
Abstract
Splicing is an important eukaryotic mechanism for expanding the transcriptome and proteome, influencing a number of biological processes. Understanding its regulation and identifying small molecules that modulate this process remain a challenge. We developed an assay based on time-resolved fluorescence resonance energy transfer (TR-FRET) to detect the interaction between the protein NHP2L1 and U4 RNA, which are two key components of the spliceosome. We used this assay to identify small molecules that interfere with this interaction in a high-throughput screening (HTS) campaign. Topotecan and other camptothecin derivatives were among the top hits. We confirmed that topotecan disrupts the interaction between NHP2L1 and U4 by binding to U4 and inhibits RNA splicing. Our data reveal new functions of known drugs that could facilitate the development of therapeutic strategies to modify splicing and alter gene function.
Introduction
Most genes in the human genome comprise multiple exons interspersed with introns, necessitating splicing to form mature mRNA and protein products. 1 The process of alternative splicing is a central mechanism for the regulation of gene expression, allowing increased proteomic complexity in higher eukaryotes. 2
Splicing is catalyzed by a large ribonucleoprotein complex called the spliceosome, which is formed by the interaction of small nuclear ribonucleoprotein particles (snRNPs) U1, U2, U4, U5, and U6, appropriate regions of the pre-mRNA and numerous non-snRNP proteins. 3
NHP2L1 (15.5K) is one of the non-snRNP proteins4,5 that binds to and stabilizes a kink turn (K turn) in the U4 5′ stem loop (U4 5′SL) and is required for the subsequent recruitment of the human PrP31 protein to U4/U6.6,7 This interaction between NHP2L1 and U4 has been shown to play a critical role in the late stage of spliceosome assembly. 3
We developed an assay based on the time-resolved fluorescence resonance energy transfer (TR-FRET) method to detect the interaction between NHP2L1 and U4 5′SL. We used this assay to identify small molecules that interfere with this interaction in a high-throughput screening (HTS) campaign, and validated the results by orthogonal methods. Topotecan and other camptothecin derivatives were among the top hits, and we confirmed that topotecan disrupts the interaction between NHP2L1 and U4 5′SL by binding to U4, and documented that this disruption leads to inhibition of RNA splicing.
Materials and Methods
Reagents
Cy5-U4 5′SL (5′Cy5-AUCGUAGCCAAUGAGGUUUAUCCGAGGCGCGAU) and biotin-U4 5′SL (5′Biosg-AUC GUAGCCAAUGAGGUUUAUCCGAGGCGCGAU) were synthetized by Integrated DNA Technologies (Coralville, IA). Histidine-tagged NHP2L1 (His-NHP2L1) was obtained as described below. Terbium-labeled anti-His-tag antibody (Tb-anti-His antibody), bovine serum albumin (BSA), and dithiothreitol (DTT) were purchased from Invitrogen (Waltham, MA). Tris-HCl (pH 7.6, 1 M) was purchased from TEKNOVA (Hollister, CA). NaCl (5 M) and DMSO were purchased from Fisher Scientific (Pittsburgh, PA). Triton X-100, (S)-camptothecin, and 9-NO2-10-OH-(S)-camptothecin were purchased from Sigma (St. Louis, MO). Topotecan, SN-38, 9-NH2-(S)-camptothecin, 10-OH-(S)-camptothecin, and irinotecan were purchased from Cayman Chemical (Ann Arbor, MI). N-Desmethyl topotecan and 7,11-diethyl-10-OH-(S)-camptothecin were purchased from Toronto Research Chemicals (Toronto, ON) tRNA was purchased from Roche Diagnostics (Basel, Switzerland). Low-volume, 384-well black assay plates were obtained from Corning, Inc. (Tewksbury, MA)
Chemical Library
The St. Jude Children’s Research Hospital Food and Drug Administration (FDA) drug and bioactive library was assembled from four commercial suppliers, MicroSource (Gaylordsville, CT), Prestwick Chemical (San Diego, CA), Sigma, and Selleckchem (Houston, TX), with a total of 10,173 compounds (4617 unique small molecules), as previously described.8–11
Protein Expression and Purification
NHP2L1 was cloned into pET-15b vector by using XhoI and BamHI restrictions sites. This plasmid was used to transform BL21 (DE3) cells (Novagen) and grown in Luria-Bertani (LB) medium at 37 °C in the presence of ampicillin (100 µg mL–1) to an optical density at 600 nM (OD600) of 0.6. Protein was induced by addition of 0.2 mM isopropyl-β-
TR-FRET Assay
Various concentrations of Cy5-U4 5′SL were incubated with 2 nM terbium-anti-His in 20 µL binding buffer (20 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% Triton X-100, 0.5 mg/mL tRNA, 0.01% BSA, and 2 mM DTT) with or without 2 nM His-NHP2L1 in low-volume, 384-well black plates with a final DMSO concentration of 0.5% for each well. After centrifugation, shake, and room temperature incubation, the TR-FRET signals (fluorescence emission ratio of 10,000 × 665 nm/620 nm) for each well were collected with a PHERAstar FS (BMG Labtech, Durham, NC) using a 340 nm excitation filter, 100 µs delay time, and 200 µs integration time. The reactions were incubated between 15 and 300 min. Data were plotted with GraphPad Prism 6.07 (GraphPad Software, Inc., La Jolla, CA) by fitting into the one site a total binding equation to derive the Kd value of the interaction between Cy5-U4 5′SL and His-NHP2L1.
