 7.5), 100 µM ascorbate, 10 µM Fe(II), 10 µM [Lys(Me3)9]-histone H3 (1–21) peptide, and 1× acetoacetyl-CoA. After incubation at 23 °C for 60 min, 5 µL of Succinate Detection Reagent I was then added to the reactions and incubated for 60 min, followed by addition of 10 µL of Succinate Detection Reagent II and additional incubation for 10 min.
For mode of action experiments, the inhibitor IOX-1 was serially diluted twofold from 50 to 0.024 µM in a 10 µL reaction containing 0.159 µM JMJD2C in 50 mM HEPES (pH 7.5), 100 µM ascorbate, 0.6 µM Fe(II), 10 µM [Lys(Me3)9]-histone H3 (1–21) peptide, 1× acetoacetyl-CoA, 0.01 % Tween-20, and 50 µg/mL BSA, and α-KG was used at a final concentration of 20 or 1 µM. The inhibitor JIB 04 was serially diluted in similar conditions, except for α-KG, which was kept constant at 10 µM, and Fe(II) was used at a final concentration of 1 or 0.1 µM. For all reactions, after incubation at 23 °C for 60 min, 10 µL of Succinate Detection Reagent I was then added to the reactions and incubated for 60 min, followed by addition of 20 µL of Succinate Detection Reagent II and additional incubation for 10 min. Luminescence was then recorded in a TECAN Infinite M1000 PRO microplate reader (TECAN, Männedorf, Switzerland).
LOPAC Library Screening and Automation
The LOPAC 1280 library is a collection of 1280 pharmacologically active compounds from 56 pharmacological classes with well-characterized activities (Sigma-Aldrich). Assay-ready plates were created by acoustic droplet ejection technology using an Echo 555 instrument (Labcyte, Sunnyvale, CA). LOPAC compounds where transferred at native 10 mM concentrations in 5 nL volumes per well into 384-well low-volume plates in quadruplicate and screened at a final compound concentration of 10 µM. For screening against the JMJD2C enzyme, in addition to the wells containing LOPAC + JMJD2C, each 384-well plate contained 32 positive (0.1% DMSO in buffer + JMJD2C) and 32 negative (0.1% DMSO-only in buffer) control wells. Assay reagents (4.5 µL reaction containing 0.159 µM JMJD2C in demethylase/hydroxylase buffer with 1× acetoacetyl-CoA and 10 µM [Lys(Me3)9]-histone H3 (1–21)) were added by a Combi_nL instrument (Thermo Fisher Scientific, Waltham, MA). For screening against Succinate-Glo reagents to detect false hits, a similar scheme was used, except in the wells containing LOPAC, 5 µM succinate in buffer was used instead of the full JMJD2C reaction, and each 384-well plate also contained 32 positive (0.1% DMSO in buffer + 5 µM succinate) and 32 negative (0.1% DMSO-only in buffer) control wells. Reagent additions were time stacked to match the time required for plate luminescence reading, resulting in equal incubation times for all plates. After incubation at 23 °C for 60 min, 4.5 µL of Succinate Detection Reagent I was added to the reactions and incubated for 60 min at 23 °C, followed by addition of 9 µL of Succinate Detection Reagent II and additional incubation for 10 min at 23 °C. The reagents were also added to the plates using the Combi_nL instrument. After incubation, the luminescence was recorded using a TECAN Spark 20M multifunctional reader with Te-Cool temperature control option (22 °C).
Signal Detection and Data Analysis
All 96-well assay plates were read using the GloMax 96 Microplate Luminometer from Promega. The instrument was set to 0.5 s integration time. The same setting was used for 384-well plates when luminescence was read in a TECAN Infinite M1000 PRO or TECAN Spark 20M multifunctional reader with Te-Cool temperature control option microplate readers. To plot and analyze the data and calculate all enzyme reaction biochemical values, both Microsoft Excel and GraphPad Prism, version 7 software were used. IC50 values were determined by using a nonlinear regression fit to a sigmoidal dose–response (variable slope).