HTS Using TR-FRET Assay
For the primary screen, stock compound (the St. Jude Children’s Research Hospital bioactive and FDA drug library with a total of 10,173 compounds and 4617 unique ones) solutions (10 mM in DMSO) or DMSO alone (vehicle control) were transferred to the individual wells in low-volume, 384-well black assay plates containing 20 µL of complete U4-NHP2L1 interaction mixture (2 nM terbium-anti-His, 2 nM His-NHP2L1, and 2 nM Cy5-U4 5′SL in binding assay buffer) or partial U4-NHP2L1 interaction mixture (complete U4-NHP2L1 interaction mixture without His-NHP2L1) by using a V&P (San Diego, CA) 384-well pin tool at 30 nL/well to give a final compound concentration of 15 µM. The final DMSO concentration was 0.15% for each well. The DMSO control wells with complete U4-NHP2L1 interaction mixture and the DMSO wells with partial U4-NHP2L1 interaction mixture were used as negative (0% inhibition) and positive (100% inhibition) controls, respectively. After a 45 min room temperature incubation, the TR-FRET signals (fluorescence emission ratio of 10,000 × 665 nM/620 nm) for each well in individual assay plates were collected. Compounds with percent inhibition of ≥40% were selected for the dose-responsive analysis (10 concentrations, following a 1:3 serial dilution scheme with final concentrations ranging from 3.5 nM to 70 µM, in triplicate) under similar assay conditions as the primary screen with a final DMSO concentration of 0.7% for all assay wells. The activity data for each small molecule were normalized to those of positive and negative controls and fit into sigmoidal dose–response curves, if applicable, to derive IC50 values with GraphPad Prism 6.07. To confirm the activity of topotecan and other camptothecin derivatives, powder compounds were solubilized in DMSO as 10 mM stocks and dilutions of individual chemicals (16 concentrations, following a 1:2 serial dilution scheme with final concentrations ranging from 3.1 nM to 100 µM, in triplicate) were used in the TR-FRET assay. The DMSO control (1.0%) wells with and without 2 nM His-NHP2L1 were used as negative control (0% inhibition) and positive control (100% inhibition), respectively.
Chemical Similarity Clustering
The hierarchical molecular network graph relates biological activity in the NHP2L1 primary assay to chemical structure. Leaf nodes represent the actual molecules tested and are color-coded according to percent inhibition (blue = higher inhibition). Intermediate nodes represent scaffolds at various levels of chemical abstraction, and edges between nodes signify chemical similarity. To define the network, input molecules were first abstracted to Murcko scaffolds 12 using the Generate Fragments module in Pipeline Pilot (v. 9.2.0, Biovia, Inc., San Diego, CA). Murcko scaffolds were clustered according to Tanimoto similarity using the FCFP_4 fingerprint (cluster center selection method = maximum dissimilarity; maximum distance = 0.6; distance metric = Euclidean; recenter count = 10). In our experience, the above two-step procedure does the best job of clustering together compounds with high global similarity. However, Murcko fragmentation reduces molecules to contiguous ring systems plus chains that link two or more rings, and will sometimes fail to identify relationships between molecules that are not similar according to a global chemical fingerprint, but that nonetheless share a common substructure. To address this limitation, we further abstracted the Murcko scaffolds using the Schuffenhauer algorithm, 13 which hierarchically decomposes molecules to a single-ring system according to a defined set of rules. The network graph relating molecules and intermediate scaffolds was visualized using Cytoscape (v. 3.4.0). 14
Electrophoretic Gel Mobility Shift Assay
NHP2L1 and biotinylated U4 5′SL were mixed in 10 µL of buffer reaction (containing 20 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% Triton X-100, 2 mM DTT, and 0.5 g/L tRNA) and incubated at room temperature for 45 min. Reactions were loaded onto 6% DNA retardation gel (Invitrogen) and separated in 0.5× TBE buffer (Invitrogen) at 100 V for 60 min at room temperature and transferred to a nylon membrane. After cross-linking, the complexes were detected using the chemiluminescent nucleic acid detection module (Thermo Scientific, Waltham, MA) according to the manufacturer’s instructions.
Biotinylation of NHP2L1
NHP2L1 was minimally biotinylated by reaction with EZ-Link Sulfo-NHS-LC-LC-Biotin (Thermo Scientific). 15 The biotin reagent was added to the protein at a 0.5:1 molar ratio, and the reaction was incubated on ice for 3 h. Unconjugated biotin was removed by processing the reaction sample through two Zeba Spin Desalting Columns (Thermo Scientific) that had been equilibrated with storage buffer (20 mM Bis-Tris, pH 6.5, 100 mM NaCl, 2 mM TCEP, and 20% glycerol). The biotinylated protein was aliquoted, flash-frozen in liquid N2, and stored at −80 °C for later use.