Results and Discussion
Bioluminescent Succinate Detection Principle and Formats
The succinate detection assay is performed in two steps after the completion of the Fe(II)/2-oxoglutarate-dependent oxygenase reaction; an equal volume of the first step reagent (Succinate Detection Reagent I) is added to the demethylase/hydroxylase reaction to convert the produced succinate to ATP in a coupled enzyme reaction. In the second step, the newly formed ATP is detected by the addition of twice the amount of the luciferin/luciferase reagent (Succinate Detection Reagent II) to produce bioluminescence ( Fig. 1 ). The amount of light generated is proportional to the succinate produced and the activity of the Fe(II)/2-oxoglutarate-dependent oxygenase. It should be noted that in order to convert succinate to ATP, the reaction requires acetoacetyl-CoA as a substrate for the converting enzyme. Acetoacetyl-CoA was found to increase the background of the assay if it is mixed in Succinate Detection Reagent I and left for a longer period of time before the reagent is added to the samples containing succinate. To prevent this, the protocol was optimized in a way that acetoacetyl-CoA is added to Succinate Detection Reagent I just prior to its addition to the succinate sample, or it can be added to the Fe(II)/2-oxoglutarate-dependent oxygenase reaction. We tested the effect of acetoacetyl-CoA on the activity of several JMJC demethylases and Fe(II)/2-oxoglutarate-dependent oxygenases and no adverse effect was observed when acetoacetyl-CoA was part of the enzymatic reaction instead of Succinate Detection Reagent I ( Suppl. Fig. S1 ). We have optimized the time required for the conversion of succinate to ATP in the first step and ATP to light in the second step and found that an average of 60 and 10 min, respectively, is sufficient to convert succinate concentration up to 15 µM ( Fig. 2A,B ). This detection range meets the requirement of a wide range of enzyme activities (data not shown). The addition pattern for optimal performance follows the 1:1:2 ratio of the demethylase/hydroxylase reaction–Succinate Detection Reagent I–Succinate Detection Reagent II, with volume ratios of 25:25:50 µL used for 96-well plates and 10:10:20 or 5:5:10 µL used for 384-well plates ( Fig. 2C ).
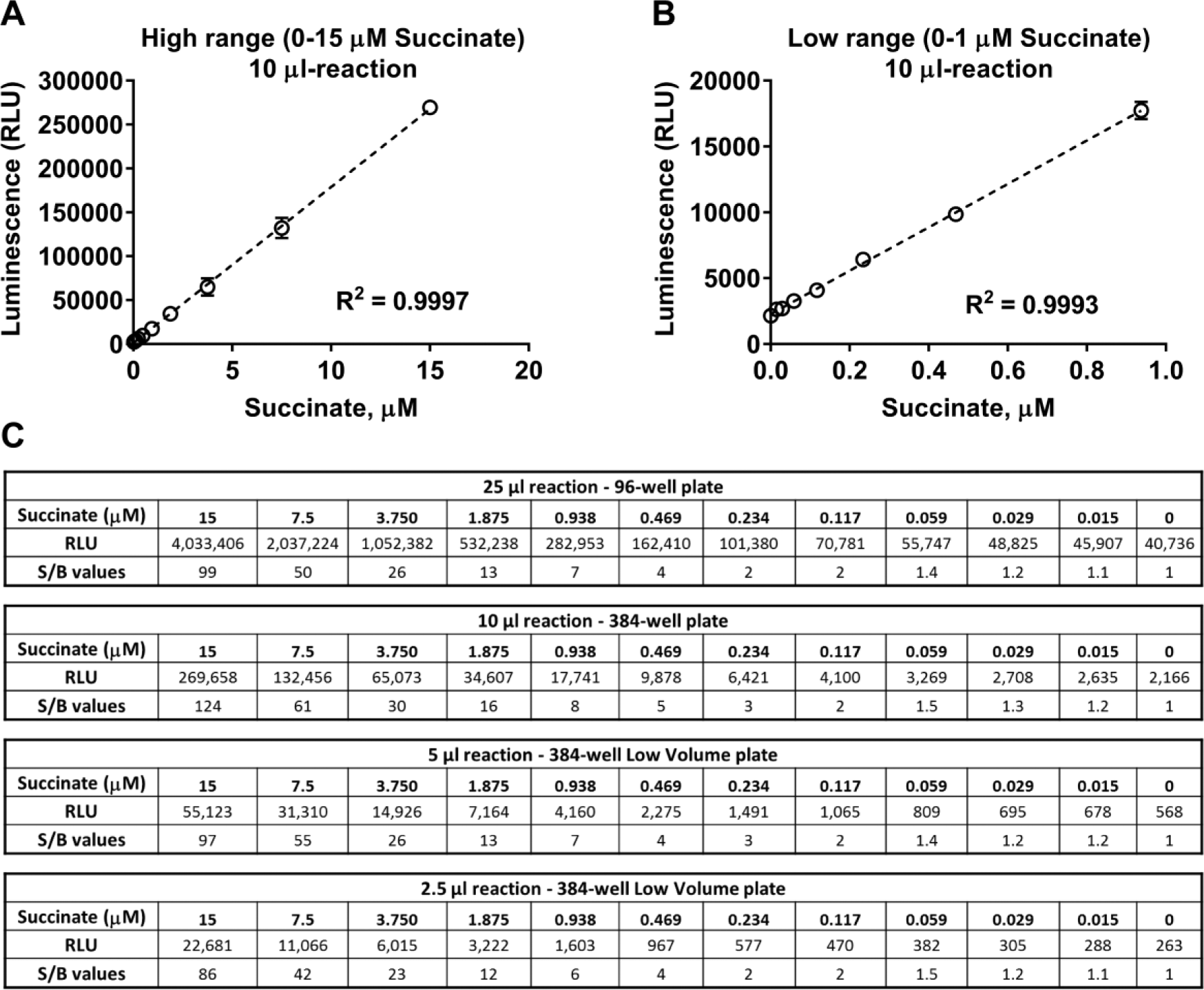
Linearity and sensitivity of bioluminescent succinate detection assay. Succinate titration curves were performed in 384-well plates in the concentration ranges shown, and the Succinate-Glo assay was performed at the 1:1:2 volume ratio of the reaction to the reagents as described in Materials and Methods. (
Assay Sensitivity and Linearity
In order to assess the linearity and sensitivity of the bioluminescent succinate detection, we performed a standard curve of succinate and detected the light generated by each concentration following the assay procedure described in Materials and Methods. Figure 2 shows the standard curves generated, the luminescence relative light units (RLU), and the signal-to-background ratios (S/Bs) for each succinate concentration. There is a linear response with increasing concentrations of succinate up to 15 µM ( Fig. 2A ) with an R2 value of 0.999 and a limit of detection of approximately 120 nM with an S/B value of 2 ( Fig. 2B,C ). The stability of the signal was monitored by recording the luminescence emitted from the same standard curve every hour, and it was found that the RLU signals remain stable for at least 3 h at room temperature ( Suppl. Fig. S2 ).
The results of succinate standard curves show that the range of detection and the sensitivity would meet the requirements of a suitable assay for a broad range of enzyme activities, and because of its homogeneous feature (add and read with no washes or liquid transfers), it is an adequate assay for high-throughput screening.
Characterization of Fe(II)/2-Oxoglutarate-Dependent Oxygenase Activities
Because the assay detects succinate production, it would be capable of monitoring the activity of all Fe(II)/2-oxoglutarate-dependent oxygenases that generate succinate as a reaction product. To evaluate bioluminescent succinate detection as a reliable readout for demethylase/hydroxylase enzymatic activities and ensure the universality of the assay, we tested several members of the dioxygenase superfamily. Represen-tative members of the KDM histone demethylase subfamilies, such as KDM3 (JMJD1A and JMJD1B), KDM4 (JMJD2A, JMJD2B, JMJD2C, JMJD2D, and JMJD2E), KDM5 (JARID1A, JARID1B, and JARID1C), KDM6 (UTX and JMJD3), and KDM7 (JHDM1D and PFH8), as well as representatives of DNA hydroxylases/demethylases (TET1 and ALKBH3), RNA hydroxylases/demethylases (FTO), and prolyl-hydroxylases (EGLN1), were incubated with their respective substrates, and succinate formation was detected. We found that in the presence of the corresponding substrate, a substantial amount of succinate was formed by the histone demethylases and Fe(II)/2-oxoglutarate-dependent hydroxylases in a concentration-dependent manner, indicating that succinate detection can be used for monitoring such enzymatic activities regardless of the substrate methylation state or structure (i.e., peptide, DNA, or RNA) (
Fig. 3
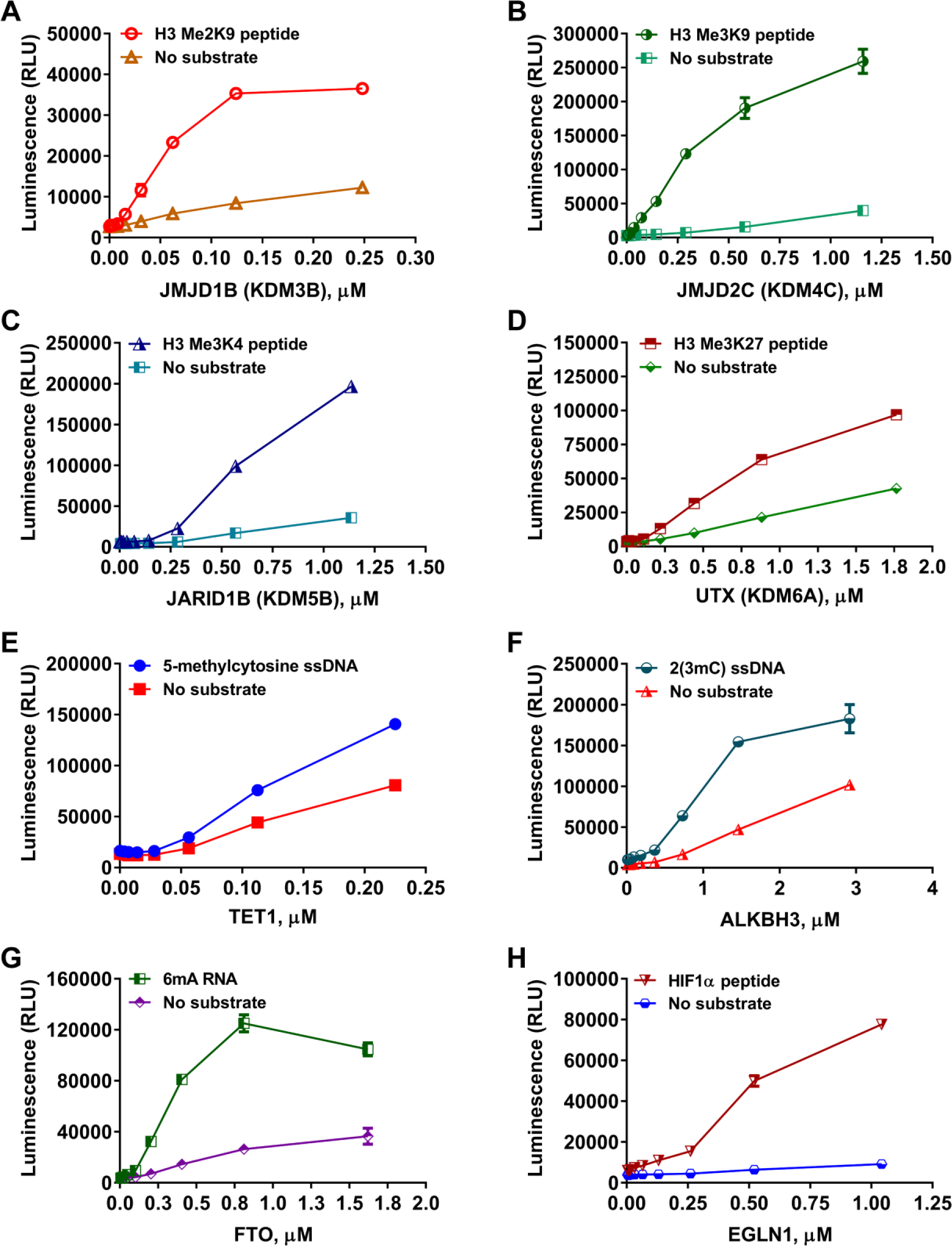
Universality of bioluminescent succinate detection assay toward JumonjiC histone demethylases and Fe(II)/α-KG-dependent hydroxylases. Enzyme titrations of representative members of the JMJC histone demethylase and of the Fe(II)/α-KG-dependent hydroxylase superfamily in the presence or absence of 10 µM of the indicated substrates. (