Surface Plasmon Resonance Experiments
Surface plasmon resonance (SPR) experiments were performed at 20 °C on a SensiQ Pioneer optical biosensor (SensiQ Technologies, Oklahoma City, OK). Neutravidin (Thermo Scientific) was immobilized on carboxylated polysaccharide-coated gold chips (COOH5 chips; SensiQ Technologies) by routine amine coupling chemistry in immobilization buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, and 0.005% Tween 20). Carboxyl groups on the hydrogel were activated with N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS), and neutravidin was injected in 10 mM sodium acetate, pH 5.0, until immobilization levels of ~10,000–11,500 RU/channel were achieved. Any remaining active sites were blocked with ethanolamine. Biotin-NHP2L1 and U4 5′SL-biotin (Integrated DNA Technologies) were injected on separate channels until 1330 and 1014 RU were captured, respectively. One channel on the chip was immobilized with neutravidin without adding protein or RNA and was used as a reference cell. Topotecan was prepared in binding buffer (20 mM Bis-Tris, pH 6.5, 200 mM NaCl, 2 mM TCEP, 0.01% Triton X-100, 10% glycerol, and 5% DMSO) at a concentration of 50 µM and was injected in duplicate at a flow rate of 200 µL using the OneStep injection feature, which exploits Taylor dispersion to generate a concentration gradient that provides a full titration of analyst in a single injection.16,17 Buffer-only (blank) injections were included in the experiment to account for instrumental noise. The data were processed, double-referenced, solvent corrected, and analyzed using the software package Qdat (v. 2.6.1.8, SensiQ Technologies). Equilibrium dissociation constants (Kd) were determined by globally fitting the data to a 1:1 equilibrium affinity model.
Nuclear Magnetic Resonance Spectroscopy and Resonance Assignments
Uniformly labeled NHP2L1 with 15 N or with 15 N and 13 C was produced in MOPS minimal medium supplemented with 15 NH4Cl (Cambridge Isotope Laboratories, Inc., Tewksbury, MA) or 15 NH4Cl and 13 C6H12O6 (Sigma). Cells were harvested and lysed and proteins were purified following the same procedures as mentioned above in the “Protein Expression and Purification” section. Samples were prepared for nuclear magnetic resonance (NMR) spectroscopy in a buffer consisting of 100 mM sodium chloride, 100 mM sodium phosphate, pH 5.0, 10% D2O, and 2% DMSO-d6. 15 N isotope–labeled NHP2L1 alone or in the presence of unlabeled U4 5′SL (stoichiometric ratio NHP2L1:U4 5′SL of 1:1) was titrated with increasing amounts of topotecan to stoichiometric ratios (NHP2L1 or NHP2L1 + U4: topotecan) of 1:0, 1:2, 1:10, and 1:20. Spectral changes were monitored by one-dimensional (1D) 1 H and two-dimensional (2D) [ 1 H, 15 N] heteronuclear single-quantum correlation (HSQC) or transverse relaxation-optimized spectroscopy (TROSY) spectra. Unlabeled U4 5′SL was titrated with increasing amounts of topotecan to stoichiometric ratios (U4: topotecan) of 1:0, 1:1, 1:2, 1:5, and 1:10. Spectral changes were monitored by 1D 1 H NMR spectra. NMR experiments were recorded at 25 °C on either a 600 or 800 MHz Bruker (Billerica, MA) Avance spectrometer equipped with 5 mm triple resonance cryoprobe (TCI) using a single-axis pulse field gradient. Although the assignments of NHP2L1 were closed to the data in BMRB ID 7249, 18 several resonances were shifted because of the pH (5.0) and the buffer conditions. Therefore, we assigned the backbone resonances of NHP2L1 from the analysis of 2D [ 15 N, 1 H] HSQC along with HNCA and HNCACB spectra on 13 C, 15 N-labeled NHP2L1. 2D [ 15 N, 1 H] HSQC or TROSY spectra were used for monitoring chemical shift perturbations of NHP2L1 resonances, U4 5′SL at 100 µM concentration, and various concentrations of topotecan using 64 scans. Only the 2D [ 15 N, 1 H] TROSY of the NHP2L1 complex with U4 and topotecan at a molar ratio of 1:1:20 was measured using 400 scans. Binding of U4 with topotecan was monitored using both 1D and 2D [ 1 H, 1 H] TOCSY (60 ms mixing time) and nuclear Overhauser enhancement spectroscopy (NOESY) (100 ms mixing time) spectra at 100 µM U4 and varying concentrations of topotecan (data not shown). 1D waterlogsy spectra were measured using 100 µM topotecan with and without NHP2L1 (5 µM) with 128 scans. All the NMR data were processed using Topspin 3.2 software and analyzed using Topspin or CARA. 19
Cell Culture
Human embryonic kidney 293 cells
NHP2L1 Overexpression
HEK293 cells were infected with lentiviral transduction particles overexpressing green fluorescent protein (GFP) or NHP2L1-GFP. GFP-positive cell clones were isolated by cell sorting.