Because this assay can detect the activity of any enzyme that produces succinate, regardless of the substrate chemical structure, we tested several JumonjiC histone demethylases using full-length histone H3 with diverse methylation positions and states. Succinate formation was well detected when protein substrates were used in the demethylase reaction ( Suppl. Fig. S4 ). It is worth noting that succinate formation was also detected across all enzymes tested in the absence of substrate with varying lower levels of activity than in the presence of the substrate ( Fig. 3 and Suppl. Figs. S3 and S4 ). This is possibly due to the uncoupled decarboxylation of 2-oxoglutarate, as previously reported.28,29
Determination of Enzyme Kinetic Parameters
In order to characterize the biochemical activity of any enzyme, it is important to learn about its behavior toward different substrates and identify its optimal substrate. First, we monitored succinate formation to define substrate specificity for one representative of the JumonjiC histone demethylases. The JumonjiC histone demethylase JMJD2C was screened against three histone H3 peptides with different methylation states ( Fig. 4A ). In agreement with published literature, JMJD2C demonstrated a higher preference for the trimethylated histone H3K9(Me3) over the dimethylated H3K9(Me2) peptide substrate. In contrast, no increased activity was observed when the unmethylated histone H3 peptide substrate was used in comparison with the control with no peptide ( Fig. 4A ). Because this assay can be used with a wide range of substrates and substrate concentrations, we measured substrate Km values. The results shown in Figure 4B,C are representative of data for JMJD2C, with histone H3 Me3K9 peptide and α-KG showing Km values of 4.5 and 2 µM, respectively.
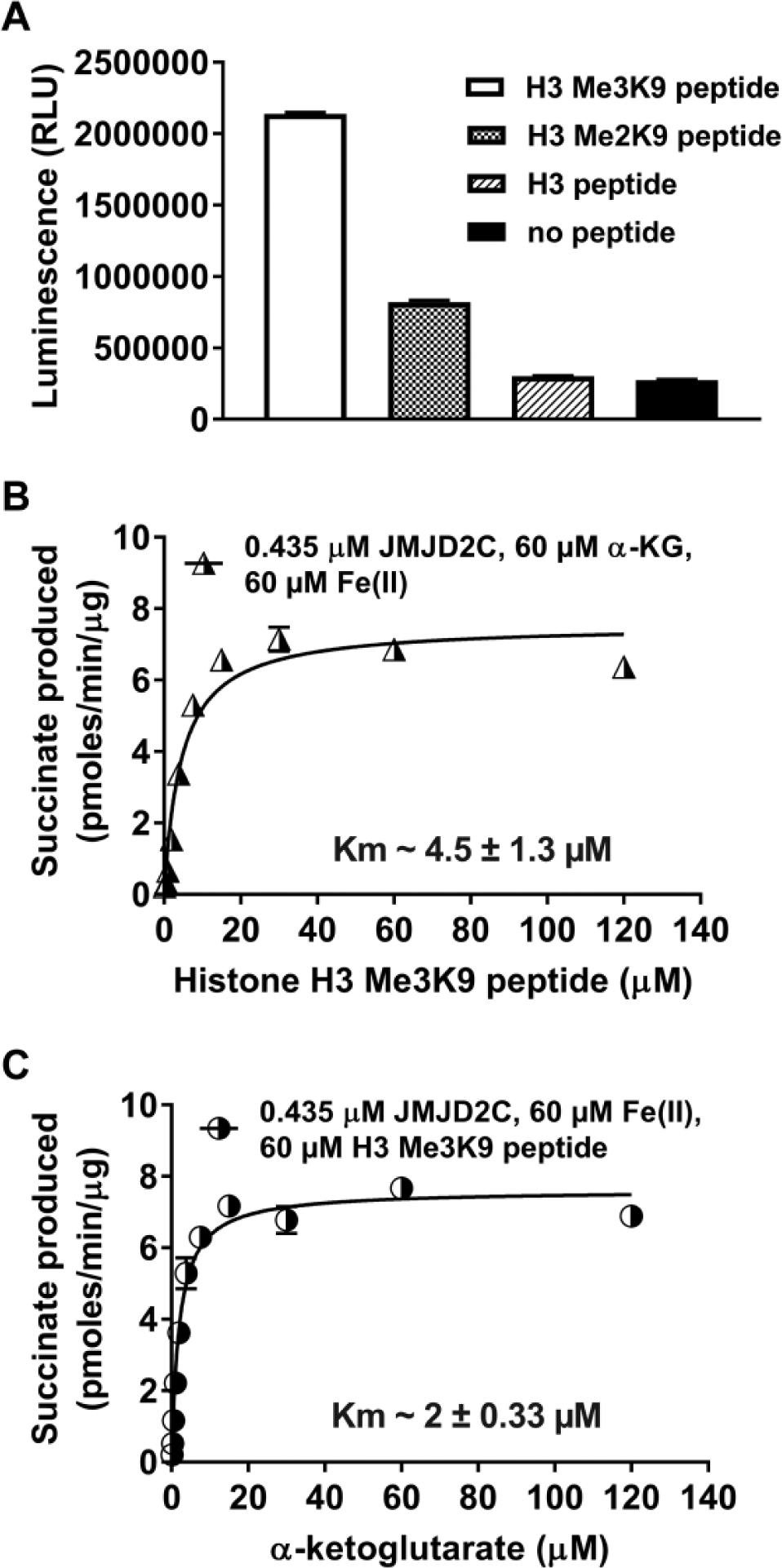
Substrate specificity and kinetic analysis of JMJD2C demethylase reactions. (
The bioluminescent succinate detection assay monitored several Fe(II)/2-oxoglutarate-dependent oxygenases with different activity levels and substrate requirements ( Figs. 3 and 4 and Suppl. Figs. S3 and S4 ). This demonstrates the universality of the assay and an advantage over assays that have different reagent requirements, depending on the substrate modification (e.g., methylation-specific antibodies or labeled substrates). To perform these assays using existing systems, it is necessary to modify or label either the substrate or the reagent needed to capture or monitor the product. These requirements are obviated for the bioluminescent succinate detection assay, which makes it easy to use, and because of its universality, the assay saves not only time but also money for formatting the different reagents specific to the enzyme to be studied. Moreover, in many enzyme reactions where the substrate is not known, such as for a novel demethylase or hydroxylase, monitoring succinate formation is also advantageous, as the enzymes can produce some levels of succinate in the absence of substrate, allowing some initial studies of these enzymes. The assays that detect the substrate modification cannot be used in this instance due to the absence of required substrate information. Furthermore, the detection of these substrate-independent enzyme activities could be used in inhibitor screening or other substrate determinations for Fe(II)/2-oxoglutarate-dependent oxygenases similarly to what was shown previously for kinases or glycosyltransferases using bioluminescent ADP or UDP detection assays, respectively.30,31
Enzyme Inhibition Assays
To evaluate the potential for inhibitor identification using the bioluminescent succinate detection assay, we evaluated JMJD2C inhibition in the presence of increasing concentrations of the histone demethylase JMJD inhibitor IOX-1. 32 As shown in Figure 5A , JMJD2C was inhibited by IOX-1 in a dose-dependent fashion, with an IC50 of 0.65 µM, which is similar to what was previously reported, 32 demonstrating succinate detection as a useful strategy for inhibitor identification.