Splicing Assay
The HEK293 cells were transfected with pTN23 splicing reporter using Gene Jammer (Stratagene, La Jolla, CA). The plasmid pTN23 contains reporter genes for β-galactosidase and luciferase separated by an exon–intron–exon cassette. 20 Cells were harvested 24 h after transfection with or without topotecan and lysed in reporter lysis buffer. Luciferase and β-galactosidase activities were measured using the dual-light system (Applied Biosystems, Waltham, MA). The ratio of luciferase to β-galactosidase activities was determined.
Western Blot Analysis
Cell lysates were separated by electrophoresis on a sodium dodecyl sulfate (SDS)–polyacrylamide gel, and the proteins were then electroblotted onto a Hybond P polyvinylidene fluoride (PVDF) membrane. Protein expression was analyzed using different antibodies.
Antibodies
Primary antibodies were purchased for GAPDH and GFP (B2) from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase–conjugated secondary antibodies were purchased from Dako.
Results
Detection of NHP2L1 and U4 Interaction by Using the TR-FRET Assay
The interaction between NHP2L1 and U4 5′SL was previously characterized by using techniques such as NMR, 6 crystallography,3,6 coimmunoprecipitation, 21 and electrophoretic mobility shift assay (EMSA), 6 but those techniques are not suitable for a high-throughput screen. We therefore developed a TR-FRET assay suitable for HTS to identify small molecules capable of disrupting the interaction between NHP2L1 and U4 5′SL. TR-FRET obeys the same principles as standard FRET assays: when a suitable pair of fluorophores are brought within close proximity of one another, excitation of the first fluorophore (the donor) results in energy transfer to the second fluorophore (the acceptor). However, compared with conventional FRET techniques, TR-FRET has the advantage of a much lower background. 