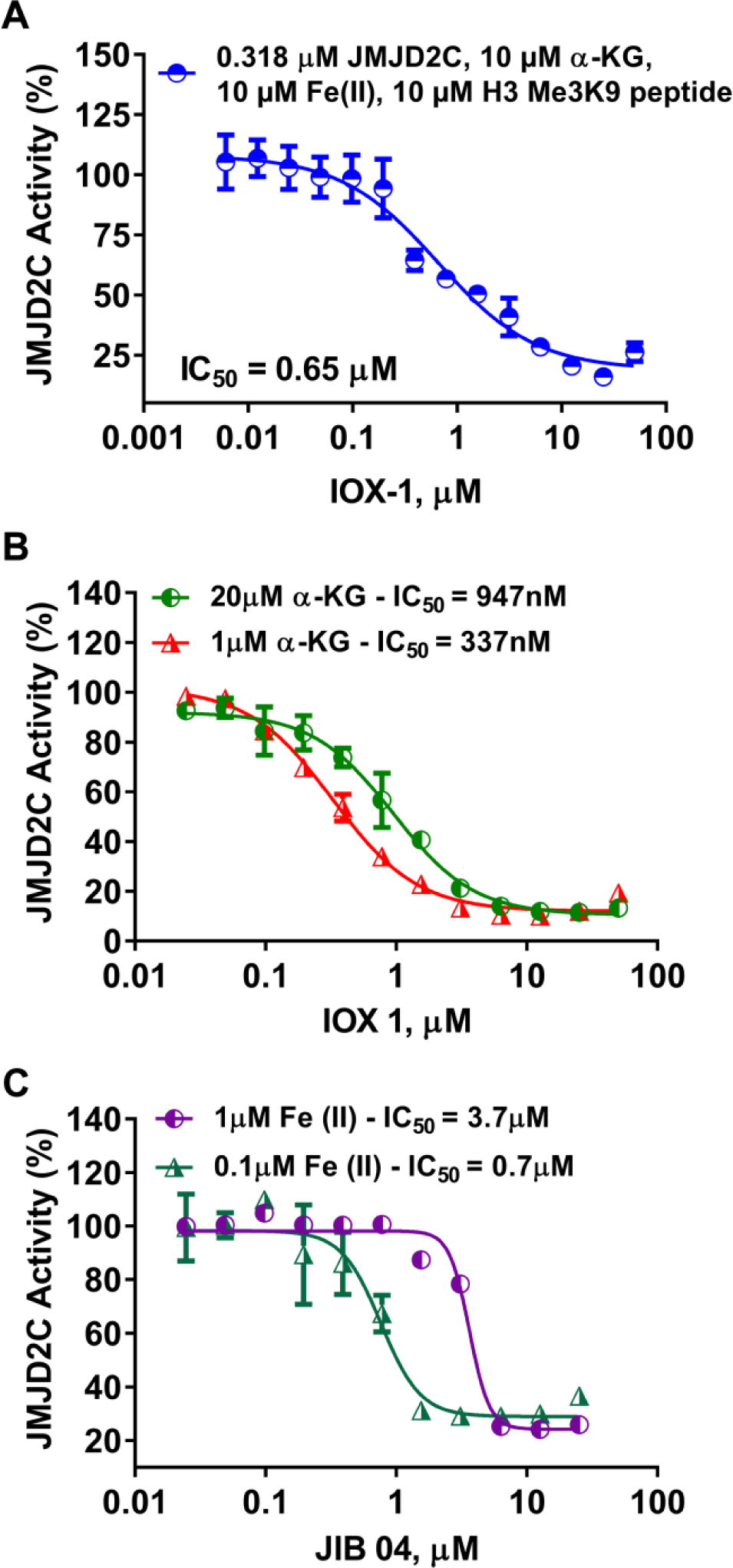
Detection of JMJC demethylase inhibitor effect and mode of action. (
To study inhibitor mode of action, the inhibitor can be titrated in the presence of different concentrations of the substrates or cofactors. If IC50 is higher at a greater concentration of a substrate or cofactor, this indicates that the inhibitor binds the same site as the agent that causes the increase. Because the bioluminescent assay does not require modification of any of the substrates or cofactors needed in the reaction, it is ideal for determining if a Fe(II)/2-oxoglutarate-dependent dioxygenase inhibitor is competitive with substrate, Fe(II), or α-KG. To test this, we used two inhibitors, IOX-1, a known α-KG competitive inhibitor of JMJD2C, and JIB 04, which is known to be an Fe(II) pocket competitive inhibitor. Serial dilutions of these compounds were incubated with JMJD2C, and the reaction was initiated by the addition of a substrate mixture containing low or high concentrations of 2-oxoglutarate (1 or 20 µM) or Fe(II) (0.1 or 1 µM). As predicted, Figure 5B,C showed that there is a shift in the inhibition curves and an increase in IC50 values when the 2-oxoglutarate or Fe(II) concentrations increased, confirming previous reports and demonstrating the utility of the assay for this type of mode of action study.16,33
During drug discovery endeavors, it is always necessary to assess the selectivity of the lead compounds for the enzyme target to predict any side effects later in the clinic. Bioluminescent succinate detection is ideal for selectivity profiling because it is universal and can detect the activity of enzymes with different reaction condition requirements simultaneously with the same reagents. Other approaches can be challenging to set up for profiling experiments, as they require different modified substrates or antibodies for each assay. To test the ease of use of the bioluminescent assay in a profiling experiment, we used three published JumonjiC histone demethylase and Fe(II)/2-oxoglutarate-dependent hydroxylase inhibitors against one enzyme each from two subfamilies. The JumonjiC histone demethylase JMJD2C and the RNA hydroxylase FTO were incubated with the inhibitors daminozide, 2,4-PDCA, and IOX-1.17,32,34 With the exception of the plant growth regulator daminozide (KDM2/7 inhibitor), which did not inhibit JMJD2C and FTO as expected ( Suppl. Fig. S5C ), 34 the other compounds successfully inhibited the activities of these enzymes with different potencies ( Suppl. Fig. S5A,B ). In addition, 2,4-PDCA demonstrated a higher potency for JMJD2C over FTO, with an IC50 of 47.89 nM versus 1.27 µM, respectively, confirming the succinate assay capability of determining compound selectivity ( Suppl. Fig. S5A ). Overall, these results demonstrate the usefulness of monitoring succinate formation as a method for identifying novel demethylase/hydroxylase inhibitors, for determining inhibitor mode of action and potency, and for inhibitor profiling studies.
Compound Library Screening
Since it was demonstrated that succinate detection is suitable for measuring Fe(II)/2-oxoglutarate-dependent dioxygenase inhibition, we tested it in a compound library screen to determine its suitability for inhibitor discovery. We were particularly interested in determining the false hit rate. To screen for chemicals that could inhibit the enzyme-coupled reactions responsible for the conversion of succinate to ATP, compounds from the LOPAC 1280 library (10 µM final concentration) were incubated with Succinate-Glo in assay buffer containing all components required for the demethylase/hydroxylase reaction, and the assay reaction was initiated by adding 5 µM succinate. The assay was performed in quadruplicate using 384-well low-volume assay plates. As shown in Figure 6A , only four compounds (0.31% false hits) impaired the bioluminescent succinate detection above 50%. The four false hits are inhibitors of several luciferase-based assays previously tested (data not shown). The structure of these compounds is shown in Suppl. Table S1 . The very few false hits observed with the LOPAC 1280 library indicate that the assay is resistant to a diverse set of pharmacologically active compounds. In addition, the coefficient of variation (CV) between wells was determined to be 3.8%, indicating limited well-to-well variability. Moreover, to demonstrate the robustness of this assay in a screening setting, the statistical Z′ factor parameter was determined. This parameter takes into account the standard deviation and the means of the positive and negative controls, and assays with Z′ values higher than 0.50 are considered robust. 35 The Z′ factor observed for the assay was 0.84 ( Fig. 6A ). Together, these results indicate that the bioluminescent succinate detection assay is robust and reproducible with minimal compound interference, and therefore it is a suitable method for high-throughput screening applications.
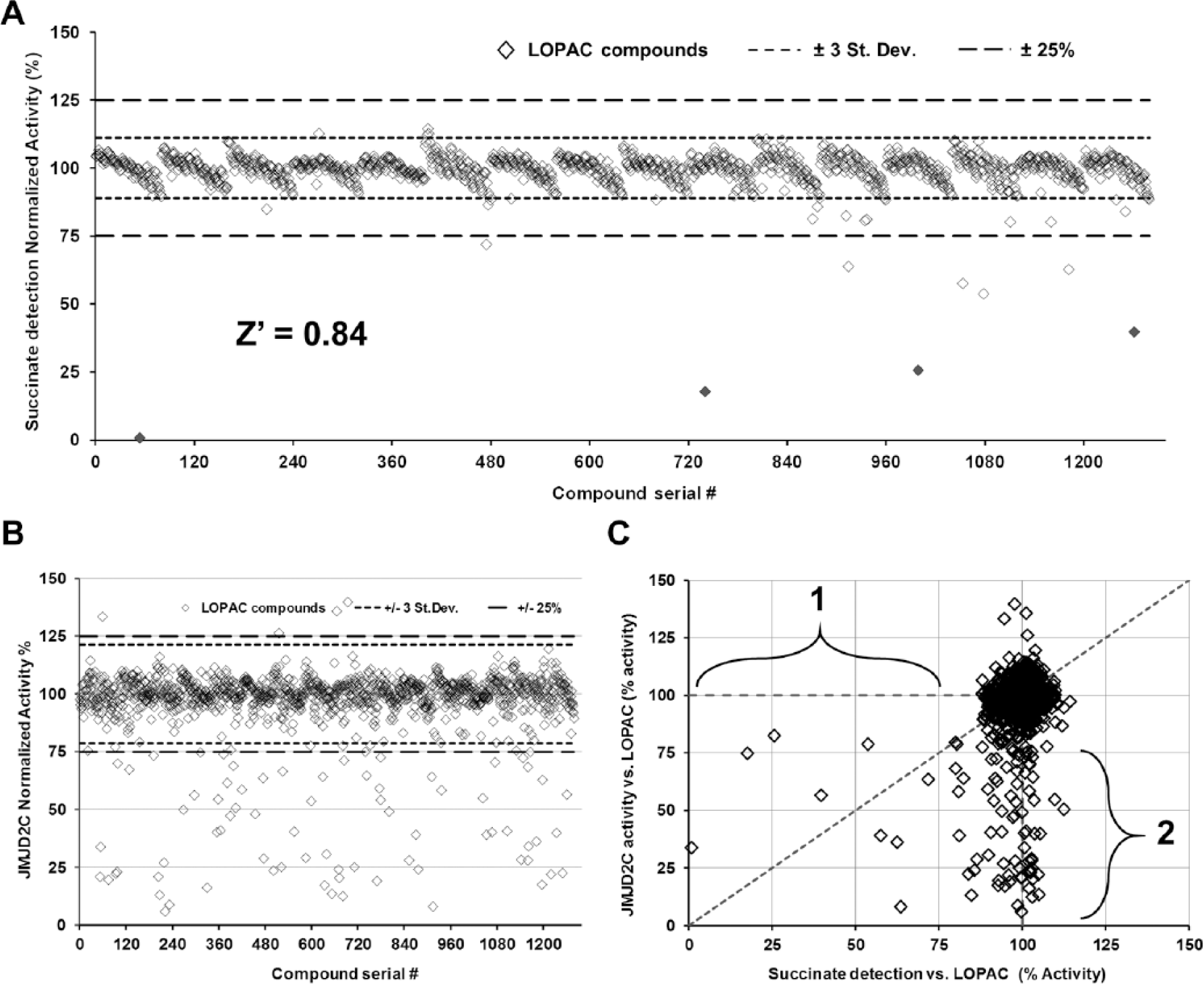
Determination of the robustness of the bioluminescent succinate detection assay for high-throughput screening. (
Finally, in order to validate Succinate-Glo utility as a method for high-throughput screening for enzyme inhibitors, we performed an assay using the LOPAC 1280 library against JMJD2C demethylase. The assay successfully identified several compounds that affected JMJD2C activity (
Fig. 6B,C
Publicly available data about these compounds were searched on the PubChem Compound database (https://www.ncbi.nlm.nih.gov/pccompound/). 38 The search results indicate that the majority of the hits tested have been previously reported against another JMJC demethylase, JMJD2E (PubChem Assay ID 2147). 33 Interestingly, there was no prior description of hispidin as a JumonjiC histone demethylase inhibitor. In order to confirm these compounds, including hispidin, as JMJD2E inhibitors, we tested them against JMJD2E. As predicted, including hispidin, all compounds inhibited JMJD2E with IC50 values similar to those of JMJD2C ( Suppl. Fig. S6 ). These results also showed that the Succinate-Glo assay can be automated using liquid dispensing instruments to dispense the small volumes required for high-throughput screening. This allows identification of inhibitors from compound libraries in a robust and easy fashion.