22 We used TR-FRET to monitor the interaction between NHP2L1 and U4 5′SL using terbium anti-His-tag antibody, which recognizes His-NHP2L1 and Cy5-U4 5′SL. The TR-FRET signals were stable from 30 to 300 min ( Suppl. Fig. S1 ) and were not affected by up to 5% DMSO. We selected 45 min of reaction for the further tests. Using this method, we were able to detect specific TR-FRET signals by subtracting background from a reaction mixture without His-tag NHP2L1 (used as control) ( Fig. 1A ), which confirmed the specific interaction of NHP2L1 and U4.
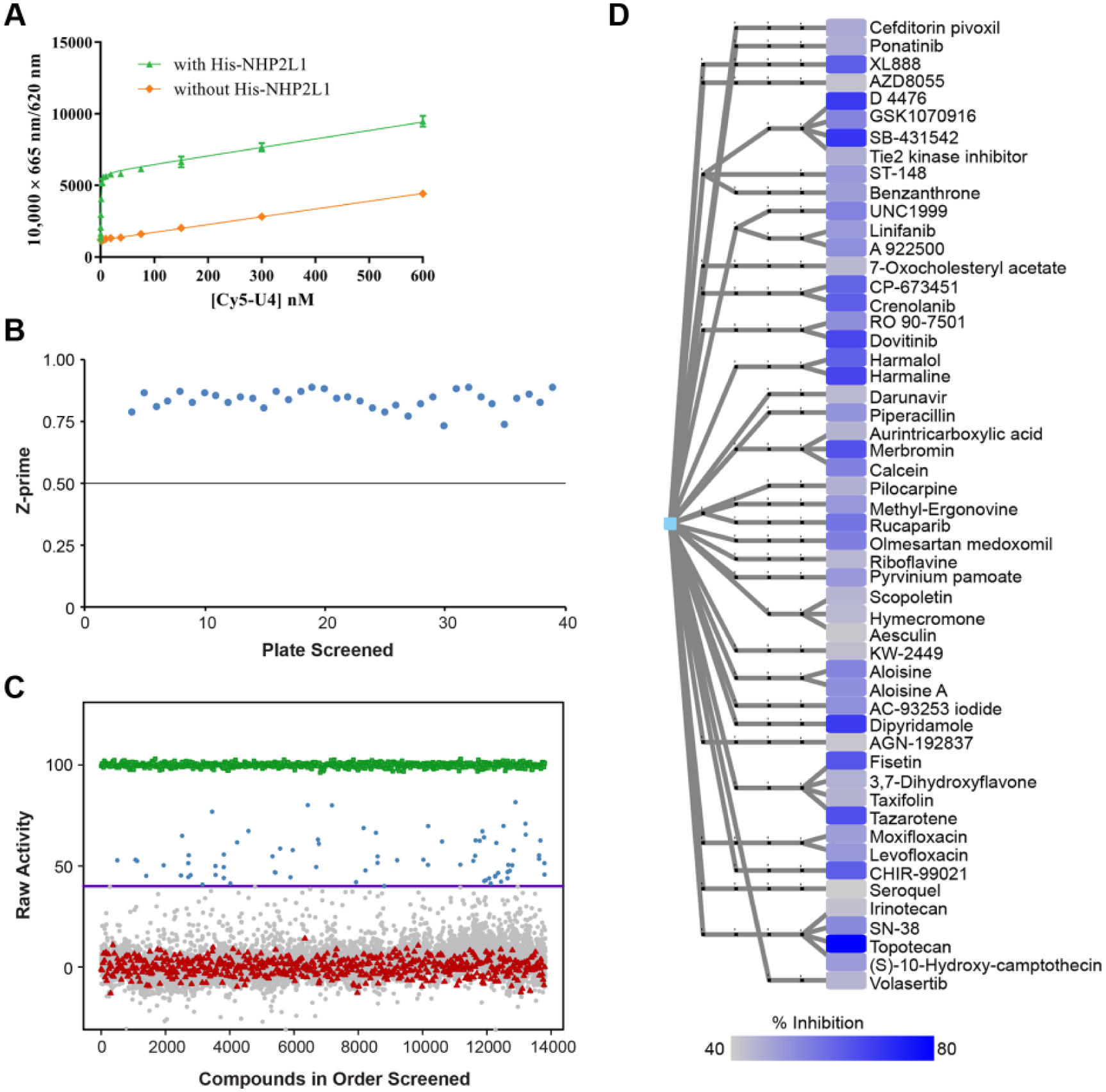
Specific interaction of NHP2L1 protein and U4 and results of the primary screen of small-molecule inhibitors of NHP2L1-U4 interaction. (
High-Throughput Screen of Small Molecules Interfering with NHP2L1 and U4 Interaction
To identify small molecules interfering with NHP2L1 and U4 interaction, a total of 10,173 bioactive compounds (4617 unique molecules), described in Materials and Methods, were first tested at a single concentration of 15 µM in the primary screening. The consistency and reproducibility of each assay plate were measured by the Z′ factor, which was 0.83 in average (
Fig. 1B
) beyond the minimum value (0.5) required for a valid assay.
23
Forty-six compounds (31 unique) achieved 50% inhibition, and 75 compounds (53 unique) achieved 40% inhibition (
Fig. 1C
), with topotecan hydrochloride giving the highest percentage of inhibition, 81.6% (
Suppl. Table S1