We characterized a homogeneous bioluminescent succinate detection method and demonstrated its utility with JumonjiC histone demethylases and other representative members of the Fe(II)/2-oxoglutarate-dependent dioxygenase superfamily. The assay is performed in a two-step “add-mix-read” format, converting the succinate product into ATP, which is subsequently detected by a thermostable luciferase system to generate a bioluminescent signal. The method detects succinate from nanomolar concentrations to 15 µM, and its signal is stable for at least 3 h at room temperature.
We were able to detect enzymatic activities from several members of the Fe(II)/2-oxoglutarate-dependent dioxygenase superfamily, demonstrating that succinate detection can be used as a universal method regardless of substrate requirements. We also demonstrated that it could be used to determine substrate requirements and preferences, as in the case of JumonjiC histone demethylases, as well as to determine the apparent kinetic values of the substrates used in the demethylase/hydroxylase reactions. In addition, the bioluminescent succinate detection method can be useful for inhibitor mode of action studies, as well as profiling experiments. Furthermore, its robustness and resistance to compound interference against the LOPAC 1280 library exemplified the utility of this assay for high-throughput screening applications. Finally, primary hits identified against JMJD2C were validated in reconfirmation and counterscreen experiments, and in query searches using the PubChem database, and were also tested on JMJD2E demethylase. To our knowledge, this is the first report that hispidin is a JumonjiC histone demethylase inhibitor.
A variety of methods are currently available for detecting JumonjiC histone demethylase activities, including fluorescent-based enzyme-coupled methods for formaldehyde detection, homogeneous time-resolved fluorescence (HTRF)– and AlphaScreen-based methods, mass spectrometry–based methods, and enzyme-linked immunosorbent assay (ELISA)/Western blot–based assays. Some of the drawbacks of the aforementioned methods are the risk of assay interference due to compound fluorescence, the requirement for expensive instrumentation and training of personnel, low assay throughput, and the necessity of multiple steps. Most importantly, very few options exist for a robust assay that could be used with Fe(II)/2-oxoglutarate-dependent dioxygenases other than JumonjiC histone demethylases. Therefore, results from our group and others 39 demonstrate the usefulness of a bioluminescent-based succinate detection method for in vitro characterization of virtually any Fe(II)/2-oxoglutarate-dependent dioxygenase, and this approach could have a significant impact on subsequent studies of members of this enzyme superfamily.
Footnotes
Acknowledgements
We thank Jim Cali for his critical reading of the manuscript and Kevin Hsiao for invaluable input and support throughout this study.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
All authors are employees of Promega Corporation, which manufactures and markets the Succinate-Glo assay kits used in the research reported here. The authors declared no other potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.