). Hierarchical cluster analysis was performed in order to visualize the distribution of these 53 compounds (
Fig. 1D
), revealing chemical diversity among these small molecules, with an average number of neighbors per node of 1.987. The 53 unique molecules (providing ≥40% inhibition at 15 µM) were validated by dose-responsive analysis covering 10 1:3 serially diluted concentrations. The concentration of each molecule required for 50% disruption of the complex NHP2L1-U4 (IC50) was determined (
Suppl. Table S1
). The dose response screening confirmed the primary screening hits with topotecan and other campthothecin derivatives (SN-38 and 10-OH-(S)-camptothecin) among the most potent inhibitors (
Fig. 2A
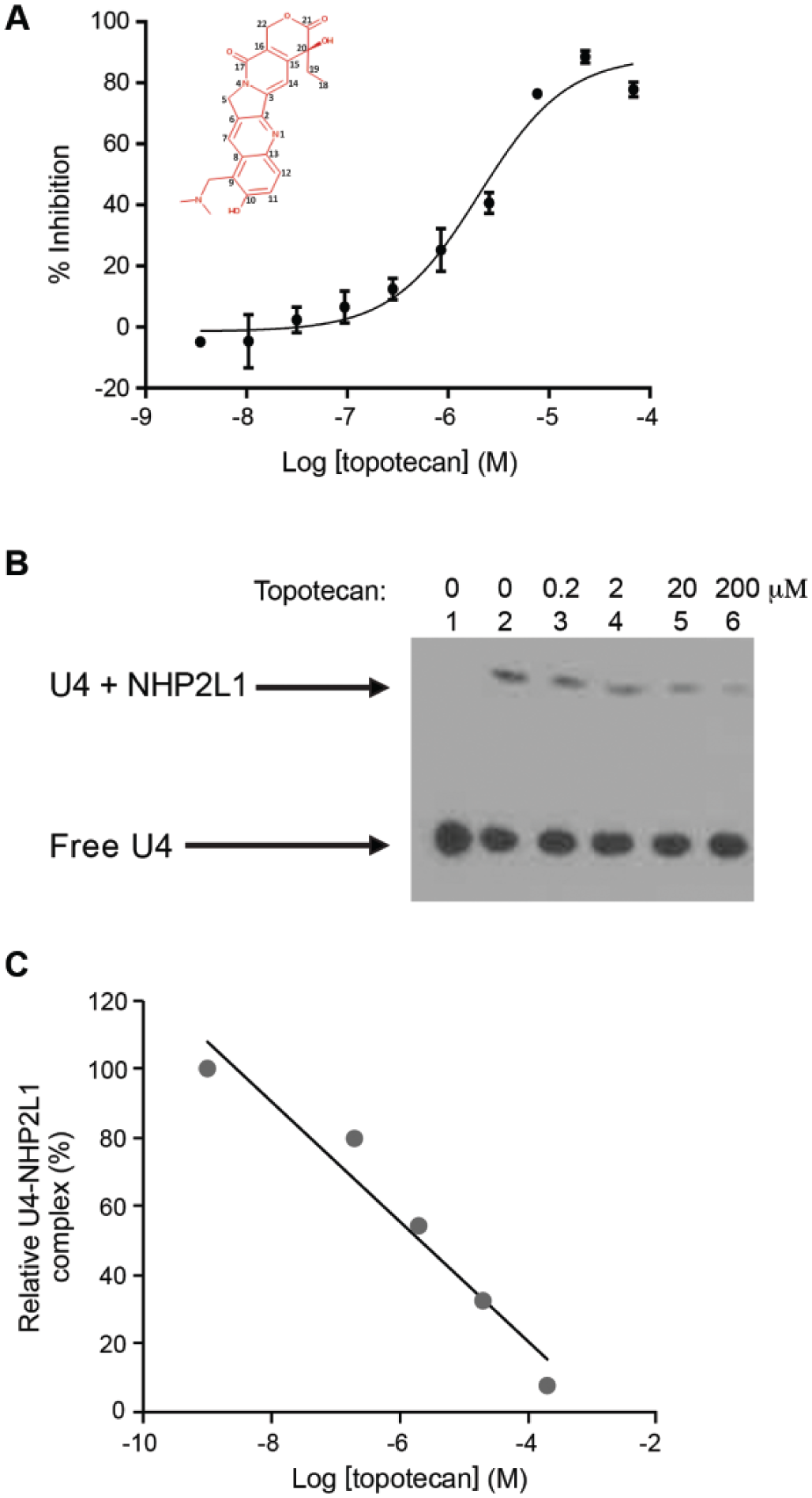
Analysis of the effect of topotecan on NHP2L1-U4 interaction. (
Characterization of Small Molecules Interfering with NHP2L1 and U4 Interaction
To confirm our findings, we used an orthogonal assay to test the inhibition of the NHP2L1-U4 interaction by topotecan. EMSA documented that NHP2L1 binds to U4 5′SL, as shown by the shift in the EMSA, and documented greater inhibition with increasing concentrations of topotecan, confirming results obtained by TR-FRET (
Fig. 2B,C
). To determine how topotecan interferes with the NHP2L1-U4 5′SL interaction, we determined whether topotecan binds to U4 5′SL and/or to NHP2L1 protein. SPR with immobilized NHP2L1 or U4 showed that topotecan binds to U4 in a time-dependent manner (
Fig. 3A
), with markedly less binding to NHP2L1 (
Fig. 3B
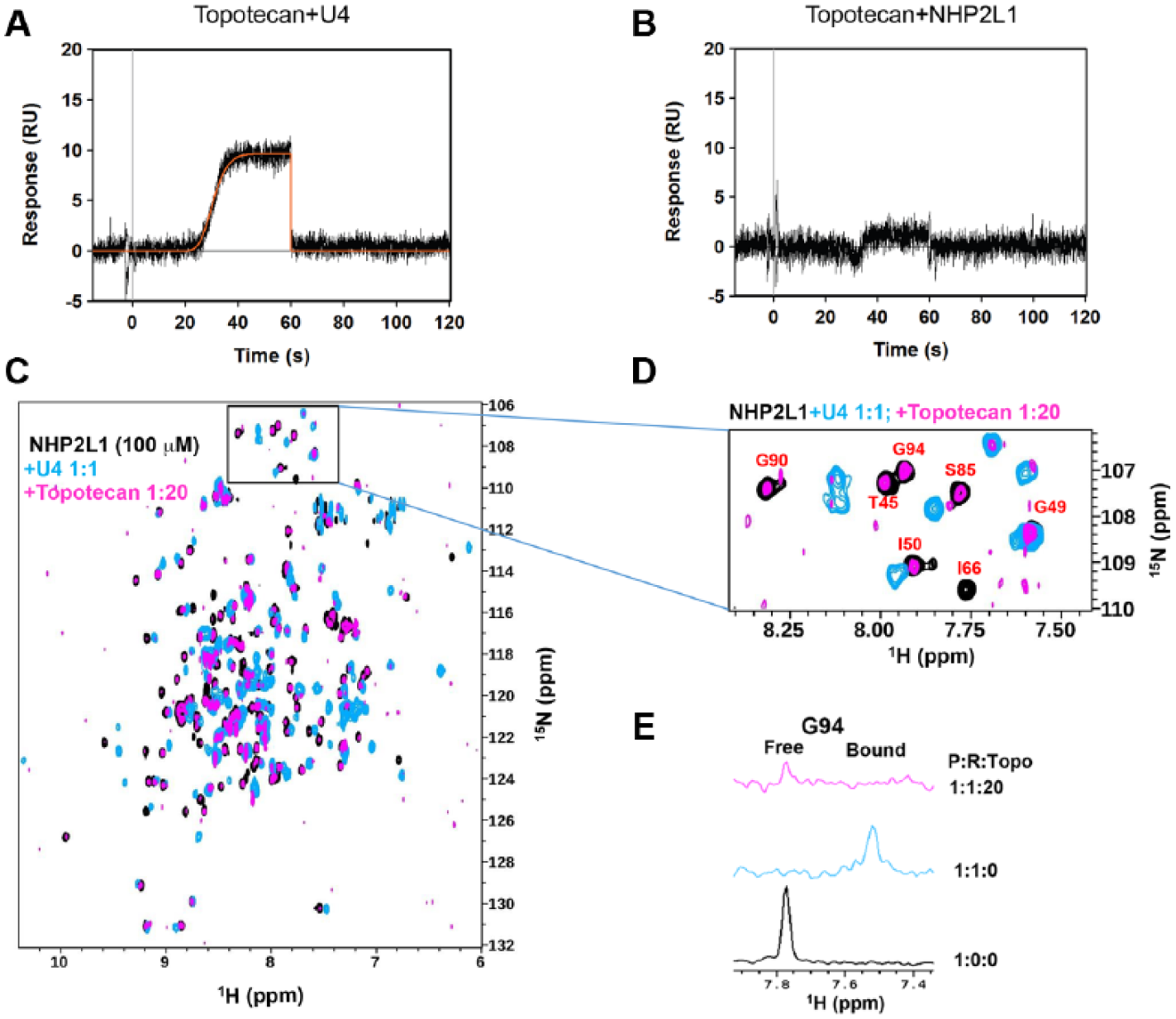
NMR and equilibrium analysis of topotecan binding to NHP2L1 protein or to U4 5′SL by SPR. Topotecan was injected at a maximum concentration of 50 µM using the OneStep gradient injection mode over surfaces of U4 5′SL (
Inhibition of NHP2L1 and U4 Interaction and Splicing
Because the interaction of NHP2L1 and U4 is required for splicing, we assessed the effect of topotecan on pre-mRNA splicing, using a previously reported splicing reporter vector, pTN23. 20 This construct contains reporter genes for β-galactosidase and luciferase separated by an exon–intron–exon cassette. Although β-galactosidase is constitutively expressed, luciferase is only expressed when splicing occurs. The ratio of luciferase to β-galactosidase activities is used to quantify splicing efficiency. These data show that topotecan significantly decreased splicing efficiency (p < 0.001) without significantly changing β-galactosidase activities (p = 0.4) ( Fig. 4A ). Overexpression of NHP2L1 ( Fig. 4B ) significantly reduced the effect of topotecan on splicing (p = 0.04) ( Fig. 4C ), consistent with topotecan inhibiting splicing by interfering with the NHP2L1 U4 interaction.
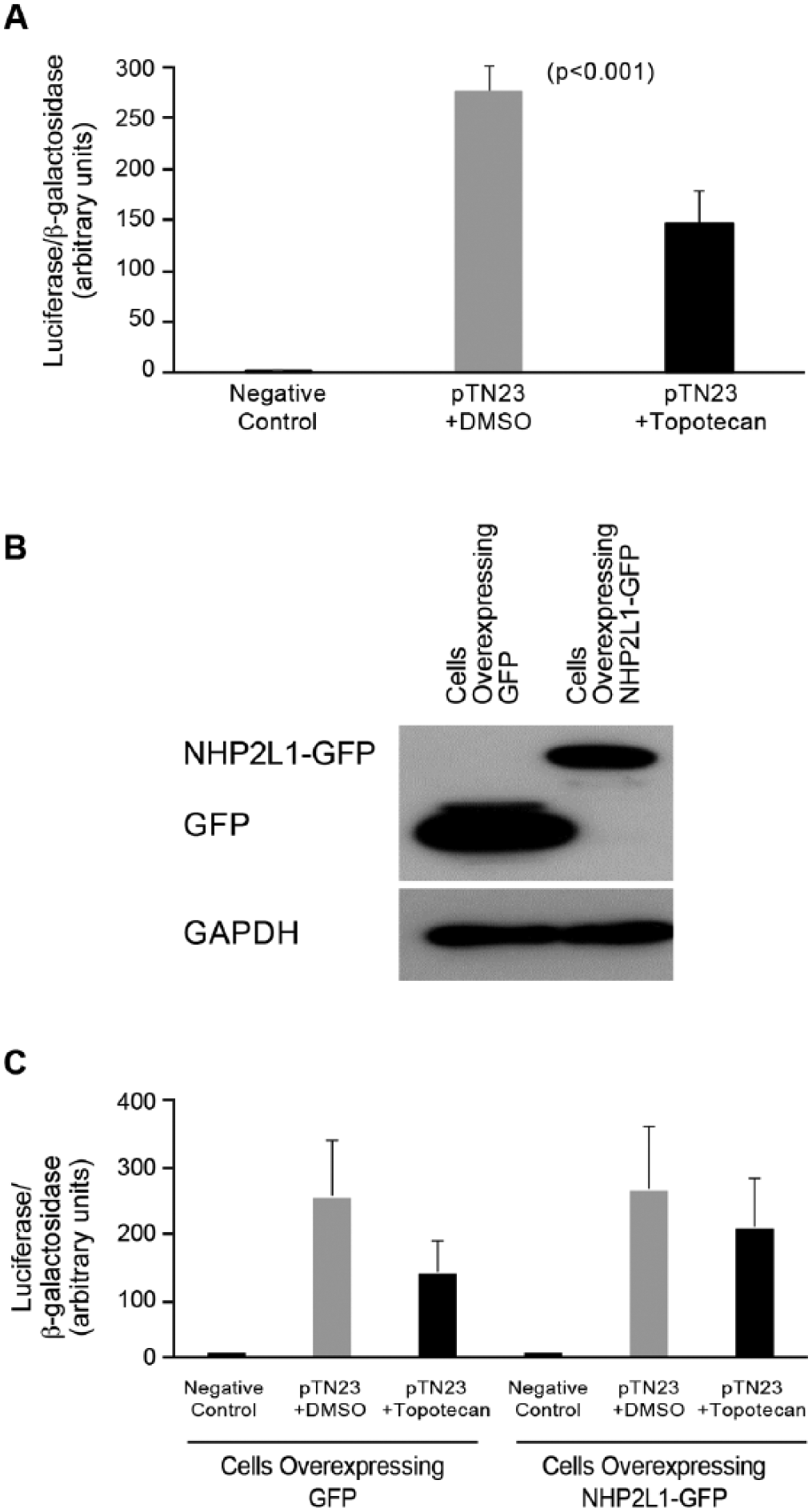
Inhibition of NHP2L1-U4 interaction and splicing. (
Discussion
Protein–nucleic acid interactions are essential for multiple cellular processes, and small-molecule disruptors of these interactions could be powerful tools for investigation of these biological processes and could have potential as therapeutics. 25
In the present study, we have successfully developed a TR-FRET assay to assess the interaction between NHP2L1 protein and U4 (RNA), two key players in splicing. This assay confirms the specific and high binding affinity of NHP2L1 to U4, as previously shown,3,6,21 and provides a tool for the discovery of small molecules that inhibits this process.
The identification of compounds that perturb protein–nucleic acid interactions is extremely challenging, 25 and there is a paucity of reports of small-molecule inhibitors of any protein–DNA 26 or protein–RNA interactions. 27
By using a high-throughput TR-FRET screen, we have identified for the first time several small-molecule inhibitors that disrupt the complex formed by NHP2L1 and U4. These small molecules are chemically diverse, reflecting the overall diversity of the compound library, a good predictor for library performance.28–30 Moreover, the presence of several distinct chemical small molecules among our top hits suggests that there may be different mechanisms by which they interfere with NHP2L1 and U4. It is possible that some could bind to the protein and/or the RNA with different interaction sites, as previously shown in other protein and RNA interactions. 27 It will be important for future experiments to determine precisely how the different small molecules disrupt the interaction between NHP2L1 and U4, and if they inhibit a broader set of nucleic acid protein interaction. We have focused on our top hit topotecan, and our results show that it binds to the U4 (RNA), consistent with previous findings that very few compounds target the protein in an RNA–protein complex. 31 Topotecan and related camptothecin derivatives are widely used anticancer agents that exert their pharmacologic effects primarily by inhibiting topoisomerase I.32,33 Topoisomerase interacts with DNA, whereas NHP2L1 interacts with RNA, and the mechanism by which topotecan and camptothecin derivatives inhibit topoisomerase I likely differs from the way they interfere with NHP2L1 and U4. Indeed, topotecan does bind to DNA alone at high concentrations but not to topoisomerase alone; 34 rather, it stabilizes the transient covalent topoisomerase I–DNA complex. 35 Our new findings show that topotecan binds to the NHP2L1-U4 complex (as shown by NMR) and binds U4 alone, but it disrupts this interaction instead of stabilizing it. The other difference is that while only the 10-OH camptothecin derivatives, namely, SN38, topotecan, and N-demethyl-topotecan, are able to disrupt the interaction of U4 and NHP2L1, camptothecin and its derivative, irinotecan, which are known to inhibit topoisomerase I,34,36–39 were less effective at disrupting the NHP2L1 and U4 interaction. Attempts to cocrystallize topotecan and NHP2L1-U4 did not produce crystals, precluding further structural studies at this time.
The current study has limitations, including that we used only the pTN23 splicing reporter vector and not endogenous RNA to show the effects of topotecan on splicing. Also, further in vivo studies are needed in order to determine the concentrations of topotecan and the other small molecules that are required to alter the in vivo function of NHP2L1-U4 and, by extension, splicing, and to determine the impact of this inhibition on the known and potentially new therapeutic effects of these small molecules.
In summary, we have identified the first documented inhibitors of the interaction between NHP2L1 and U4, providing new insights into how topotecan interferes with this interaction. Taken together, our data reveal new functions of known drugs, which could facilitate the development of therapeutics that modify splicing and thereby gene function.
Footnotes
Acknowledgements
We thank Darcie Miller, Sivaraja Vaithiyalingam, Amanda Nourse, and Duane Currier for their technical assistance. We thank Elizabeth Stevens for preparation of the figures. We thank Ian C. Eperon (University of Leicester) for the pTN23 plasmid. We thank the shared research resources of the St. Jude Children’s Research Hospital Comprehensive Cancer Center.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported in part by National Institutes of Health grants R37 CA36401 (W.E.E. and M.V.R.), U01 GM92666 (M.V.R. and W.E.E.), and P50 GM115279 (W.E.E. and M.V.R.); Comprehensive Cancer Center grant CA21765, from the National Cancer Institute; and the American Lebanese Syrian Associated Charities (ALSAC).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.